
Abstract
Emerging evidence points towards a regulatory role of the circadian clock in alternative splicing (AS). Whether alterations in core-clock components may contribute to differential AS events is largely unknown. To address this, we carried out a computational analysis on recently generated time-series RNA-seq datasets from three core-clock knockout (KO) genes (ARNTL, NR1D1, PER2) and WT of a colorectal cancer (CRC) cell line, and time-series RNA-seq datasets for additional CRC and Hodgkin’s lymphoma (HL) cells, murine WT, Arntl KO, and Nr1d1/2 KO, and murine SCN WT tissue. The deletion of individual core-clock genes resulted in the loss of circadian expression in crucial spliceosome components such as SF3A1 (in ARNTLKO), SNW1 (in NR1D1KO), and HNRNPC (in PER2KO), which led to a differential pattern of KO-specific AS events. All HCT116KO cells showed a rhythmicity loss of a crucial spliceosome gene U2AF1, which was also not rhythmic in higher progression stage CRC and HL cancer cells. AS analysis revealed an increase in alternative first exon events specific to PER2 and NR1D1 KO in HCT116 cells, and a KO-specific change in expression and rhythmicity pattern of AS transcripts related to cancer hallmarks genes including FGFR2 in HCT116_ARNTLKO, CD44 in HCT116_NR1D1KO, and MET in HCT116_PER2KO. KO-specific changes in rhythmic properties of known spliced variants of these genes (e.g. FGFR2 IIIb/FGFR2 IIIc) correlated with epithelial-mesenchymal-transition signalling. Altogether, our bioinformatic analysis highlights a role for the circadian clock in the regulation of AS, and reveals a potential impact of clock disruption in aberrant splicing in cancer hallmark genes.
Similar content being viewed by others


Introduction
Alternative splicing (AS) is a central event in transcriptional regulation leading to distinct mRNAs being generated from a single gene, and is thus a major contributor for protein diversity1,2. Approximately 95% of human genes undergo AS in a tissue type-specific manner3, and AS resulting products lead to the production of proteins involved in numerous biological processes including cell cycle4 and metabolism5. Splicing factors (SFs) mark the splice sites, facilitate spliceosome assembly during the splicing process, and can regulate AS decisions and thus the resulting splicing product(s)6. Dysregulations in splicing account for the production of non-functional, or with the incorrect function, protein products with an impact on the onset and progression of several diseases among which cancer7,8,9,10,11,12. A systematic evaluation of 11 different tumour types revealed a subset of transcript isoform switches associated with gain and/or loss of protein domains, which are associated with the hallmarks of cancer13. Known examples of AS failures related to cancer include the aberrant splicing in the Vascular Epithelial Growth Factor (VEGF), essential for angiogenesis, resulting in one transcript isoform with anti-angiogenic properties in cancer14. Another example is CD44, which encodes for a cell-surface protein and can act as both tumour-suppressing, as well as invasiveness-promoting molecule, depending on its different splice variants15. In addition, alterations in the expression of several SFs can impact the splicing of their target genes, for e.g., the differential expression of the splicing factor Serine and Arginine Rich Splicing Factor 6 (SRSF6) affects the splicing of several oncogenes including the Insulin Receptor (INSR), MAPK Interacting Serine/Threonine Kinase 2 (MKNK2) and Discs Large MAGUK Scaffold Protein 1 (DLG1) leading to the production of oncogenic isoforms, or to the reduction of the tumour-suppressing isoforms of those genes16. Also, aberrant AS in other members of the SR splicing factor and SR-like families, including SRSF4 and Transformer 2 Beta Homolog (TRA2β) have been shown to regulate cancer proliferation and metastasis by affecting the splicing of genes involved in epithelial-mesenchymal transition (EMT, e.g., CD44) in breast cancer17. Mounting evidence suggests as well an association between AS regulation and the circadian clock in various organisms in particular in mammals18,19.
Circadian rhythms are driven by an endogenous oscillator with an approximate period of 24 h and regulate the timing of cellular processes in most organisms20. These rhythms are generated by a Transcriptional-Translational Feedback Loop (TTFL) in which Brain and muscle ARNT-Like 1 (BMAL1), also known as ARNTL forms a heterodimer with Circadian Locomoter Output Cycles Kaput (CLOCK) and regulates the transcriptional activation of Period 1/2/3 (PER 1/2/3), and Cryptochrome 1/2 (CRY 1/2), which inhibit BMAL1-CLOCK-mediated transcription21. In addition, Nuclear Receptor Subfamily 1 Group D Member 1, 2 (NR1D1/2) and RAR Related Orphan Receptor A, B, C (RORA/B/C) regulate BMAL1 transcription and contribute to the fine-tuning of its expression. The circadian clock regulates the expression of genes and proteins related to several cancer hallmarks like cell proliferation, metabolism, and DNA damage22. Disruption of circadian rhythms is associated with increased risk of cancer, including prostrate, breast, and colon cancer23.
Recent work points to a complex cross-talk between the circadian clock and AS events underlying different cancer types24,25,26,27,28,29,30,31. Changes in amplitude and peak phase of SFs including hnRNPs and RBM proteins in colorectal cancer (CRC) cells were correlated with altered splicing outcome of target genes like VEGFA and CD4424. A later study in the same cell lines reported rhythmic alternative splicing events of cancer-related genes like Cold inducible RNA binding protein (CIRBP) and Poly(RC) Binding Protein 2 (PCBP2)25. Clock-mediated alternative splicing events in the cell differentiation marker Interferon Regulatory Factor 4 (IRF4) were reported in an in vitro model of Hodgkin lymphoma (HL)26. In the same HL cell lines, clock-mediated changes in the expression of transcript isoforms were reported in elements associated with the tumour necrosis factor (TNF) pathway, which controls cell proliferation, migration, and apoptosis27. Rhythmic alternative splicing of the splicing factor U2AF26 controls the stability of Per1 in mice30. A splice variant of human BMAL1 (hBMAL1a) is known to act opposite to its canonical isoform (hBMAL1b) and functions as a negative regulator of the circadian clock31. However, the impact in alternative splicing events resulting from clock alterations in cancer remains unexplored.
In this study, we analysed time-course RNA-seq datasets generated from three HCT116 core-core knockouts (ARNTL, PER2, NR1D1) and wild type (WT) cells, to investigate the impact of clock disruption on AS events in CRC cell lines. In addition, we retrieved publicly available time-course RNA-seq datasets from human CRC cell lines (SW480, SW620), HL cell lines (HDMYZ, L1236), and mouse Arntl KO, Nr1d1/2 KO, and SCN (Suprachiasmatic nucleus) WT tissue. Our study revealed that deletion of core-clock genes (ARNTL, PER2 or NR1D1) altered the circadian rhythmic pattern of SFs, as well as the resulting AS events in a KO-specific manner. We observed a common rhythmicity loss of crucial SFs such as U2 Small Nuclear RNA Auxiliary Factor 1(U2AF1) in all three HCT116KO, as well as in SW620, and L1236 cells. Moreover, we found a phase shift in circadian transcripts of SFs such as Receptor of Activated C Kinase 1 (RACK1), Heterogeneous nuclear ribonucleoprotein D (HNRNPD) involved in pre-mRNA processing and Serine and Arginine rich splicing factor 5 (SRSF5), component of the spliceosome machinery, in HCT116_ARNTLKO, HCT116_NR1D1KO, and HCT116_PER2KO vs. WT, respectively. This led to a distinct pattern of AS events (specifically in gain and loss of splicing events) in HCT116KO datasets vs. HCT116_WT. Differential rhythmicity analysis revealed that the majority of phase shifted transcript pairs consisted of protein-coding biotypes. In HCT116KO cells, genes with phase-shifted transcript pairs were related to apoptotic signalling (HCT116_ARNTLKO), macrophage proliferation (HCT116_NR1D1KO), and negative regulation of chromatin organization (HCT116_PER2KO). Moreover, we identified differentially rhythmic transcript isoform pairs of target genes associated with cancer hallmarks. These included Casein Kinase 2 Alpha 1 (CSNK2A1), related to invasion, metastasis, tumour proliferation, in HCT116_WT and HCT116KO. We further identified phase shifted transcript pairs from cancer hallmarks-related genes e.g., transcripts pairs from MET Proto Oncogene (MET), which controls several cancer hallmarks, in HCT116_PER2KO. Finally, we observed KO-specific altered expression and rhythmicity pattern of transcripts associated with cancer hallmarks related genes such as in Fibroblast Growth Factor Receptor 2 (FGFR2), which promotes tumour progression seen in HCT116_ARNTLKO, Caspase 8 (CASP8) which regulates extrinsic apoptotic pathway seen in HCT116_NR1D1KO, and HRas proto-oncogene, GTPase (HRAS) involved in cell proliferation, growth and apoptosis detected in HCT116_PER2KO. The evaluation of coding exon sequences for selected cancer hallmarks revealed the altered rhythmicity in spliced variants associated with cancer progression such as skipped exon 14 variant of MET. Our findings point to a direct impact of circadian clock disruption in AS events related to cancer hallmarks, which may support cancer onset or progression.
Results
Rhythmicity analysis of mammalian RNA-seq datasets showed a KO-specific effect in the circadian phenotype both at the gene and at the transcript levels
In mammals, many different biological processes like metabolism or the immune system are regulated by the endogenous circadian clock32. To investigate the effect of clock disruption on the circadian phenotype (circa 24 h rhythmicity) and the subsequent effects in biological processes, we analysed recent datasets from our group for three core-clock knockout (KO) cell lines (HCT116_ARNTLKO, HCT116_NR1D1KO, and HCT116_PER2KO) and compared their gene expression pattern to the corresponding HCT116_WT (Fig. 1a, Supplementary Fig. 1d). Previous in vitro and in vivo studies showed an effect of ARNTL, PER2 and NR1D1 KO on the rhythmic expression of core-clock genes (mostly via bioluminescence recordings, or mouse studies)33,34, which are in agreement with our findings regarding the loss (ARNTL KO) and period alterations (PER2 and NR1D1 KO) in the respective oscillation profiles. Although, it is important to notice that the assessment of rhythmicity via in vitro bioluminescence measurements of ARNTL promoter activity, or via in vivo analysis of movement in mice, is not directly comparable with RNA-seq time course measurements.

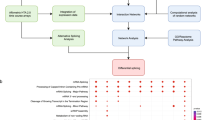
a Schematic representation of the pipeline for RNA-seq data analysis. The input data consisted of 222 samples from eight mammalian time-course RNA-seq data. Murine datasets from Per1/2 KO (GSE171975) and Cry1/2 KO (GSE135898) were only analysed at the gene level. b–d Classification of 24 h rhythmic features in HCT116 datasets at (b) gene level and (c) transcript level showed a decline in the total number of circadian expressed genes/transcript-sets and changes in their biotypes due to core-clock disruption. d Scatter plot represents each of the HCT116 WT and KO cells according to the total number of 24 h rhythmic genes (x-axis) and the total number of 24 h rhythmic transcripts (y-axis).
To pinpoint specific changes due to the KO of core-clock genes, we additionally gathered publicly available time-course RNA-seq datasets for human CRC cell lines (SW480: derived from the primary tumour, SW620: derived from a metastasis site, from the same patient), HL cell lines (HDMYZ: stage III, L1236: stage IV), murine WT and Arntl KO, murine WT and Nr1d1/2 KO, murine SCN tissue, murine WT and Cry1/2 KO, and murine WT and Per1/2 KO, resulting in a total of 222 samples (Fig. 1a). To examine if the rhythmicity pattern varies when analysed at the transcript level vs gene level, we extracted 24 h rhythmic sets both at gene and transcript levels in six-mammalian time course RNA-seq datasets (Fig. 1b–d, Supplementary Figs. 1–3). A higher number of 24 h rhythmic features were found at transcript level vs gene level in all datasets. The number of circadian (~24 h period) expressed genes and transcripts decreased in all three HCT116KO vs. WT (Fig. 1b–d). At the gene level, we identified 1261, 443, 866 and 525 circadian gene sets in HCT116_WT, HCT116_ARNTLKO, HCT116_NR1D1KO, and HCT116_PER2KO, respectively (Fig. 1b; left panel). We observed KO-specific loss in circadian expression of crucial genes such as Keratin 8 (KRT8), which maintains cellular structural integrity, Transmembrane and Coiled-Coil Domain Family 1 (TMCC1), which promotes endoplasmic reticulum-associated fission, and HECT and RLD Domain Containing E3 Ubiquitin Protein Ligase 3 (HERC3), which enables protein-transferase activity in HCT116_ARNTLKO, HCT116_NR1D1KO, and in HCT116_PER2KO, respectively. At transcript level, we observed 5499 (corresponding to 4063 genes), 3039 (2485 genes), 3519 (2899 genes), 2850 (2345 genes) circadian transcripts in HCT116_WT, HCT116_ARNTLKO, HCT116_NR1D1KO, and HCT116_PER2KO, respectively (Fig. 1c; left panel). Among KO-specific loss of circadian expressed transcripts, we saw a loss of circadian transcript expression of C-Terminal Binding Protein 1 (CTBP1), which is a transcriptional repressor, HRAS an oncogene involved in signal transduction pathways, and Calmodulin 1 (CALM1), which controls several proteins including enzymes and ion channels, through calcium-binding. All these genes modulate cellular proliferation in HCT116_ARNTLKO, HCT116_NR1D1KO, and in HCT116_PER2KO, respectively.
Moreover, differences in the percentage of biotypes (e.g., protein-coding biotype) were also observed between circadian gene and transcript sets (Fig. 1b, c; right panel). For example, 1089 genes (~86%) and 3509 transcripts (~63%) with protein-coding biotype were circadian expressed in HCT116_WT. The comparison of rhythmicity at gene and transcript levels in HCT116 datasets showed a roughly linear relationship between the number of circadian genes and circadian transcripts (Fig. 1d). Similar differences in gene- and transcript-level rhythmicity phenotype were also seen in other human CRC cell lines (SW480; SW620), HL cell lines (HDMYZ; L1236), murine WT and Arntl KO, murine WT and Nr1d1/2 KO, and murine SCN WT tissue datasets (Supplementary Figs. 2, 3). Within the set of circadian rhythmic genes, 846 genes (~67%) in HCT116_WT, 192 genes (~43%) in HCT116_ARNTLKO, 521 genes (~60%) in HCT116_NR1D1KO, and 274 genes (~52%) in HCT116_PER2KO were rhythmic both at gene and transcript levels (Supplementary Fig. 1a). We further analysed circadian discrepancies between the expression pattern of some genes and their corresponding transcripts, for e.g., HIF1A was found to be circadian expressed whereas its transcripts were arrhythmic in HCT116_WT. Here, the summarization of arrhythmic transcripts resulted in a rhythmic gene. Minichromosome Maintenance Complex Component 2 (MCM2) was rhythmic both at gene and transcript levels in HCT116_WT. In contrast, Interleukin 18 (IL18) was identified as arrhythmic at the gene level whereas its transcripts were circadian expressed in HCT116_WT. In this case, the summarization of rhythmic transcripts resulted in an arrhythmic gene (Supplementary Fig. 1a; lower panel). Further, we analysed the changes in the number of transcripts per expressed gene and the number of ~24 h transcripts per gene in HCT116KO vs. WT (Supplementary Fig. 1b, c). The number of expressed transcripts per gene showed variation in all three HCT116KO vs. WT. For instance, 28 transcripts of Heterogeneous Nuclear Ribonucleoprotein H1 (HNRNPH1), an RNA binding protein, were expressed in HCT116_WT. Whereas, 25 transcripts from HNRNPH1 were expressed in HCT116_ARNTLKO, 21 transcripts in HCT116_NR1D1KO, and 24 transcripts in HCT116_PER2KO. Out of the expressed transcripts of HNRNPH1, 5 transcripts in HCT116_WT and 1 transcript in HCT116_PER2KO were circadian expressed. Altogether, our analysis confirmed previous findings from our group25, and showed that indeed a transcript-level analysis can provide new results that an be masked by gene-level analysis. Moreover, our analysis also shows that the KO of core-clock genes altered the ratio, as well as the circadian rhythmicity expression pattern of genes and transcripts in a KO-specific manner. Hence, we focused on transcript level in our subsequent analysis.
Alterations in core-clock elements influenced the rhythmic expression of splicing factors
To investigate possible alteration in SFs rhythmicity resulting from clock disruption, we examined the expression of SF transcripts and their rhythmic properties (Fig. 2). We compiled a total of 534 human SF genes from previously published studies24,25,26,35,36, Spliceosome database37, and SpliceAid 2 database38 (Supplementary file 1), and generated orthologous sets of human SFs for mouse (521 genes) using Ensembl Biomart39 (Supplementary file 2), and mapped the curated list of SFs against the circadian transcripts extracted from the RNA-seq datasets as depicted in the phase sorted heatmaps and acrophase bin plots (Fig. 2). We saw a decrease in the number of circadian expressed SF transcripts and also changes in their acrophase distribution in HCT116KO compared to HCT116_WT cells (Fig. 2a; upper panel). HCT116_PER2KO showed the lowest number of circadian rhythmic SF transcripts (143 transcripts; 104 genes) followed by HCT116_NR1D1KO (188 transcripts; 135 genes), HCT116_ARNTLKO (204 transcripts; 150 genes), and HCT116_WT (341 transcripts; 216 genes) datasets. Both HCT116_NR1D1KO and HCT116_PER2KO showed a shift in the SFs acrophase when compared with HCT116_WT cells.

Median normalized phase-sorted heatmaps (left) and acrophase bins (right) of SFs circadian transcripts (tr.) in (a) HCT116_WT and HCT116KO cells (upper panel), SW480 and SW620 cell lines (lower left panel), HDMYZ and L1236 cell lines (lower right panel), b murine WT and Arntl KO from distal bronchiolar epithelium tissue (upper left panel) and from primary tracheal epithelial cells (upper right panel), murine WT and Nr1d1/2 KO from epithelial cells (lower left panel), and murine SCN WT tissue (lower right panel).
To find a possible association between the decrease in the number of rhythmic SF transcripts and cancer progression, we analysed other CRC cell lines (SW480, SW620) and HL cell lines (HDMYZ, L1236). Indeed, we observed a decrease in the number of rhythmic SF transcripts in the metastasis-derived colon cancer (SW620: 216 transcripts; 153 genes) and in stage IV HL (L1236: 215 transcripts; 156 genes) as compared with the primary colon cancer (SW480: 378 transcripts; 212 genes) and stage III HL (HDMYZ: 325 transcripts; 201 genes) cell lines, respectively (Fig. 2a; lower panel). Also, the shift in acrophase distribution of circadian rhythmic SF transcripts was observed in both SW620 and L1236 cell lines compared with their controls. To further examine the impact of each KO on SFs rhythmicity, we examined rhythmic changes in SFs transcripts within murine RNA-seq datasets (Fig. 2b). In contrast, slightly higher number of rhythmic SF transcripts were observed in Arntl KO model of distal bronchiolar epithelium (208 transcripts; 151 genes) and primary tracheal epithelial cells (48 transcripts; 44 genes) compared with their WT (175 transcripts; 138 genes and 46 transcripts; 45 genes, respectively) (Fig. 2b; upper panel). Whereas, Nr1d1/2 KO mouse model showed lower number of rhythmic SF transcripts (101 transcripts; 87 genes) compared with WT (107 transcripts; 87 genes) (Fig. 2b). SCN tissue, the master clock, showed the highest number of rhythmic SF transcripts (767 transcripts; 384 genes) in mouse WT datasets (Fig. 2b; lower panel, for the complete list of circadian transcripts and their acrophases see Supplementary Fig. 4). In addition, we also observed loss in 24 h rhythmic SFs due to Cry1/2 deletion (Supplementary Fig. 5b; left panel) and Per1/2 deletion (Supplementary Fig. 5b; right panel) vs. their controls in mouse datasets.
The KOs resulted in a shift in the acrophase bin distribution of circadian rhythmic SF transcripts in particular for NR1D1 and PER2 KO. We further carried out pairwise comparisons of circadian SF transcripts between HCT116_WT and HCT116KO to identify commonly circadian rhythmic SF transcripts, and to obtain differentially rhythmic SF transcripts (phase shift ≥3 h) in the HCT116KO compared with HCT116_WT (Fig. 3a–c). A total of 25 circadian SFs transcripts (23 genes) were found rhythmic in HCT116_WT and HCT116_ARNTLKO, out of those, 7 transcripts showed differential rhythmicity (Fig. 3a). Besides alterations in the expression profile of SF transcripts, we observed phase shift in other transcripts, as well as loss of rhythmic oscillations in PER1, PER2 and DBP gene with ARNTL disruption in HCT116 cells (Supplementary Fig. 6a).

a–c Circular plots depict the distribution of peak phases of overlapping differentially rhythmic SF transcripts between (a) HCT116_WT and HCT116_ARNTLKO, (b) HCT116_WT and HCT116_NR1D1KO, and (c) HCT116_WT and HCT116_PER2KO. d–h Categorial heatmaps represent the loss of rhythmic SFs in (d) HCT116KO vs. WT, (e) SW620 vs. SW480, (f) L1236 vs. HDMYZ, (g) mouse Arntl KO vs. WT in distal bronchiolar epithelium (left) and primary epithelial cells (right), and (h) Nr1d1/2 KO vs. WT. Green colour indicates SFs with at least one ~24 h transcript whereas grey colour indicates SFs with no rhythmic transcript. The numbers above the categorial heatmaps indicate the total number of SFs (genes) with rhythmic transcript(s).
Differential rhythmicity analysis between HCT116_WT and HCT116_ARNTLKO cells resulted in a higher number of transcripts with phase difference greater than 9 h whereas for most differentially rhythmic splicing factor transcripts a phase difference between 6 and 9 h was observed (Supplementary Fig. 6b–f).
For HCT116_WT vs. HCT116_NR1D1KO, we found 33 circadian rhythmic transcripts (28 genes) and 11 circadian rhythmic transcripts to be differentially rhythmic (Fig. 3b). The comparison of HCT116_WT and HCT116_PER2KO resulted in a total of 28 circadian expressed transcripts (26 genes) and out of those 4 transcripts showed differential rhythmicity (Fig. 3c). Only a single transcript from the gene survival of motor neuron 1 (SMN1), which plays a catalyst role in the assembly of small nuclear ribonucleoproteins (snRNPs) was found to be rhythmic in all HCT116 WT and KO datasets and it showed the same phase, but different amplitude within HCT116KO. Out of seven differentially rhythmic transcripts in HCT116_ARNTLKO vs. WT, were SFs like RACK1, which is a component of the ribosomal subunit and Splicing factor proline and glutamine rich (SFPQ), essential at the early stages of spliceosome formation (Fig. 3a). For HCT116_NR1D1KO, differential rhythmic SFs transcripts were involved in pre-mRNA processing like HNRNPD and translational repressor like Cold Inducible RNA Binding Protein (CIRBP) (Fig. 3b). Whereas in HCT116_PER2KO, we found differentially rhythmic transcripts of SFs like SRSF5, which is a part of the spliceosome machinery (Fig. 3c).
To further understand the discrepancies in SFs rhythmicity, we extracted a list of SFs with at least one circadian transcript in HCT116_WT, SW480, HDMYZ, murine WT (from Arntl KO), murine WT (from Nr1d1/2 KO), and mapped them with their corresponding knockout/altered condition (e.g. primary tumour- vs. metastasis-derived cells). We produced categorical heatmaps to visualize the loss of rhythmically expressed SFs in each dataset compared with controls (Fig. 3d–h). 216 SFs with at least one rhythmic transcript were observed in HCT116_WT, and less than 50% were rhythmic in HCT116KO (Fig. 3d; left panel). Out of 216 SFs with at least one circadian transcript in HCT116_WT, we observed 85 SFs in HCT116_ARNTLKO, 79 SFs in HCT116_NR1D1KO, and 66 SFs in HCT116_PER2KO with at least one circadian transcript. Among the three HCT116KO cells, there were 69 common SFs with no rhythmic transcript (Fig. 3d; right panel). Similarly, the human CRC cell line (SW620), HL cell line (L1236), murine Arntl KO, and murine Nr1d1/2 KO also showed a loss of rhythmic SFs compared with their controls (Fig. 3e–h). All three HCT116KO cells showed loss of rhythmicity for U2AF1, an important RNA splicing mediator gene, which also lost rhythmicity in SW620 and L1236 cells, as compared to their corresponding lower grade cancer cell lines. We observed KO-specific loss of rhythmicity in different SF transcripts such as RNA Binding Motif Protein 5 (RBM5), Splicing factor 3a subunit 1 (SF3A1), and Serine and Arginine Rich Splicing Factor 3 (SRSF3), involved in the spliceosome machinery, uniquely in HCT116_ARNTLKO. While, HCT116_NR1D1KO resulted in rhythmicity loss of SNW Domain Containing 1 (SNW1), another spliceosome component, and HCT116_PER2KO resulted in the loss of rhythmic transcripts of HNRNPC, involved in pre-mRNA processing. Taken together, our results point to alterations in rhythmic properties of SFs in HCT116 cell lines, which were KO-specific and were also present in datasets derived from other cells and tissues, upon perturbation of the core-clock.
Disruption of circadian clock elements influenced alternative splicing events across human and murine datasets
Aberrant AS events have been reported to be associated with different types of cancer40. We investigated the impact of clock disruption in our datasets on AS events with a potential impact in cancer onset and progression. We used SUPPA to calculate the proportion spliced-in (PSI) of seven basic modes of AS: (1) Alternative 3′ splice site (A3-event), (2) Alternative 5′ splice site (A5-event), (3) Alternative first exon (AF-event), (4) Alternative last exon (AL-event), (5) Mutually exclusive event (MX-event), (6) Retention intron event (RI-event), and (7) Skipping exon event (SE-event).
We determined the overall number of genes showing significant AS events (0.1< PSI < 0.9), and found that most genes had a commonly predominant AS event (SE-event) in all human RNA-seq datasets (Supplementary Fig. 7). To further evaluate the changes in AS events caused by the KO of core-clock genes, we classified the set of genes that were alternatively spliced in the WT, but not in the KOs (following the PSI criteria), as “event loss” in its corresponding KO group. Similarly, uniquely identified sets of genes alternatively spliced in a KO cell line were labelled as “event gain” (see “Methods” for details). Bar plots were used to depict the total number of genes in the categories loss and gain for each type of splicing event (Fig. 4). We mapped the list of AS genes with the genes containing at least one circadian transcript and their biotypes. For better visualization, we separated the genes according to whether or not they contained transcripts with protein-coding biotype. All AS events in HCT116KO cells except for the AF-event in HCT116_NR1D1KO and HCT116_PER2KO showed higher number of genes present in the loss of splicing events vs. gain of splicing events (Fig. 4a–c). However, a large increase in the number of genes with AF-gain was observed in both HCT116_NR1D1KO (gain vs. loss: 75.8% vs. 24.4%) and HCT116_PER2KO (gain vs. loss: 74.2% vs. 25.7%) (Fig. 4b–c). Interestingly, higher number of genes with gain in splicing events were observed in the metastasis CRC cell line (SW620) and in stage IV HL cell line (L1236) (Fig. 4d–e). We further investigated if the same genes were alternatively spliced through AF-event (gain) in both HCT116_NR1D1KO and HCT116_PER2KO. Subsequently, we filtered for genes with at least one protein-coding transcript, carried out a functional enrichment analysis, and found biological processes such as protein acetylation enriched in common AF-gain candidates, positive regulation of cellular response to TGF-beta stimulus enriched in HCT116_NR1D1KO unique candidates, and endothelial cell proliferation unique AF-event in HCT116_PER2KO candidates (Supplementary Fig. 8).

Bar plots depict the total number of genes (x-axis) with loss and gain of splicing (a–c) in the HCT116_ARNTLKO, HCT116_NR1D1KO, and HCT116_PER2KO vs. HCT116_WT cells, (d) in the SW620 vs. SW480 cells, (e) in the L1236 vs. HDMYZ cells, (f) in Arntl KO from distal bronchiolar epithelium (left) and from primary tracheal epithelial cells (right) vs. their WT, and (g) in Nr1d1/2 KO vs. WT. In each barplot, the number of genes containing transcripts with circadian expression and protein-coding biotype (dark blue), protein-coding biotype (blue), and different biotypes (light blue) is indicated.
A similar number of genes were observed in loss and gain of splicing events within the mouse Arntl KO dataset (Fig. 4f). In the case of Nr1d1/2 KO model, a slightly higher number of genes were associated with a loss of splicing events than to a gain of splicing events (Fig. 4g). However, an increase in gain of AF-event as seen in HCT116_NR1D1KO was not observed in the mouse Nr1d1/2 KO. Taken together, we observed a distinct pattern in the number of genes being alternatively spliced in HCT116KO vs. WT pointing to clock knockout specific alterations in alternative splicing.
Further, we carried out pairwise differential splicing analysis between KO and WT (Supplementary Fig. 9). Each differentially spliced candidate showed significantly (p < 0.05) higher change in transcript abundance in the KO than in the corresponding WT condition. Both HCT116_NR1D1KO and HCT116_PER2KO showed higher number of differentially spliced genes than in HCT116_ARNTLKO cells (Supplementary Fig. 9a–c). Besides, the highest number of differentially spliced genes were observed in the SE-event for all cancer cell lines (Supplementary Fig. 9a–e). In murine KO models, a smaller number of differentially spliced candidates were observed (Supplementary Fig. 9f–g). We then searched for common differentially spliced candidates between the human and mouse KO datasets. Common differentially spliced candidates were observed only within SE-event. Sorting Nexin 3 (SNX3), which is involved in intracellular trafficking was found to be differentially spliced in both HCT116_ARNTLKO cells and mouse Arntl KO from distal bronchiolar epithelium compared to their controls. SNX3-201 and SNX3-204 isoforms were present in HCT116_ARNTLKO. Among the common differentially spliced candidates between HCT116_NR1D1KO and mouse Nr1d1/2 KO, were Prolyl 4-Hydroxylase Subunit Alpha 1 (P4HA1), associated with oxidoreductase activity and Leucine Rich Repeat Containing 8 VRAC Subunit A (LRRC8A), which is an essential component of the volume-regulated anion channel, and plays a role in cell adhesion and cellular trafficking. LRRC8A-202 and LRRC8A-203 transcripts contributed to the observed differential splicing of LRRC8A and P4HA1-202 and P4HA1-203 transcripts contributed to differential splicing of P4HA1. While in the mouse Nr1d1/2 KO, Lrrc8a-201 and Lrrc8a-202 isoforms contributed to the observed differential splicing of Lrrc8a and P4ha1-202 transcript contributed to differential splicing of P4ha1. Taken together, our results showed a core-clock KO-specific impact on AS events within the datasets analysed.
Core-clock KO in HCT116 cells resulted in the occurrence of differentially rhythmic transcript pairs associated with cancer hallmarks
Based on the differences in SFs rhythmicity and local AS events, as seen in HCT116KO, we would expect clock dependent changes in the rhythmicity of alternatively spliced transcripts. We used data from circadian transcripts to examine whether transcript isoforms from the same gene in each condition show phase-shifted rhythmic expression. For that, we extracted rhythmic and phase-shifted transcript pairs (Fig. 5). In short, we compared each rhythmic transcript with all other rhythmic transcripts of the same gene, in a pairwise manner, within the same cell line datasets. Differentially rhythmic transcript pairs (q < 0.05) were obtained for phase differences larger than 3 h. The scatter plot depicts the distribution of differentially rhythmic transcript pairs according to their phase difference and amplitude ratio (Fig. 5a). All HCT116KO cells showed a decrease in the number of differentially rhythmic transcript pairs vs. HCT116_WT (Fig. 5b). 1021 transcript isoform pairs (592 unique genes) showed differential rhythmicity in HCT116_WT and out of those, 750 transcript pairs (469 unique genes) showed a phase shift >3 h. The lowest number of differentially rhythmic transcript pairs were seen in HCT116_ARNTLKO (336 isoform pairs; 255 unique genes) and out of those, 272 transcript isoform pairs (220 unique genes) showed the pre-defined phase shift (Fig. 5b). 398 transcript isoform pairs (297 unique genes) were differentially rhythmic in HCT116_NR1D1KO and out of those, 295 pairs (228 genes) had a phase shift larger than 3 h. HCT116_PER2KO cells showed 342 differentially rhythmic isoform pairs (241 unique genes), and 275 pairs (196 unique genes had a phase shift larger than 3 h (Fig. 5b). To further explore the existence of different phase shifts within differentially rhythmic isoform pairs, we compared the peak phases of isoform pairs (Fig. 5c). A higher number of differentially rhythmic transcript pairs showed phase-difference ≥9 h (Fig. 5c; right panel) as compared with phase-difference ≥3 h and <6 h (Fig. 5c; left panel) and phase-difference ≥6 h and <9 h (Fig. 5c; middle panel) in all HCT116 datasets. To further understand the role of phase-shifted isoform pairs, we explored their biotypes (Fig. 5d). Transcript isoform pairs with protein-coding biotypes were observed as the largest pair in all HCT116 datasets. Among phase-shifted transcript pairs with protein-coding biotypes, we observed 328 pairs in HCT116_WT, 122 pairs in HCT116_ARNTLKO, 140 pairs in HCT116_NR1D1KO, and 123 pairs in HCT116_PER2KO. Also, transcript pairs with different biotypes such as nonsense-mediated decay or retained intron were phase-shifted in HCT116 datasets (Fig. 5d). For instance, three transcript pairs from Baculoviral IAP Repeat Containing (BIRC5), which promotes cell proliferation and prevents apoptosis, were differentially rhythmic only in HCT116_WT cells. Out of the three transcript pairs, two pairs consisted of protein-coding biotypes and one pair consisted of protein-coding and nonsense-mediated decay biotype. We observed two phase-shifted transcript pairs from Polycomb Group Ring Finger 2 (PCGF2), which controls cell proliferation specific to HCT116_ARNTLKO. Both the transcript pairs of PCGF2 consisted of protein-coding biotypes. In HCT116_NR1D1KO, we also saw KO-specific phase-shifted transcript pairs with retained intron and nonsense-mediated decay biotypes from ATP Binding Cassette Subfamily A Member 2 (ABCA2), a lipid transporter. HCT116_PER2KO showed two unique phase-shifted transcript pairs with protein-coding and retained intron biotypes from Chromodomain Helicase DNA Binding Protein 8 (CHD8), a transcription suppressor gene.

Differential rhythmicity analysis between rhythmic transcripts within the same cell line was carried out using DODR. a The scatter plot depicts the distribution of differentially rhythmic transcript pairs according to their phase difference and amplitude ratio in HCT116. b Decline in differentially rhythmic isoform pairs observed in all HCT116KO. c The chord diagram represents the peak phases of transcript pairs across HCT116 under different phase shift cut-offs. d The chord diagram depicts the biotypes of phase shifted transcript pairs. e The circular plot shows the association of genes containing phase shifted transcript pairs with hallmarks of cancer.
A functional enrichment analysis for genes with phase-shifted isoform pairs was carried out for HCT116 datasets (Supplementary Fig. 10). Genes with phase-shifted isoform pairs in HCT116_WT were enriched in processes like DNA damage response and G1/S cell cycle transition (Supplementary Fig. 10). Whereas, HCT116_ARNTLKO phase-shifted candidates were uniquely enriched in processes like regulation of apoptotic signalling and EMT. HCT116_NR1D1KO phase-shift candidates were uniquely enriched in processes like macrophage proliferation and intracellular cholesterol transport. HCT116_PER2KO candidates were uniquely enriched in processes like regulation of intracellular transport and negative regulation of chromatin organization (Supplementary Fig. 10).
Subsequently, we mapped the genes with phase-shifted isoform pairs (phase-diff ≥3 h) with a cancer hallmark list of genes obtained from the cancer hallmark genes database41 (Fig. 5e). A total of 76 genes in HCT116_WT, 31 genes in HCT116_ARNTLKO, 38 genes in HCT116_NR1D1KO, and 30 genes in HCT116_PER2KO with differentially rhythmic isoform pairs, were related to cancer hallmarks (Fig. 5e). Transcript pairs from CSNK2A1 were observed in all HCT116 datasets. However, different phase-shifted transcript pairs were seen in HCT116_WT and HCT116KO. In HCT116_WT, we observed phase-shift transcript pairs from SMAD2, a cancer hallmark gene. Also, transcript pairs from AKT2, involved in all cancer hallmark processes except genome instability were phase-shifted in HCT116_WT. However, differentially rhythmic transcript pairs of both SMAD2 and AKT2 were not found in the KO cells. Phase-shifted transcripts pairs from MET, which controls several cancer hallmarks were seen in HCT116_WT and HCT116_PER2KO. In particular, MET-202 and MET-206 in HCT116_WT and MET-203 and MET-206 in HCT116_PER2KO cells were differentially rhythmic. In HCT116_ARNTLKO, we observed a phase-shift in differentially rhythmic transcript pairs (from cancer hallmark genes) including HIF1A and FGFR2 that were neither differentially rhythmic in HCT116_WT nor in other HCT116KO (Fig. 5e). Similarly, we observed phase-shifted transcript pairs from cancer hallmarks genes like HRAS and CD63 unique to HCT116_PER2KO. Moreover, we observed phase-shifted transcript pairs related to cancer hallmark genes like CASP8 and Mitogen Activated Protein Kinase 3 (MAPK3) that were unique to HCT116_NR1D1KO (Fig. 5e).
The association of specific cancer hallmarks genes with differentially rhythmic isoform pairs due to different clock alterations in HCT116 motivated the subsequent analysis of alternatively spliced candidates and uniquely spliced candidates in all HCT116KO cells. Therefore, we intersected the list of candidates that were differentially spliced, and found in the subsets of loss or gain of splicing event (rhythmic at least in HCT116_WT), and further shortlisted the candidates related to cancer hallmarks. For better visualization, we filtered cancer hallmarks genes for which at least two transcripts were rhythmic in HCT116_WT for loss of splicing event (Fig. 6a). Whereas, candidates with at least two rhythmic transcripts in KO were shortlisted for gain of splicing event. The complete list of candidates categorized based on cancer hallmarks is provided in Supplementary file 3.

Commonly spliced and uniquely spliced candidates in HCT116KO were mapped to the cancer hallmarks gene list. a Circular plots depict the association between spliced candidates and different cancer hallmarks (b) HNRNPM, a spliceosome machinery component lost its circadian expression in HCT116KO cells. HCT116KO cells also showed discrepancies in the mean expression level of HNRNPM transcripts. Expression of uniquely alternatively spliced candidate transcripts in (c) HCT116_PER2KO, (d) HCT116_ARNTLKO, and (e) HCT116_NR1D1KO were plotted. Genomic region plots of MET, FGFR2 and LRRC8A transcripts represent differences in their exon composition (marked in red) compared to canonical forms. Circadian rhythmic transcripts were plotted using harmonic regression fit and arrhythmic transcripts were plotted using Loess fit in R.
HNRNPM, a component of spliceosome machinery and involved in pre-mRNA processing showed either a change in the rhythmic properties or a loss of rhythmicity in HCT116KO vs. HCT116_WT cells (Fig. 6b). Moreover, we observed differences in the mean expression level of HNRNPM transcripts among HCT116 datasets. Both HCT116_NR1D1KO and HCT116_PER2KO resulted in the loss of rhythmicity of transcripts from HNRNPM. On the contrary, HCT116_ARNTLKO resulted in the gain of one circadian HNRNPM transcript (HNRNPM-215) vs. HCT116_WT. When analysing knockout-specific spliced candidates, we observed cancer hallmarks genes like MET as uniquely spliced in HCT116_PER2KO (Fig. 6c). The genomic region plots of MET transcripts (all protein-coding biotypes) represent the difference in coding exon composition compared to the canonical form of MET (MET-202). Moreover, the comparison of the coding exon sequence between the canonical form and other transcripts showed missing/skipping of exon 1- exon 21 (in MET-206), missing of exon 10 (in MET-201), and missing/skipping of all exons between 1 to 21 except exon 10,11 (in MET-203). The AS transcripts of cancer hallmarks genes like FGFR2 showed differential rhythmicity unique to HCT116_ARNTLKO (Fig. 6d). Specifically, HCT116_ARNTLKO showed changes in the expression of two of the FGFR2 transcripts (FGFR2-202 and FGFR2-211; both protein-coding). The genomic plots for FGFR2 transcripts show the loss of coding exon region in FGFR2-202 and FGFR2-211 vs. FGFR2-206 (canonical form). The comparison of coding exons sequence between FGFR2-206 and FGFR2-202/-211 showed exclusion of exon 9 in FGFR2-202 while exclusion of exon 8 in FGFR2-211 (Fig. 6d). LRRC8A was differentially spliced in both HCT116_NR1D1KO vs. HCT116_WT and mouse Nr1d1/2 KO vs. WT. LRRC8A-201 was not circadian in HCT116_WT, but it was circadian expressed in HCT116_NR1D1KO (Fig. 6e). CD44 was differentially spliced only in HCT116_NR1D1KO vs. HCT116_WT, however, the resulting transcripts were rhythmic only in HCT116_WT. We also observed gain in rhythmic transcripts of cancer hallmark genes unique to specific HCT116KO vs. HCT116_WT. For instance, rhythmic transcripts from Succinate Dehydrogenase Complex Flavoprotein Subunit A (SDHA), a complex of mitochondrial respiratory chain were unique to HCT116_ARNTLKO vs. HCT116_WT. SDHA-206 (retained intron) was rhythmic in HCT116_WT whereas SDHA-202 (retained intron) and SDHA-211 (protein-coding) were rhythmic only in HCT116_ARNTLKO. In case of HCT116_NR1D1KO, we observed a new rhythmic transcript of Diacylglycerol Kinase Zeta (DGKZ), a regulator of intracellular signalling. DGKZ-215 and DGKZ-218 (both with a retained intron event) were rhythmic in HCT116_WT whereas DGKZ-201 (protein-coding) was rhythmic only in HCT116_NR1D1KO. Similarly, we saw that a new rhythmic transcript from Platelet Derived Growth Factor Subunit (PDGFA), which is essential for cell survival was unique to HCT116_PER2KO. PDGFA-201 (protein-coding) was rhythmic in HCT116_WT and PDGFA-203 (protein-coding) was only rhythmic in HCT116_PER2KO. Altogether, our findings showed that each of three core-clock knockouts resulted in aberrant alternative splicing of different cancer hallmarks related genes, pointing to a role for the circadian clock in the regulation of alternative splicing with potential consequences in tumorigenesis.
Discussion
Pre-mRNA splicing contributes to generate diversity in the products of more than 95% human genes and leads to alternatively spliced transcripts that encode distinct proteins. This mechanism is often used to maintain cellular homeostasis and to regulate cell differentiation and development42,43. Previous studies have pointed to a regulation of AS via the circadian clock in cancer, suggesting a temporal pattern of AS, which affected the expression of alternatively spliced target genes in a time-dependent manner24,25,26,27. Interestingly, the distinct temporal AS pattern and the time-dependent expression of target genes were shown to correlate with the circadian phenotype of the investigated cancer cell lines. For example, SW480 colon cancer cells with a robust circadian clock showed twice as many genes with circadian alternative exons (59 genes) compared to the SW620 cell line (29 genes), the metastatic counterpart of SW480 from the same patient with a less robust circadian rhythm24. In addition, many of the affected alternatively spliced target genes with circadian AS patterns were involved in various key cancer pathways including VEGFA (involved in angiogenesis) and CD44 (involved in EMT and metastasis), indicating a relevant role for temporal AS in mediating cancer progression.
In the current study, we evaluated the effect of circadian regulation of AS in the colon cancer cell line HCT116 with a robust circadian clock, as well as in core-clock knockout mutants of HCT116, and investigated the potential clock-AS interplay in cancer promoting properties. We also investigated our results in different colon cancer cell lines (SW480 and SW620), HL cells and healthy mouse tissues for a more general understanding of the interplay between alterations of the circadian clock and resulting AS events.
Our analysis showed that the alterations in core-clock components directly influenced the rhythmicity pattern of splicing factor transcripts, which subsequently impacted AS events. The SFs whose transcripts showed altered rhythmicity pattern in HCT116KO included RACK1, SFPQ (altered in HCT116_ARNTLKO), CIRBP, HNRNPD (altered in HCT116_NR1D1KO), and SRSF5 (altered in HCT116_PER2KO). Both RACK1 and SFPQ regulate numerous cancer-related cellular processes.
RACK1 is critical for cell proliferation and transcription44, while SFPQ is a multi-functional protein that regulates several processes such as RNA post-transcriptional activity45, splicing regulation46, and DNA repair47. Aberrant expression of SFPQ is associated with aetiology of colorectal cancer48. A previous study by Pellarin et al reported the reduction in platinum chemotherapy induced apoptosis due to SFPQ via alternative splicing of CASP9 in human epithelial ovarian cancer samples49. Our analysis showed that ARNTL deletion in HCT116 cells resulted in a phase shift (>6 h) within SFPQ transcripts (Fig. 3, Supplementary Fig. 6), pointing towards its regulation via a core-clock component. Similarly, phase shift in RACK1 transcript in HCT116_ARNTLKO vs. WT suggests that its circadian variation is linked uniquely with ARNTL. A previous study using mouse fibroblasts reported the recruitment of RACK1 receptor in a circadian manner into the nuclear BMAL1 complex, and the overexpression of RACK1 suppressed CLOCK-BMAL1 transcriptional activity50. Similarly, BMAL1 was also shown to regulate the expression of Sfpq in rat pituitary cells51, thereby corroborating our findings.
In addition to the observed phase shift in SFs due to the KO of ARNTL, we also observed phase shifts among other transcripts. CIRBP, an RNA binding protein regulates several processes like cell proliferation and circadian gene expression52 while HNRNPD regulates mRNA stability of genes involved in the cell cycle53. The phase shift in rhythmic expression of CIRBP and HNRNPD transcripts seen in HCT116_NR1D1KO suggests an effect of NR1D1 depletion on the spliceosome machinery. Indeed, Cirbp was reported to regulate sleep and circadian clock via Nr1d1 in mice54 and Hnrnpd was rhythmic in mice cells26. Yet, to our knowledge, no studies were found reporting NR1D1-mediated HNRNPD expression modulation. Likewise, the phase shift detected in SRSF5 transcript in HCT116_PER2KO pointed towards its regulation via PER2. Aberrant expression of SRSF5 is associated with different cancer types and their severity55, however, no direct role of PER2-mediated SRSF5 rhythmicity was reported before. Moreover, we observed loss of rhythmicity in several relevant components of the spliceosome complex including U2AF1 and MBNL2 (required for spliceosome binding to the pre-mRNA branch site and a modulator of AS, respectively) with deletion of clock components in all three HCT116KO cells. Mutations in U2AF1 contribute to cancer progression and have been reported in several different cancer types including CRC56, while MBNL2 was reported as tumour suppressor in hepatocarcinogenesis57. Accordingly, the loss of U2AF1 and MBNL2 rhythmic transcripts, as seen in HCT116KO, point towards their possible contribution to metastasis formation. Indeed, our results also showed the loss of U2AF1 and MBNL2 rhythmicity in SW620 (metastasis CRC) and L1236 (stage IV HL).
Several studies reported that alterations in the spliceosome machinery lead to aberrant AS patterns12,58,59,60,61,62,63,64. In agreement with these data and our findings for HCT116 cells, we also found loss of rhythmicity in SFs genes in recently published RNA-seq datasets of murine Per1/2 KO (GSE17197565) and murine Cry1/2 KO (GSE13589866) (Supplementary Fig. 5). These results highlight the relevance of core-clock elements in the regulation of genes involved in splicing. Indeed, aberrant pre-mRNA splicing and alterations in splicing factors are known to act as oncogenic drivers and contribute to tumour progression67. Our results show the existence of alterations in rhythmic properties of SFs transcripts due to the KO of core-clock elements, which was reflected on the resulting AS transcripts. For instance, the pre-spliceosomal complex element RBM5 regulates AS of CASP9, an apoptosis-related gene4,68 and showed altered rhythmic properties in HCT116_ARNTLKO vs. HCT116_WT cells. RBM5, a tumour suppressor gene and splicing factor, improves the production of mRNAs by recognizing incorrect 3′splice sites of epidermal growth factor receptor (EGFR) pre-mRNA, thereby inhibiting the proliferation of tumour cells69. Previous studies in different cancer types suggested RBM5 as a potential target to prevent tumorigenesis70,71. In our study, we observed a regulation of RBM5 expression via the circadian clock, which led to a loss of its rhythmicity due to ARNTL disruption.
The impact of circadian dysregulation in the splicing machinery was also seen through the loss/gain of AS events in all HCT116KO cells. Among the different splicing events, exon skipping is the most common splicing event in human tissues72, as also observed in our study. The unique AF-event gain as seen in HCT116_NR1D1KO and HCT116_PER2KO indicates that the deletion of either PER2 or NR1D1 in HCT116 alters AS by preferring AF-event to the other six modes of AS.
By comparing our results to datasets from mouse models, we found SNX3 (common between HCT116_ARNTLKO and murine Arntl KO from distal bronchiolar epithelium tissue) and P4HA1, LRRC8A (common between HCT116_NR1D1KO and murine Nr1d1/2 KO) among the common differentially spliced candidates between HCT116KO and murine KO models, however resulting in different sets of alternatively spliced transcripts. These differences suggest that the deletion of ARNTL or NR1D1 in HCT116 is likely to have a different biological impact (protein product) compared to the same KOs in murine models. Differential splicing of SNX3 in HCT116_ARNTLKO and P4HA1, LRRC8A in HCT116_NR1D1KO point towards splicing alterations, which are KO specific. SNX3, a unique mediator of WNT protein secretion, was reported to mediate EMT and metastasis in CRC cells including HCT11673. P4HA1, a catalytic enzyme, was reported to regulate cell proliferation in CRC cells via HIF1A and WNT signaling6. While, LRRC8A, a main regulatory subunit of VRAC (volume-regulated anion channel), was found to be upregulated in colon cancer patients and might contribute to metastasis74. However, the direct correlation between these genes and core-clock components remains unexplored.
Following the temporal regulation of different AS events in HCT116 datasets, our analysis revealed several transcript pairs that showed differential rhythmicity. Moreover, we also showed the occurrence of new KO-specific alterations in transcripts rhythmicity vs. WT. In HCT116_WT, phase-shifted isoforms were found to be enriched in DNA damage response and G1/S cell cycle transition. This suggests a temporal regulation of DNA repair and cell cycle pathways that might be regulated by circadian AS events. Indeed, a previous study reported splicing as an emerging pathway contributing to DNA damage response75. In the KO cells, we observed different enriched processes among phase-shifted isoforms such as apoptotic signalling and EMT (HCT116_ARNTLKO), macrophage proliferation and intracellular cholesterol transport (HCT116_NR1D1KO), intracellular transport and chromatin organization (HCT116_PER2KO). These results highlight the individual impact of each of the core-clock KOs in cellular functioning. For instance, we identified phase shifted transcript pairs from different cancer hallmark genes such as BIRC5 in HCT116_WT, PCGF2 in HCT116_ARNTLKO, ABCA2 in HCT116_NR1D1KO, and CHD8 in HCT116_PER2KO. BIRC5, an immune-related gene was found to be highly expressed in different tumour types and promotes cell proliferation76. Moreover, Birc5 was reported rhythmic in colon mucosa cells and its silencing resulted in an increased sensitivity of HCT116 cells to CDK inhibitors77. PCGF2, a ring finger protein, was reported to negatively regulate granulocyte differentiation in human HL-60 cells78. While, increased expression of ABCA2, a member of ATP transporters, was reported in different cancer types79. A previous report on CHD8, a negative regulator of Wnt signalling, showed that loss of its expression might be an indicator of aggressiveness in gastric cancer80. However, the interdependence of PCGF2, ABCA2, and CHD8 on specific core-clock elements was not reported. We also observed phase-shifted isoforms of some gene such as CSNK2A1 in all HCT116 datasets, however, different transcript pairs were phase-shifted in each KO, pointing to a KO-specific effect. Moreover, we saw the loss of differentially transcript pairs of cancer hallmark genes including SMAD2 and AKT1 in all HCT116KO. We also observed differential rhythmicity in certain cancer hallmark genes, that were not circadian in HCT116_WT. For instance, HIF1A and FGFR2 showed differentially rhythmic transcript pairs unique to HCT116_ARNTLKO. HIF1A is the main element of the HIF1-pathway, which plays a crucial role in adaptive responses of tumour cells to hypoxia and promotes tumour progression and metastasis via oncogenic growth factors such as TGFß (transforming growth factor beta) and EGF (epidermal growth factor)81. FGFR2 is an EMT-associated gene which encoded for two isoforms, IIIb and IIIc, characteristic to epithelial and to mesenchymal cells, respectively82. The discrepant expression pattern, as well as rhythmic properties of these two FGFR2 spliced variants in HCT116_ARNTLKO vs WT suggests that ARNTL deletion in HCT116 might promote metastasis via EMT.
Similarly, we observed differentially rhythmic transcript pairs of HRAS (proto-oncogene and member of the RAS-pathway involved in cell proliferation) and CD63 unique to HCT116_PER2KO. CD63 is a member of the tetraspanin family involved in cell differentiation and migration, which was found to be a prognostic marker in colorectal and esophageal cancer83,84. In HCT116_NR1D1KO, we observed unique differentially rhythmic transcript pairs of CASP8 and MAPK3. CASP8 is an apoptosis-related cysteine peptidase and an essential part of the death-inducing signalling complex (DISC)85. In cancer, it promotes proliferation and angiogenesis through the activation of NF-kB in glioblastoma86 and breast cancer87. MAPK3 encodes for a member of the MAP kinase family (aka extracellular signal-regulated kinases (ERK)) and regulates various cellular processes such as proliferation, differentiation, and cell cycle progression in response to a variety of extracellular signals. These results point towards a cross-talk between aberrant AS events and cancer hallmarks related to each of the HCT116KO.
We also observed cancer hallmark genes like MET, whose different transcript pairs were phase shifted in HCT116_WT and HCT116_PER2KO. The MET oncogene encodes for a receptor tyrosine kinase with pleiotropic functions in initiating and sustaining neoplastic transformation, as well as in cancer cell survival and tumour dissemination88. MET is known to undergo AS and its AS isoform (lacking exon 14) is known to inhibit HGF-induced tyrosine phosphorylation of Met, as well as cell proliferation and migration in skeletal muscle myoblasts89.
We further compared our list of AS spliced candidates to a list of known cancer hallmark genes41. Of these, ACTB was differentially spliced in all HCT116KO vs WT, however rhythmicity of ACTB transcripts were lost in all three KOs. This suggests that the loss in circadian profile of ACTB transcripts may contribute towards metastasis due to clock alterations. A previous study reported that the abnormal expression and polymerization of ACTB contributes to invasiveness and metastasis of different cancer types90. Besides different cancer hallmarks related to AS candidates, we also observed discrepant expression of HNRNPM SF transcripts in all three HCT116KO vs. WT. Among different KO-specific AS candidates, FGFR2 (in HCT116_ARNTLKO), MET (in HCT116_PER2KO), CD44 (in HCT116_NR1D1KO) lost/gained rhythmicity in the KOs. In HCT116_ARNTLKO, FGFR2 IIIb isoform (exon 9 exclusion and exon 8 inclusion; FGFR2-202) was circadian while in HCT116_WT FGFR2 IIIc (exon 8 exclusion and exon 9 inclusion; FGFR2-211) was circadian. These discrepancies were better seen when we analysed the average expression of both isoforms in WT and HCT116_ARNTLKO cells. The lower average expression of FGFR2 IIIb in HCT116_ARNTLKO while higher expression of FGFR2 IIIc point to possible alterations in EMT signalling. Matsuda Y et al. reported increased FGFR2 IIIc expression among colorectal carcinomas samples and a human anti-FGFR2 IIIc monoclonal antibody was reported to inhibit growth in colorectal carcinoma cells91. These findings suggest that FGFR2 IIIc could be a promising therapeutic target for colorectal carcinoma.
CD44 encodes for a family of cell adhesion molecules involved in homotypic and heterotypic interactions with extracellular matrix components, as well as EMT92. It promotes proliferation and invasiveness of cells via recruiting ERM proteins (Ezrin, Radixin und Moesin) under certain conditions. However, CD44 can also act as a tumour suppressor, for example when cells reach confluent growth conditions, and thus inhibit cell growth. The prognostic value of the CD44 variant 6 (CD44v6) in CRC was debated for years due to contradictory results93,94,95. The function of CD44 in a cell is determined by the CD44 isoform pattern expressed15. Several studies showed the relevance of CD44v6 as an independent negative prognostic factor and a promising therapeutic target in CRC96,97. In our study, we showed that NR1D1 disruption in HCT116 cells results in aberrant alternative splicing of CD44, which points to a clock specific regulation of CD44 splicing, and could potentially play a role in CD44-targeted therapy regimens. Interestingly, several CD44-targeted drugs have been approved for clinical trials, which highlights the importance of timing treatment in cancer therapy targeting CD44 (reviewed in98). Our results suggest that the perturbations in core-clock elements result in different AS outcomes for this gene.
Furthermore, certain genes showed a gain of new rhythmic transcripts in the KO vs. WT. These included SDHA (HCT116_ARNTLKO vs. WT), DGKZ (HCT116_NR1D1KO vs. WT), and PDGFA (HCT116_PER2KO vs. WT). SDHA is responsible for transferring electrons from succinate to ubiquinone (coenzyme Q), and acts as a tumour suppressor and inhibitor of angiogenesis in paraganglioma99. DGKZ attenuates protein kinase C activity by regulating diacylglycerol levels in intracellular signalling cascade and signal transduction. It has proven to be associated with various signalling pathways, including ERK and MYC and acts as a potential oncogene in osteosarcoma100. PDGFA is a member of the PDGF as well as VEGF signalling. Paracrine PDGF signalling is commonly observed in epithelial cancers, where it triggers stromal recruitment and may be involved in EMT affecting tumour growth, angiogenesis, invasion, and metastasis. Overexpression of PDGF signalling was shown to drive tumour cell growth and to promote tumorigenesis in colorectal cancer, breast cancer, lung cancer and sarcomas (reviewed in ref. 101). The gained rhythmicity of a coding transcript of SDHA due to ARNTL deletion, DGKZ due to NR1D1 deletion, and PDGFA due to PER2 deletion in HCT116 may lead to aberrant protein functions resulting from clock-KO-specific splicing. However, the biological role of these transcripts remains to be elucidated.
The relevance of timing treatment for increasing tolerability and efficacy while minimizing toxic side-effects has been largely demonstrated for numerous anticancer agents in experimental models, as well as in clinical settings including clinical studies in patients with metastatic colorectal cancer102,103,104. Aberrant alternative splicing events and circadian clock disruption are reported in several different cancer types, adding an additional complexity to the genetic landscape of cancer67,105. The results of our study highlight the importance of timed AS, especially in genes regulating cancer hallmarks that are shown to be suitable drug targets. Our data point to the regulation of AS patterns in cancer via the circadian clock. It would be important to further explore these findings and to consider drug timing, at least for drugs targeting such genes, in future clinical studies.
Altogether, the results of our study suggest an interplay between circadian clock elements and AS in cancer, with distinct and unique roles for the core-clock genes ARNTL, PER2 and NR1D1 in regulating SF rhythmicity and AS events in cancer hallmark genes, with relevance in cancer onset and progression.
Methods
Cell culture
Human colorectal carcinoma cell line HCT116 (ATCC® CCL-247™, Gaithersburg, MD, USA) was cultured in Dulbecco’s Modified Eagle Medium (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% Fetal Bovine Serum (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) and 1% Penicillin–Streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) in an incubator with 5% CO2 at 37 °C. LUNA™ Automated Cell Counter (Logos Biosystems, Anyang, South Korea) was used for cell counting and morphology analysis. Cell lines were tested for mycoplasma by using the Mycoplasma check service of Eurofins Genomics (Eurofins Genomics, Ebersberg, Germany).
CRISPR-Cas9 Knockout generation
A CRISPR-Cas9 mediated approach was applied to generate the core-clock knockout cell lines in HCT116. Briefly, HCT116_WT cells were seeded in 6-well plates at a density of 4 × 105 cells/well and transfected with CRISPR-Cas9 plasmids containing GFP marker and guided RNAs targeting multiple exons of ARNTL, PER2 or NR1D1 genes, respectively. Cell transfection was performed using FuGENE HD Transfection Reagent (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s instructions. 48 h post-transfection, CRISPR/Cas9 GFP-positive cells were single-cell sorted using an S3e cell sorter (Bio-Rad laboratories, Hercules, CA, USA) into 96-well plates. Colonies were expanded for subsequent testing and successful knockout colonies were used for the time-course RNA-seq experiment.
For each knockout condition, several single clones were investigated on RNA gene expression level to characterize and confirm the knockout. All KO cell lines displayed significantly reduced target gene expression compared to WT (Supplementary Fig. 1d). The off-target activity was investigated using Off-Spotter106 and the Welcome Trust Sanger Institute Genome Editing database (WGE)107 to search for the most likely potential off-target sites using gRNA sequences. Off-target sites with up to three mismatches within protein-coding regions were Sanger-sequenced and compared to WT. All investigated potential off-target sites in KO cells showed 100% sequence similarity to the WT, indicating absent off-target modifications. For regions where it was not possible to design specific primers amplifying the off-target site, we compared band sizes on gel electrophoresis.
Sample preparation and RNA extraction
HCT116 cells were seeded in triplicates in 12-well plates with a density of 2 × 105 cells per well. On the next days, cells were synchronized by changing the media. Sampling was started 9 h after synchronization and samples were taken every 3 h for a time-series of 45 h. To prepare the cells for RNA extraction, media was discarded and cells were washed with phosphate buffer saline (PBS) and lysed using RLT Plus buffer (Qiagen, Hilden, Germany) directly on the plate. A total of 64 samples were obtained from the HCT116 datasets (WT and three KOs).
Total RNA was isolated using the RNeasy Plus Mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s guidelines. Genomic DNA was digested from the cells using gDNA eliminator columns provided with the kit (Qiagen, Hilden, Germany). RNA was eluted in 30 µL RNase-free water. RNA concentration was measured using a Nanodrop 1000 (Thermo Fisher Scientific, Waltham, MA, USA). RNA was then stored at −80 °C until further use.
Data generation
High-quality RNA was used to generate mRNA libraries and prepared using the TruSeq Stranded mRNA Sample Preparation Kit (Illumina, San Diego, CA, USA) according to the sample protocol guidelines and sequenced on an Illumina NextSeq 500 platform to an average depth of 100 M 75-bp paired-end reads at the European Molecular Biology Laboratory (EMBL) GeneCore Facility (Heidelberg, Germany).
Acquisition of additional RNA-seq datasets
Using a strict criterion of “paired-end RNA-seq”, “less than or equal to 4 h interval”, “mammalian tissue”, “circadian rhythm”, we obtained a total of 134 samples from six different datasets. Published paired-end RNA-seq for Per1/2 was not available, therefore we included for this dataset recently published single-end Per1/2 KO (24 samples). Out of seven different datasets, two datasets were retrieved from human model system and five datasets were retrieved from the mouse model system.
Based on human model system, RNA-seq datasets were collected derived from: (1) human CRC cell lines (SW480: primary tumour, SW620: metastatic tumour; Accession number: E-MTAB-7779) from the same patient, and (2) Hodgkin’s lymphoma (HL) cell lines of cancer progression stages (HD-MY-Z: stage IIIB, L-1236: stage IV; Accession number: GSE16206) from different patients. CRC cells (SW480, SW620) and HL cells (HDMYZ, L1236), which represent different progression grades of CRC and HL, respectively, display various clock phenotypes (Supplementary Fig. 2e). These datasets were chosen to find a possible correlation between clock disruption and cancer progression stage.
Based on mouse model system, we retrieved RNA-seq datasets derived from (1) mouse embryonic stem cells in WT condition and the Nr1d1/2 KO (Accession number: GSE125696) generated using CRISPR/Cas9, (2) laser micro-dissected SCN tissue of male C3H/HeH wild type mice (age: 10–12 weeks; Accession number: GSE72095), (3) distal bronchiolar epithelium tissue isolated using laser capture microdissection from WT mice and Arntl KO mice bearing the targeted deletion in mouse club cells (age: 10–20 weeks; Accession number: E-MTAB-6384), and primary tracheal epithelial cells isolated from WT mice and Arntl global KO (age: 10–20 weeks; Accession number: E-MTAB-6384). In addition, we retrieved raw gene-level count data from two murine core-clock KO RNA-seq datasets from NCBI-GEO, liver tissue from WT and Per1/2 KO mice (age: 3 months old, Accession number: GSE171975; single-end RNA-seq), and liver tissue from WT and Cry1/2 KO mice under normal feeding (age: 9–14 weeks old, Accession number: GSE135898). All datasets listed in this section were downloaded from NCBI-GEO or ArrayExpress.
Synchronization and sampling protocols of used RNA-seq datasets
E-MTAB-6384 (mouse Arntl-KO lung samples pulmonary airway epithelial cells): For circadian sampling, mice were maintained in constant darkness and samples collected 1 cycle after transfer to darkness (1 day later) at circadian time (CT), which by convention anchors expected time of lights off and activity onset to CT12. Samples were taken every 4 h for 48 h in constant dark conditions108.
GSE72095 (mouse SCN samples): After 7 days of acclimatisation, mice were singly housed for 7 days prior to tissue harvesting. At each sampling time-point, pooled dissected tissue from five adult male mice were used. Samples were taken for a total of six time-points over a 12:12 LD cycle at 4 h intervals (ZT2, 6, 10, 14, 18 and 22), where ZT0 denotes the time of lights on109.
GSE125696 (Nr1d1/2 KO mouse ESCs): On differentiation day 28, cells were treated with 100 nM dexamethasone and frozen at the indicated time points. Time course RNA-Seq was performed using RNA samples at 4 h intervals over 2 days110.
Synchronization of HCT116 RNA-Seq samples: HCT116 cells were synchronized via medium exchange (which serves as a strong entrainment signal aka Zeitgeber in these cells). Previous reports on the efficacy of synchronization agents (e.g. dexamethasone, forskolin, serum shock, medium change) have shown, that medium exchange is as effective as other mentioned agents in synchronizing the circadian clock, as shown for different CRC cell lines via bioluminescence measurements of ARNTL-promoter activity111.
Curation of splicing factor list
In total 534 human spliceosome and splicing-related genes were compiled from the literature24,35,36, as well as from online databases (Spliceosome database37 and SpliceAid 238). A list of human splicing factors was mapped using Ensembl Biomart39 to obtain their orthologous sets for mouse. A total of 521 SFs were obtained for mouse. The complete list of splicing factors is provided in Supplementary files 1–2.
RNA-sequencing data processing
All RNA-seq datasets were processed and analysed according to the following protocol unless otherwise stated. Quality assessment of raw reads was carried out using fastqcr (version 0.1.2) R package which easily parse, aggregate, and analyse FastQC112 for large number of samples. Based on FastQC reports, over-represented sequences and residual adapter sequences were trimmed from raw reads using Trimmomatic (version 0.39113) using default settings. After trimming, only paired-end reads were used for further downstream analysis. Paired-end reads from human datasets were aligned to human genome (Homo sapiens GRCh38, Ensembl release 92) and from mouse datasets were aligned to mouse genome (Mus musculus GRCm38, Ensembl release 102) using STAR aligner (version 2.6.0114). Based on STAR transcriptome alignment, transcript-level abundances were quantified in transcripts per million (TPM) using Salmon (version 0.10.2115). Afterwards, tximport R package116 was used to import transcript level abundance, estimate counts and transcript lengths. Tximport summarizes the quantification results into matrices for downstream analyses. Tximport can be used to obtain both transcript level (txOut = TRUE) and gene-level (txOut = FALSE) summarization. Here, we used transcript-level counts for detailed alternative splicing analysis. Nevertheless, gene-level summarization was also used for its comparison with transcript-level analysis. Trimmed mean of M-values (TMM) method of averaging was used as the normalization factor to scale up the raw library size using edge R package (version 4.0117). Counts per million (CPM) function was used as a descriptive measure of transcript expression. All transcript feature with at least 0.5 CPM on average over all time points (specific to datasets) were retained and renormalized using selected features. The complete pipeline for the analysis is depicted in Fig. 1.
Raw unlogged gene-level count data from Per1/2 KO (GSE171975; single-end RNA-seq) and Cry1/2 KO (GSE135898; paired-end RNA-seq) were downloaded from NCBI-GEO and used for rhythmicity analysis.
Rhythmicity analysis
Unlogged CPM values were used to detect rhythmic signals from time-series datasets. 24 h rhythmicity was evaluated through a non-parametric method using RAIN R package (version 1.24.0118). Rhythmic gene/transcript-sets were obtained using a cut-off of q < 0.05. The acrophase and relative amplitude were estimated for rhythmic gene-/transcript-sets using Cosinor119 within Discorhythm R package (version 1.6.0120). The rhythmic gene/transcript-sets were further filtered using an additional cut-off of relative amplitude (rAMP) ≥ 0.1.
Alternative splicing analysis using SUPPA
To calculate the local alternative splicing events based on the expression of transcripts in each dataset, we used SUPPA2121. SUPPA2 is helpful in studying splicing at the local alternative splicing level or at the transcript isoform level. We analyzed seven alternative splicing events types; (1) Alternative 3′ splice site (A3-event) where 3′ site acts as an acceptor, (2) Alternative 5′ splice site (A5-event) where 5′ site acts as a donor, (3) Alternative first exon (AF-event) where the first exon is retained after splicing, (4) Alternative last exon (AL-event) where the last exon is retained after splicing, (5) Mutually exclusive event (MX-event) where one of two exons is retained, (6) Retention intron event (RI-event) where the intron is confined within mRNA, and (7) Skipping exon event (SE-event) where an exon may be spliced out or retained.
For each gene in a given tissue or condition, the average percent spliced-in (PSI) value was calculated. Alternative splicing of a gene in a particular dataset is considered only if it fulfils the criteria of PSI < 0.9 and > 0.1. Using the PSI cut-off, we detected specific local events which only happened either in the WT or in the KO datasets. To classify these events, we grouped the local events as gain or loss in the KO with respect to its control. If a KO contained a gene which is spliced by a particular local event and the WT did not show the gene in that particular event, the event would match to gain in the KO. Similarly, if the control group contained a gene which is alternatively spliced by a particular local event and the corresponding KO did not show the gene in that particular event, the event would correspond to loss in the KO.
Besides, we also used SUPPA2 to carry out the differential splicing analysis where a pairwise comparison between the KO and WT was made. The differentially spliced candidates were obtained using p-val < 0.05.
Differential rhythmicity analysis
The detection of differentially rhythmic transcripts isoform within each dataset was carried out using DODR R package (version 0.99.2122). All 24 h rhythmic transcript sets were used for this analysis. 24 h differentially rhythmic transcript isoform pairs per single tissue/condition type were obtained using FDR corrected p-value cut-off meta. p < 0.05. To further examine the transcript isoform with more prominent changes in HCT116, phase difference cut-off ≥3 h was applied.
Acquisition of list of cancer hallmark genes
To understand the direct influence of clock alterations on cancer hallmarks, we retrieved the complete gene list categorized according to processes from Cancer Hallmark Genes database41.
The CHG database41 was established by systematically integrating manually curated hallmarks genes from the confirmed literature and KEGG pathways123. In parallel, based on pre-defined keywords related to hallmarks, a text mining software Lucene (https://lucene.apache.org/) was used to identify hallmark specific pathways in the literature and in the KEGG database. To establish the CHG database Zhang et al. used: (1) 301 KEGG pathways for Lucene search and the extraction of pathways genes, (2) Gene variant data (7075 mutation samples, 6177 methylation samples, 9445 copy number variation samples) from TCGA (The Cancer Genome Atlas https://www.cancer.gov/tcga) to calculate the frequency of gene variation, (3) Human protein-protein interaction data from HPRD (Human Protein Reference Database124), STRING125, BioGRID126, HTRIdb127 to integrate a gene interaction network. CHG provides the corresponding gene sets for ten cancer hallmarks: (1) Activating invasion and metastasis (1,149 genes), (2) Enabling replicative immortality (308 genes), (3) Evading growth suppressors (709 genes), (4) Evading immune destruction (609 genes), (5) Genome instability and mutation (231 genes), (6) Inducing angiogenesis (507 genes), (7) Reprogramming energy metabolism (461 genes), (8) Resisting cell death (1192 genes), (9) Sustaining proliferative signaling (1314 genes), and (10) Tumor promoting inflammation (648 genes).
Functional enrichment analysis
Gene ontology analysis for selected gene sets was carried out using an R based package ClusterProfiler (version 4.0128). Enriched biological processes were obtained for each of the gene sets. Dot plots were used to visualize the top 10 enriched biological processes in the case of differentially rhythmic isoform pairs. The enriched biological processes for the candidate cancer hallmarks markers were manually curated after removing the redundant biological processes. The visualization of cancer hallmarks connected with selected candidate markers was carried out using an R based package GO plot (version 1.0.2129).
Data availability
The datasets analysed during the current study are available as follows: recently generated RNA-sequencing data for HCT116 WT cells and KOs in this study have been deposited in the ArrayExpress repository (Accession number: E-MTAB-9701). Other datasets used in this manuscript are available as follows: SW480 and SW620 cell lines, ArrayExpress, E-MTAB-7779; HD-MY-Z and L-1236 cell lines, Gene Expression Omnibus (GEO) GSE16206; mouse Arntl-KO lung samples Pulmonary airway epithelial cells, ArrayExpress, E-MTAB-6384; mouse SCN samples, GEO, GSE72095; Nr1d1/2 KO mouse ESCs, GEO, GSE125696; murine Per1/2 KO liver samples, GEO, GSE171975; murine Cry1/2 KO liver samples, GEO, GSE135898.
Code availability
All programs, packages and functions used in this manuscript are freely available and are referred to in the Method section.
References
Lee, Y. & Rio, D. C. Mechanisms and regulation of alternative Pre-mRNA splicing. Annu. Rev. Biochem. 84, 291–323 (2015).
Chen, M. & Manley, J. L. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 10, 741–754 (2009).
Wang, E. T. et al. Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 (2008).
Paronetto, M. P., Passacantilli, I. & Sette, C. Alternative splicing and cell survival: from tissue homeostasis to disease. Cell Death Differ. 23, 1919–1929 (2016).
Ravi, S., Schilder, R. J. & Kimball, S. R. Role of precursor mRNA splicing in nutrient-induced alterations in gene expression and metabolism. J. Nutr. 145, 841–846 (2015).
Du, J. X. et al. Splicing factors: Insights into their regulatory network in alternative splicing in cancer. Cancer Lett. 501, 83–104 (2021).
Scotti, M. M. & Swanson, M. S. RNA mis-splicing in disease. Nat. Rev. Genet. 17, 19–32 (2016).
Venables, J. P. Aberrant and alternative splicing in cancer. Cancer Res. 64, 7647–7654 (2004).
Wang, E. & Aifantis, I. RNA splicing and cancer. Trends Cancer 6, 631–644 (2020).
Ben-Dov, C., Hartmann, B., Lundgren, J. & Valcárcel, J. Genome-wide analysis of alternative pre-mRNA splicing. J. Biol. Chem. 283, 1229–1233 (2008).
Oltean, S. & Bates, D. O. Hallmarks of alternative splicing in cancer. Oncogene 33, 5311–5318 (2014).
Bonnal, S. C., López-Oreja, I. & Valcárcel, J. Roles and mechanisms of alternative splicing in cancer—implications for care. Nat. Rev. Clin. Oncol. 17, 457–474 (2020).
Climente-González, H., Porta-Pardo, E., Godzik, A. & Eyras, E. The functional impact of alternative splicing in cancer. Cell Rep. 20, 2215–2226 (2017).
Harper, S. J. & Bates, D. O. VEGF-A splicing: the key to anti-angiogenic therapeutics? Nat. Rev. Cancer 8, 880–887 (2008).
Herrlich, P. et al. CD44 acts both as a growth- and invasiveness-promoting molecule and as a tumor-suppressing cofactor. Ann. N. Y. Acad. Sci. 910, 106–120 (2000).
Cohen-Eliav, M. et al. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J. Pathol. 229, 630–639 (2013).
Park, S. et al. Differential functions of splicing factors in mammary transformation and breast cancer metastasis. Cell Rep. 29, 2672–2688.e2677 (2019).
Shakhmantsir, I. & Sehgal, A. Splicing the clock to maintain and entrain circadian rhythms. J. Biol. Rhythms 34, 584–595 (2019).
Patke, A. et al. Mutation of the human circadian clock gene CRY1 in familial delayed sleep phase disorder. Cell 169, 203–215.e213 (2017).
Hastings, M. H., Reddy, A. B. & Maywood, E. S. A clockwork web: circadian timing in brain and periphery, in health and disease. Nat. Rev. Neurosci. 4, 649–661 (2003).
Andreani, T. S., Itoh, T. Q., Yildirim, E., Hwangbo, D. S. & Allada, R. Genetics of Circadian rhythms. Sleep. Med. Clin. 10, 413–421 (2015).
Innominato, P. F., Lévi, F. A. & Bjarnason, G. A. Chronotherapy and the molecular clock: clinical implications in oncology. Adv. Drug Deliv. Rev. 62, 979–1001 (2010).
Shafi, A. A. & Knudsen, K. E. Cancer and the Circadian clock. Cancer Res. 79, 3806–3814 (2019).
El-Athman, R., Fuhr, L. & Relógio, A. A systems-level analysis reveals circadian regulation of splicing in colorectal cancer. EBioMedicine 33, 68–81 (2018).
El-Athman, R., Knezevic, D., Fuhr, L. & Relógio, A. A computational analysis of alternative splicing across mammalian tissues reveals circadian and ultradian rhythms in splicing events. Int. J. Mol. Sci. 20, https://doi.org/10.3390/ijms20163977 (2019).
Genov, N., Basti, A., Abreu, M., Astaburuaga, R. & Relógio, A. A bioinformatic analysis identifies circadian expression of splicing factors and time-dependent alternative splicing events in the HD-MY-Z cell line. Sci. Rep. 9, 11062 (2019).
Genov, N., Basti, A., Abreu, M. & Relógio, A. Temporal splicing switches in elements of the tnf-pathway identified by computational analysis of transcriptome data for human cell lines. Int. J. Mol. Sci. 20, https://doi.org/10.3390/ijms20051182 (2019).
Pluquet, O. et al. Posttranscriptional regulation of PER1 underlies the oncogenic function of IREα. Cancer Res. 73, 4732–4743 (2013).
Koyanagi, S. et al. A molecular mechanism regulating circadian expression of vascular endothelial growth factor in tumor cells. Cancer Res. 63, 7277–7283 (2003).
Preußner, M. et al. Rhythmic U2af26 alternative splicing controls PERIOD1 stability and the circadian clock in mice. Mol. Cell 54, 651–662 (2014).
Lee, J., Park, E., Kim, G. H., Kwon, I. & Kim, K. A splice variant of human Bmal1 acts as a negative regulator of the molecular circadian clock. Exp. Mol. Med. 50, 1–10 (2018).
Duffield, G. E. DNA microarray analyses of circadian timing: the genomic basis of biological time. J. Neuroendocrinol. 15, 991–1002 (2003).
Wu, Y. et al. Systematic studies of the circadian clock genes impact on temperature compensation and cell proliferation using CRISPR tools. Biology (Basel) 10, https://doi.org/10.3390/biology10111204 (2021).
Bunger, M. K. et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 103, 1009–1017 (2000).
Relógio, A. et al. Alternative splicing microarrays reveal functional expression of neuron-specific regulators in Hodgkin lymphoma cells. J. Biol. Chem. 280, 4779–4784 (2005).
Seiler, M. et al. Somatic mutational landscape of splicing factor genes and their functional consequences across 33 cancer types. Cell Rep. 23, 282–296.e284 (2018).
Cvitkovic, I. & Jurica, M. S. Spliceosome database: a tool for tracking components of the spliceosome. Nucleic acids Res. 41, D132–141 (2013).
Piva, F., Giulietti, M., Burini, A. B. & Principato, G. SpliceAid 2: a database of human splicing factors expression data and RNA target motifs. Hum. Mutat. 33, 81–85 (2012).
Kinsella, R. J. et al. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxf.) 2011, bar030 (2011).
Wojtuszkiewicz, A. et al. Pre-mRNA splicing in cancer: the relevance in oncogenesis, treatment and drug resistance. Expert Opin. Drug Metab. Toxicol. 11, 673–689 (2015).
Zhang, D. et al. CHG: a systematically integrated database of cancer hallmark genes. Front. Genet. 11, https://doi.org/10.3389/fgene.2020.00029 (2020).
Gallego-Paez, L. M. et al. Alternative splicing: the pledge, the turn, and the prestige: the key role of alternative splicing in human biological systems. Hum. Genet. 136, 1015–1042 (2017).
Baralle, F. E. & Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 18, 437–451 (2017).
McCahill, A., Warwicker, J., Bolger, G. B., Houslay, M. D. & Yarwood, S. J. The RACK1 scaffold protein: a dynamic cog in cell response mechanisms. Mol. Pharm. 62, 1261–1273 (2002).
Bottini, S. et al. Post-transcriptional gene silencing mediated by microRNAs is controlled by nucleoplasmic Sfpq. Nat. Commun. 8, 1189 (2017).
Mora Gallardo, C. et al. Dido3-dependent SFPQ recruitment maintains efficiency in mammalian alternative splicing. Nucleic Acids Res. 47, 5381–5394 (2019).
Jaafar, L., Li, Z., Li, S. & Dynan, W. S. SFPQ•NONO and XLF function separately and together to promote DNA double-strand break repair via canonical nonhomologous end joining. Nucleic Acids Res. 45, 1848–1859 (2017).
Klotz-Noack, K. et al. SFPQ depletion is synthetically lethal with BRAF(V600E) in colorectal cancer cells. Cell Rep. 32, 108184 (2020).
Pellarin, I. et al. Splicing factor proline- and glutamine-rich (SFPQ) protein regulates platinum response in ovarian cancer-modulating SRSF2 activity. Oncogene 39, 4390–4403 (2020).
Robles, M. S., Boyault, C., Knutti, D., Padmanabhan, K. & Weitz, C. J. Identification of RACK1 and protein kinase Calpha as integral components of the mammalian circadian clock. Science 327, 463–466 (2010).
Becquet, D. et al. Evidence for an internal and functional circadian clock in rat pituitary cells. Mol. Cell Endocrinol. 382, 888–898 (2014).
Liao, Y., Tong, L., Tang, L. & Wu, S. The role of cold-inducible RNA binding protein in cell stress response. Int J. Cancer 141, 2164–2173 (2017).
White, E. J., Brewer, G. & Wilson, G. M. Post-transcriptional control of gene expression by AUF1: mechanisms, physiological targets, and regulation. Biochim. Biophys. Acta 1829, 680–688 (2013).
Hoekstra, M. M., Emmenegger, Y., Hubbard, J. & Franken, P. Cold-inducible RNA-binding protein (CIRBP) adjusts clock-gene expression and REM-sleep recovery following sleep deprivation. Elife 8, https://doi.org/10.7554/eLife.43400 (2019).
Kim, H. R. et al. SRSF5: a novel marker for small-cell lung cancer and pleural metastatic cancer. Lung Cancer 99, 57–65 (2016).
Kandoth, C. et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013).
Lee, Y. H., Jhuang, Y. L., Chen, Y. L., Jeng, Y. M. & Yuan, R. H. Paradoxical overexpression of MBNL2 in hepatocellular carcinoma inhibits tumor growth and invasion. Oncotarget 7, 65589–65601 (2016).
Anczuków, O. & Krainer, A. R. Splicing-factor alterations in cancers. RNA 22, 1285–1301 (2016).
Blijlevens, M., Li, J. & van Beusechem, V. W. Biology of the mRNA splicing machinery and its dysregulation in cancer providing therapeutic opportunities. Int. J. Mol. Sci. 22, https://doi.org/10.3390/ijms22105110 (2021).
da Silva, M. R. et al. Splicing regulators and their roles in cancer biology and therapy. Biomed. Res. Int. 2015, 150514 (2015).
Martín, E., Vivori, C., Rogalska, M., Herrero-Vicente, J. & Valcárcel, J. Alternative splicing regulation of cell-cycle genes by SPF45/SR140/CHERP complex controls cell proliferation. RNA 27, 1557–1576 (2021).
Venables, J. P. Unbalanced alternative splicing and its significance in cancer. Bioessays 28, 378–386 (2006).
Zhang, J. & Manley, J. L. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 3, 1228–1237 (2013).
Gebauer, F., Schwarzl, T., Valcárcel, J. & Hentze, M. W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 22, 185–198 (2021).
Aviram, R., Dandavate, V., Manella, G., Golik, M. & Asher, G. Ultradian rhythms of AKT phosphorylation and gene expression emerge in the absence of the circadian clock components Per1 and Per2. PLoS Biol. 19, e3001492 (2021).
Weger, B. D. et al. Systematic analysis of differential rhythmic liver gene expression mediated by the circadian clock and feeding rhythms. Proc. Natl Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2015803118 (2021).
Zhang, Y., Qian, J., Gu, C. & Yang, Y. Alternative splicing and cancer: a systematic review. Signal Transduct. Target Ther. 6, 78 (2021).
Bechara, E. G., Sebestyen, E., Bernardis, I., Eyras, E. & Valcarcel, J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol. Cell 52, 720–733 (2013).
Su, Z. et al. Lentiviral vector-mediated RBM5 overexpression downregulates EGFR expression in human non-small cell lung cancer cells. World J. Surg. Oncol. 12, 367 (2014).
Jiang, Y. et al. RBM5 inhibits tumorigenesis of gliomas through inhibition of Wnt/β-catenin signaling and induction of apoptosis. World J. Surg. Oncol. 15, 9 (2017).
Prabhu, V. V. & Devaraj, N. Regulating RNA binding motif 5 gene expression—a novel therapeutic target for lung cancer. J. Environ. Pathol. Toxicol. Oncol. 36, 99–105 (2017).
Florea, L., Song, L. & Salzberg, S. L. Thousands of exon skipping events differentiate among splicing patterns in sixteen human tissues. F1000Res 2, 188 (2013).
Pan, B. et al. SNX3 suppresses the migration and invasion of colorectal cancer cells by reversing epithelial-to-mesenchymal transition via the β-catenin pathway. Oncol. Lett. 18, 5332–5340 (2019).
Zhang, H. et al. High expression of leucine‑rich repeat‑containing 8A is indicative of a worse outcome of colon cancer patients by enhancing cancer cell growth and metastasis. Oncol. Rep. 40, 1275–1286 (2018).
Lenzken, S. C., Loffreda, A. & Barabino, S. M. L. RNA splicing: a new player in the DNA damage response. Int. J. Cell Biol. 2013, 153634 (2013).
Xu, L., Yu, W., Xiao, H. & Lin, K. BIRC5 is a prognostic biomarker associated with tumor immune cell infiltration. Sci. Rep. 11, 390 (2021).
Siffroi-Fernandez, S. et al. Functional genomics identify Birc5/survivin as a candidate gene involved in the chronotoxicity of cyclin-dependent kinase inhibitors. Cell Cycle 13, 984–991 (2014).
Jo, S. et al. Inhibition of PCGF2 enhances granulocytic differentiation of acute promyelocytic leukemia cell line HL-60 via induction of HOXA7. Biochem. Biophys. Res. Commun. 416, 86–91 (2011).
Davis, W. & Tew, K. D. ATP-binding cassette transporter-2 (ABCA2) as a therapeutic target. Biochem. Pharmacol. 151, 188–200 (2018).
Sawada, G. et al. CHD8 is an independent prognostic indicator that regulates Wnt/β-catenin signaling and the cell cycle in gastric cancer. Oncol. Rep. 30, 1137–1142 (2013).
Masoud, G. N. & Li, W. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 5, 378–389 (2015).
Warzecha, C. C., Sato, T. K., Nabet, B., Hogenesch, J. B. & Carstens, R. P. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol. Cell 33, 591–601 (2009).
Kaprio, T., Hagström, J., Andersson, L. C. & Haglund, C. Tetraspanin CD63 independently predicts poor prognosis in colorectal cancer. Histol. Histopathol. 35, 887–892 (2020).
Lai, X. et al. Decreased expression of CD63 tetraspanin protein predicts elevated malignant potential in human esophageal cancer. Oncol. Lett. 13, 4245–4251 (2017).
Tummers, B. & Green, D. R. Caspase-8: regulating life and death. Immunol. Rev. 277, 76–89 (2017).
Fianco, G. et al. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. Elife 6, https://doi.org/10.7554/eLife.22593 (2017).
De Blasio, A. et al. Unusual roles of caspase-8 in triple-negative breast cancer cell line MDA-MB-231. Int J. Oncol. 48, 2339–2348 (2016).
Comoglio, P. M., Trusolino, L. & Boccaccio, C. Known and novel roles of the MET oncogene in cancer: a coherent approach to targeted therapy. Nat. Rev. Cancer 18, 341–358 (2018).
Park, M. et al. A novel isoform of met receptor tyrosine kinase blocks hepatocyte growth factor/Met signaling and stimulates skeletal muscle cell differentiation. J. Biol. Chem. 290, 1804–1817 (2015).
Guo, C., Liu, S., Wang, J., Sun, M. Z. & Greenaway, F. T. ACTB in cancer. Clin. Chim. Acta 417, 39–44 (2013).
Matsuda, Y., Hagio, M., Seya, T. & Ishiwata, T. Fibroblast growth factor receptor 2 IIIc as a therapeutic target for colorectal cancer cells. Mol. Cancer Therapeutics 11, 2010–2020 (2012).
Isacke, C. M. & Yarwood, H. The hyaluronan receptor, CD44. Int J. Biochem. Cell Biol. 34, 718–721 (2002).
Coppola, D. et al. CD44V6 expression in human colorectal carcinoma. Hum. Pathol. 29, 627–635 (1998).
Mikami, T. et al. Decreased expression of CD44, alpha-catenin, and deleted colon carcinoma and altered expression of beta-catenin in ulcerative colitis-associated dysplasia and carcinoma, as compared with sporadic colon neoplasms. Cancer 89, 733–740 (2000).
Gotley, D. C. et al. Alternatively spliced variants of the cell adhesion molecule CD44 and tumour progression in colorectal cancer. Br. J. Cancer 74, 342–351 (1996).
Todaro, M. et al. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 14, 342–356 (2014).
Saito, S. et al. CD44v6 expression is related to mesenchymal phenotype and poor prognosis in patients with colorectal cancer. Oncol. Rep. 29, 1570–1578 (2013).
Li, M. et al. Drug delivery systems based on CD44-targeted glycosaminoglycans for cancer therapy. Carbohydr. Polym. 251, 117103 (2021).
Burnichon, N. et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 19, 3011–3020 (2010).
Yu, W., Tang, L., Lin, F., Yao, Y. & Shen, Z. DGKZ acts as a potential oncogene in osteosarcoma proliferation through its possible interaction With ERK1/2 and MYC pathway. Front Oncol. 8, 655 (2018).
Heldin, C. H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal 11, 97 (2013).
Dallmann, R., Okyar, A. & Levi, F. Dosing-time makes the poison: Circadian regulation and pharmacotherapy. Trends Mol. Med 22, 430–445 (2016).
Lévi, F., Okyar, A., Dulong, S., Innominato, P. F. & Clairambault, J. Circadian timing in cancer treatments. Annu. Rev. Pharm. Toxicol. 50, 377–421 (2010).
Giacchetti, S. et al. Phase III trial comparing 4-day chronomodulated therapy versus 2-day conventional delivery of fluorouracil, leucovorin, and oxaliplatin as first-line chemotherapy of metastatic colorectal cancer: the European Organisation for Research and Treatment of Cancer Chronotherapy Group. J. Clin. Oncol. 24, 3562–3569 (2006).
Alamoudi, A. A. Why do cancer cells break from host circadian rhythm? Insights from unicellular organisms. Bioessays 43, e2000205 (2021).
Pliatsika, V. & Rigoutsos, I. “Off-Spotter”: very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs. Biol. Direct 10, 4 (2015).
Hodgkins, A. et al. WGE: a CRISPR database for genome engineering. Bioinformatics 31, 3078–3080 (2015).
Zhang, Z. et al. Genome-wide effect of pulmonary airway epithelial cell-specific Bmal1 deletion. FASEB J. 33, 6226–6238 (2019).
Pembroke, W. G., Babbs, A., Davies, K. E., Ponting, C. P. & Oliver, P. L. Temporal transcriptomics suggest that twin-peaking genes reset the clock. Elife 4, https://doi.org/10.7554/eLife.10518 (2015).
Ikeda, R. et al. REV-ERBα and REV-ERBβ function as key factors regulating mammalian Circadian output. Sci. Rep. 9, 10171 (2019).
Fuhr, L. A. The Circadian Clock Modulates Tumour Progression and Drug Response in Colorectal Cancer Cells through Metabolic Phenotype Rewiring (Humboldt-Universität zu Berlin, 2019).
Simon, A. FastQC: a quality control tool for high throughput sequence data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Soneson, C., Love, M. I. & Robinson, M. D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
Thaben, P. F. & Westermark, P. O. Detecting rhythms in time series with RAIN. J. Biol. Rhythms 29, 391–400 (2014).
Cornelissen, G. Cosinor-based rhythmometry. Theor. Biol. Med. Model. 11, 16 (2014).
Carlucci, M. et al. DiscoRhythm: an easy-to-use web application and R package for discovering rhythmicity. Bioinformatics 36, 1952–1954 (2019).
Trincado, J. L. et al. SUPPA2: fast, accurate, and uncertainty-aware differential splicing analysis across multiple conditions. Genome Biol. 19, 40 (2018).
Thaben, P. F. & Westermark, P. O. Differential rhythmicity: detecting altered rhythmicity in biological data. Bioinformatics 32, 2800–2808 (2016).
Kanehisa, M., Sato, Y. & Kawashima, M. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 31, 47–53 (2022).
Keshava Prasad, T. S. et al. Human Protein Reference Database-2009 update. Nucleic Acids Res. 37, D767–772 (2009).
Snel, B., Lehmann, G., Bork, P. & Huynen, M. A. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 28, 3442–3444 (2000).
Stark, C. et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535–539 (2006).
Bovolenta, L. A., Acencio, M. L. & Lemke, N. HTRIdb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genomics 13, 405 (2012).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287 (2012).
Walter, W., Sánchez-Cabo, F. & Ricote, M. GOplot: an R package for visually combining expression data with functional analysis. Bioinformatics 31, 2912–2914 (2015).
Acknowledgements
We are gratefull to all members of Relógio group for their critical remarks and feedback. Vector icons used in this manuscript were obtained from Flaticon (https://www.flaticon.com/) and modified accordingly.
Funding
The work in the group of A.R. was funded by the Dr. Rolf M. Schwiete Stiftung. A.B. was additionally funded by the Berlin School of Integrative Oncology (BSIO) graduate program funded within the German Excellence Initiative, and the German Cancer Consortium (GCC). Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.R. conceptualized the study, attained the funding, carried out the investigation, provided supervision, prepared the initial draft, reviewed and revised the manuscript. D.M. carried out the transcriptome data analysis, visualization, prepared the initial draft, reviewed and revised the manuscript. A.B. carried out in vitro experiments, reviewed and revised the manuscript. All authors have read and agreed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Malhan, D., Basti, A. & Relógio, A. Transcriptome analysis of clock disrupted cancer cells reveals differential alternative splicing of cancer hallmarks genes. npj Syst Biol Appl 8, 17 (2022). https://doi.org/10.1038/s41540-022-00225-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41540-022-00225-w
This article is cited by
-
Differential expression of the circadian clock network correlates with tumour progression in gliomas
BMC Medical Genomics (2023)
