
Abstract
The urea cycle is a liver-based pathway enabling disposal of nitrogen waste. Urea cycle disorders (UCDs) are inherited metabolic diseases caused by deficiency of enzymes or transporters involved in the urea cycle and have a prevalence of 1:35,000 live births. Patients present recurrent acute hyperammonaemia, which causes high rate of death and neurological sequelae. Long-term therapy relies on a protein-restricted diet and ammonia scavenger drugs. Currently, liver transplantation is the only cure. Hence, high unmet needs require the identification of effective methods to model these diseases to generate innovative therapeutics. Advances in both induced pluripotent stem cells (iPSCs) and genome editing technologies have provided an invaluable opportunity to model patient-specific phenotypes in vitro by creating patients’ avatar models, to investigate the pathophysiology, uncover novel therapeutic targets and provide a platform for drug discovery. This review summarises the progress made thus far in generating 2- and 3-dimensional iPSCs models for UCDs, the challenges encountered and how iPSCs offer future avenues for innovation in developing the next-generation of therapies for UCDs.
Similar content being viewed by others

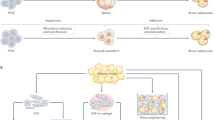

Introduction
Urea cycle disorders (UCDs) encompass a set of rare inherited disorders characterised by defects in one of the urea cycle enzymes or transporters that contribute to detoxify nitrogen waste i.e., ammonia. Ammonia is produced by the deamination of amino acids and is a highly neurotoxic molecule. The urea cycle pathway is fully expressed in the liver (Fig. 1), precisely in periportal hepatocytes following a specific metabolic zonation1. Six enzymes and two transporters have been identified as causing primary UCDs (Table 1), with an estimated incidence of 1:35,000 births in the USA and Western Europe2. UCDs cause hyperammonaemic decompensation with cerebral oedema, resulting in a range of specific symptoms from loss of appetite, vomiting, dizziness, lethargy and seizures, to coma leading to death3. Unrecognised chronic hyperammonaemia causes severe developmental delay and behavioural difficulties4,5,6. Gold standard management relies on protein-restricted diet, ammonia scavenger drugs, arginine supplementation and close monitoring of plasma ammonia, amino acids and nutritional status. Liver transplantation is currently the only curative option7,8. Gene therapy was developed targeting ornithine transcarbamylase deficiency in the late 1990s but the immunogenicity of the viral adenoviral vector led to tragic consequences9. However, in more recent years, safer approaches have been translated using adeno-associated virus (AAV) derived vectors (NTC02991144)10 or mRNA encapsulated lipid nanoparticles (NTC04442347)11. An early phase clinical trial of AAV gene therapy for ornithine transcarbamylase deficiency in adults has recently shown promise as an alternative disease-modifying therapy12. These diseases are rare, which precludes an optimal understanding of the complex pathophysiology of the disease, especially in the brain13,14,15,16,17. Indeed, the spectrum of clinical severity of the same UCD can be highly variable between individuals13,18,19. Yet, despite a common cerebral toxicity as a result of hyperammonaemia, there are pathophysiological specificities, which remain only partially understood, such as arginase deficiency and dysmyelination20, argininosuccinic aciduria and oxidative stress21,22,23,24, or Lysinuric Protein Intolerance and impaired phagocytosis25. Some mouse models fail to recapitulate the human phenotype, such as the hypomorphic Spfash mouse modelling ornithine transcarbamylase deficiency (OTCD), where a high residual ureagenesis capacity prevents hyperammonaemia, UCDs’ cardinal feature, under standard diet. In the Spfash mouse, hyperammonaemia is observed with a protein-enriched diet meaning that only partial OTCD is recapitulated26,27. In addition, traditional cell lines usually derived from immortalised cancer cell lines, have their own limitations with metabolic alterations, leading to a need for alternative modelling methods28. The parallel emergence of induced pluripotent stem cells (iPSCs) and CRISPR/Cas9 editing have provided the opportunity to model these disorders in vitro giving the potential to uncover novel mechanisms of action and new therapeutics, whilst overcoming some of the limitations posed by other models. iPSCs are derived from differentiated adult somatic cells and are characterised by their ability to propagate indefinitely and their capacity to differentiate into any cell type of the human body29,30,31. This review aims to discuss the ongoing progress and remaining limitations that have occurred in modelling these rare disorders using iPSCs.

Ammonia is converted to urea via five consecutive enzymatic reactions within the liver. The urea cycle begins in the mitochondria (blue box), where ammonia and bicarbonate are converted into carbamoyl phosphate. Carbamoyl phosphate requires allosteric activation by N-acetylglucosamine (NAG). Ornithine transcarbamylase (OTC) then catalyses formation of citrulline from carbamoyl phosphate and ornithine. Citrulline is channelled out of the mitochondria via the ornithine transporter (ORNT1), whilst citrin transports aspartate from the mitochondria to the cytoplasm. Argininosuccinate synthetase then enables the synthesis of argininosuccinate from citrulline and aspartate. Argininosuccinate is subsequently broken down into arginine and fumarate by argininosuccinate lyase, with fumarate directed to the Krebs cycle. Arginine is broken down into urea and ornithine by arginase. Ornithine is transported into the mitochondria by the ORNT1 transporter. CA5A carbonic anhydrase VA, CPS1 carbamoyl phosphate synthetase 1, NAG N-acetyl glutamate, NAGS N-acetyl glutamate synthase, OTC ornithine transcarbamylase, OTC1 ornithine transporter, ASS argininosuccinate synthetase, ASL argininosuccinate lyase, ARG arginase, NOS nitric oxide synthase.
UCD-derived iPSCs: purpose and advantages
One of the challenges in modelling liver monogenic diseases in vitro is often the lack of reliable models. Whilst immortalised human liver cancer cell lines are commonly used, such as human hepatocyte-derived Huh7 or HepG2 cell lines as surrogates of human primary hepatocytes, they differ from them in many aspects. Metabolic enzymes are invariably expressed at suboptimal levels when compared to primary hepatocytes32,33,34. In addition, immortalised cancer cell lines are often immature, have dysfunctional apoptotic pathways and have limited genotypic variability35. Primary hepatocytes are considered the gold standard and provide a more biologically relevant model, recapitulating several features of a healthy liver cell in vivo36,37. However, these cells are challenging to isolate, have limited proliferative capacity, limited viability in vitro and rapidly lose hepatic function, preventing them being maintained in culture long-term38,39,40,41. Foetal and adult human stem cells can as well be used although they can be technically challenging to isolate and culture42,43. Therefore, in the last few years, iPSCs have provided an alternative strategy to model inherited metabolic diseases, particularly UCDs (Table 2). These cells have the capacity to differentiate into all three germ layers, enabling the prospect of generating any cell type in vitro29. In addition to averting the ethical issues associated with embryonic stem cells, one major benefit of iPSCs is the opportunity to derive cells from individuals encompassing a spectrum of genetic backgrounds, generating patients’ avatars. Protocols are constantly being optimised with the aim to produce fully mature and functional cells. These protocols utilise growth factors and small molecules which replicate in vivo development. Differentiated iPSCs can be used for an organ-on-a-chip approach44,45,46. Previous work in other fields, such as cancer, has demonstrated that organ-on-a-chip approaches can yield organ-level function superior to other 2D/3D in vitro cell models47,48. In addition, unlike 2D culture systems, organ-on-a-chip can elicit more physiologically relevant pharmacological responses, allowing for more accurate predictions of drug efficacy, toxicity and pharmacokinetics49. Further, recent studies have produced multi-organ platforms, where mutiple organs are integrated on a single device, which allows modelling of the more complex in vivo environment50.
Utilising iPSCs can also have advantages over in vivo models, particularly when considering the ethical principles of the three Rs (replacement, reduction and refinement), which oversee animal research (https://nc3rs.org.uk/the-3rs)51. In vivo animal models can be labour intensive and expensive, and the resultant animal may fail to recapitulate several features of the human phenotype. In addition, the translation of drug trials to humans from animal models can be unsuccessful due to species-specific biological responses, contributing to the translational “valley of death” and high failure rates in the approval of novel therapies52. With iPSCs, mutliple cell lines derived from the same patient can be cultured simultaneously in a much smaller space, with each line having the capacity to be differentiated into several tissue types, which can also reduce the time and labour required. iPSCs are therefore more easily scalable and accessible than animal models. Experiments can be compared across different tissue types to cells that are all derived from the same genetic background. Furthermore, pharmacology and toxicology assessments can be made patient-specific to avoid possible adverse side effects.
UCD-derived iPSCs: state of the art
In the last decade, iPSCs have been increasingly used as experimental models to (i) identify pathogenicity of variant of uncertain significance (VUS), (ii) study disease mechanisms, (iii) develop and screen therapeutic strategies in vitro, with a potential for in vivo applications and iPSC transplantation to treat animal models (Table 3). As the urea cycle is only able to express in its totality in the liver, UCDs-derived iPSCs are frequently differentiated into hepatocyte-like cells. These cells are functionally assessed and compared to isogenic control lines, often generated via CRIPSR/Cas9 editing35,53.
CPS1 deficiency
This disease has been investigated using patient-derived iPSCs, enabling the results to be compared to the clinical phenotype of the affected individuals54. In this study, patient iPSC lines were genetically modified via CRISPR/Cas9 to constitutively express human codon optimised CPS1 from the AAVS1 safe harbour site and then differentiated into hepatocytes. The study aimed to assess the therapeutic potential of this approach. Despite successful differentiation, interestingly, the corrected cells failed to metabolise ammonia any more effectively than the patient cell lines. This unexpected outcome was proposed to be due to transgene promoter methylation and resultant transcriptional silencing in the undifferentiated cells. This study emphasises the requirement for thorough quality control and functional assessment of iPSCs prior to use for downstream experiments.
Ornithine transcarbamylase deficiency
Some studies develop iPSC lines from peripheral blood mononuclear cells (PBMCS), such as with neonatal-onset OTCD patients55,56 (Table 3). These studies show proof of concept that iPSCs can be generated from newly diagnosed patients and thus provide a model to develop a personalised therapeutic approach. Zabulica et al. functionally characterised iPSC-derived hepatocytes compared to isogenic controls and showed an isolated reduced OTC expression from day 8 of differentiation onwards and a significant reduction in ureagenesis. Importantly, the study identified only three potential off-target modifications of CRISPR/Cas9 editing, located in intronic or intergenic regions and highly likely to be biologically inactive53. Laemmle et al. investigated the role of aquaporin 9 (AQP9), a membrane channel protein that facilitates transit of water, glycerol, and urea, in OTCD iPSC-derived hepatocytes. In healthy control, AQP9 overexpression enabled increased ureagenesis and a phenotype closer to the in vivo environment. OTCD iPSC-derived hepatocytes recapitulated the hepatic manifestation of the patient phenotype with reduced ureagenesis and OTC activity. This phenotype was corrected after lentiviral gene therapy57.
Citrullinaemia type I
More than 130 private mutations of the ASS1 gene have been described58,59,60. The identification of variant of uncertain significance (VUS) is a large-scale problem in the era of high-throughput genomics61. The use of patients’ iPSCs can provide a unique model to perform functional studies to assess pathogenicity of a mutation with the additional possibility of screening personalised therapeutics. iPSCs carrying the disease-specific mutations can be artificially created and then used for functional studies as highlighted in citrullinaemia type I (CTLN1)62. Yoshitoshi-Uebayashi et al. differentiated iPSCs from a citrullinaemic patient and isogenic controls into functional hepatocytes, but failed to fully recapitulate a UCD phenotype, potentially due to incomplete hepatocyte differentiation. Baseline ammonia and urea levels between patient and control lines were not different but an ammonia challenge assay exhibited significantly reduced ureagenesis in ASS1-/- cells. Interestingly, the study highlighted significantly increased levels of several tricarboxylic acid (TCA) cycle-related metabolites such as pyruvate, succinate and aspartate, shedding light on aspartate accumulation caused by ASS deficiency and diversion towards TCA cycle63.
Akbari et al. differentiated iPSCs from a CTLN1 patient into endoderm-derived hepatic organoids, following a 2 week-differentiation protocol and prolonged expansion over a year64. The liver organoids for both wild-type and patient cells demonstrated similar architecture and appropriate hepatocyte differentiation confirmed by expression of hepatocyte markers, e.g., HNF4A and CK19 and functional assays, e.g., albumin production, low density lipoprotein (LDL) uptake and glycogen storage. Transcriptomic analysis was similar between wild-type and patient 3D cultures indicating that ASS1 mutation did not interfere with differentiation ability. Functionally, the CTLN1 organoids had significantly increased ammonia, with overexpression of wild-type ASS1 restoring arginine levels to wild-type levels and partially rescuing ureagenesis. These data demonstrated that hepatic organoids can recapitulate several features of the disease phenotype that can be alleviated with therapies targeting gene function64.
Argininosuccinate lyase deficiency
Argininosuccinate lyase deficiency (ASLD) is an atypical UCD due to a systemic phenotype affecting multiple organs4,21. Hence ASLD is a challenging disease to treat as liver-targeted gene therapy corrects ureagenesis but does not alleviate other phenotypic symptoms, especially within the brain65 or endothelial cells66. ASLD iPSCs have been differentiated into other tissues than hepatocytes for investigating ASLD pathophysiology, to address the consequences of arginine and downstream metabolite nitric oxide (NO) deficiencies. Kho et al. demonstrated that ASLD can present as endothelial-dependent hypertension. The authors used multiple models, in addition to clinical studies, including iPSC-derived endothelial cells67. ASLD iPSCs did not differentiate as efficiently into endothelial cells as the controls, indicating that ASL participates in vasculogenesis via NO production. ASLD iPSC-derived endothelial cells had decreased levels of intracellular NO. The cells, when injected into immunodeficient (NOD/SCID/IL2Rγ − ) mice, demonstrated a decreased ability to establish blood capillaries and arterioles in vivo. The data, combined with their results from a human clinical study, mouse model and in vitro cell models, demonstrated that endothelial dysfunction is the main cause of hypertension in ASLD. This shows the ability of iPSCs to be used to model disease and validate findings from other in vitro and in vivo models67. Jin et al. investigated the effect of ASLD in human osteoblasts, using ASLD iPSCs compared to two isogenic controls68. The ASLD-iPSC derived osteoblasts had decreased mineralisation capacity, significantly reduced expression of glycolytic genes such as SLC2A1, PKM and LDHA, or GLUT1 protein suggesting that ASLD leads to the deficient glycolysis. Treatment with NO donor S-nitroso-N-acetylpenicillamine (SNAP) during osteoblast differentiation led to an upregulation of the glycolytic genes. These findings that ASLD-iPSCs have a decreased ability to differentiate into osteoblasts and impaired glycolysis was confirmed in two additional animal models and murine osteoblastic cell lines, highlighting the reliability of iPSCs in investigating pathophysiology and identifying new therapeutic targets68.
Arginase-1 deficiency
Hui et al. studied CRISPR/Cas9-edited iPSCs lines from three patients with arginase-1 deficiency, in an attempt to restore arginase-1 activity. Corrected iPSCs were compared to unedited controls and exhibited restoration of arginase expression and ureagenesis from functional arginase. The three corrected lines were successfully differentiated over 21 days into hepatocytes, alongside their uncorrected counterparts and a control line. Off-target editing was evaluated by sequencing specific sites identified in silico being at risk of off-target effect. Neither insertion nor deletion had occurred69. Such studies provide a strategy for restoring gene function by editing paving the way for genetically modified cell therapies to treat UCDs. Arginase-1 deficiency has also been studied using iPSC-derived hepatocyte-like cells, as well as iPSC-derived macrophages. Utilising PiggyBac technology alongside CRISPR/Cas9, exons 7 and 8 of ARG1 were reincorporated into iPSCs derived from an arginase-1 deficient mouse model70, a knockout model where mutant animals die at about 2 weeks of age, in order to assess whether ARG1 function could be restored. Successful gene repair was confirmed, but ureagenesis remained suboptimal compared to adult hepatocytes, probably caused by insufficient hepatocyte differentiation. In comparison, iPSC-derived macrophages showed corrected arginase function that could be explained by a more ready differentiation into a functionally mature state71. The study demonstrated how in vitro research can be developed using iPSCs and CRISPR/Cas9 to conduct follow-up investigations in various differentiated cell types. Differentiated cells can then be transplanted in vivo to enable further maturation and long-term repopulation of hepatocytes, for instance72,73. The same group also used transcription activator-like effector nucleases (TALENs) to correct the same defect and again, differentiated these cells into hepatocyte-like cells. These cells were then transplanted back via intravenous infusion into 12-week-old arginase-1 deficient mice, in a tamoxifen-induced ARG1 knock-out mouse model. iPSC-transplanted arginase-1 deficient mice survived an additional week compared to untreated knock-out littermates without reaching statistically significant increase of survival. The treated mice had increased arginase-1 expression but ureagenesis was not comparable to that of wild-type controls. The inability to fully rescue the phenotype may be caused by the inability for transplanted cells to repopulate the periportal hepatocytes where the urea cycle is expressed, suboptimal hepatocyte differentiation, low engraftment or immunological rejection of transplanted cells74.
Citrin deficiency
Sin et al. differentiated iPSC from a patient with citrin deficiency into hepatocytes. Functional assessment confirmed that patient cells did not exhibit ureagenesis. Genes involved in mitochondrial beta-oxidation were downregulated in the patient cells, with abnormal mitochondrial structure, leading to significantly higher levels of cellular triglycerides and lipid granule levels compared to the wild-type cells. The work was further validated by a complementary Slc25a13 knockout mouse model, which recapitulated some, although not all, of the patient phenotype75.
Limitations of iPSCs for UCD research
iPSCs models present intrinsic limitations, particularly for therapeutic application and clinical translation.
One of the significant challenges faced by iPSCs research is, despite ongoing improvements in differentiation protocols, the immaturity of iPSC-derived cells, which remain closer to foetal rather than their fully differentiated counterparts76. This has been demonstrated in modelling UCDs, where both wild-type and mutated immature differentiated hepatocytes failed to achieve equivalent functionality with that of in vivo hepatocytes53,63,74. This means that results must be interpreted with caution for UCDs and particularly when modelling disease specificities associated with age. Furthermore, differentiation efficiency and reproducibility can vary due to differing protocols, genetic backgrounds, passage number and epigenetic memory77,78,79. iPSCs can also acquire mutations that confer a growth advantage, particularly p53 mutations, advocating for thorough genetic characterisation prior to translation to the clinic80. With CRISPR/Cas9 mediated reprogramming, off-target effects that could adversely alter the cell function need to be carefully assessed81. However, efforts have been made to improve identification of off-targets using in vitro assays, including circularisation for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq)82 and selective enrichment and identification of tagged genomic DNA ends by sequencing (SITE-seq)83, as well as improving the computational platforms used to design sgRNAs84. Another approach that can reduce the number of off-target effects is base editing, whereby a catalytically inactive Cas9 is employed together with cytidine deaminases to introduce site specific DNA mutations/corrections without the requirement for a double-strand break85.
Another iPSC limitation of use in research is that the cell population produced may not be representative of more complex in vivo architecture and morphology. Differentiation protocols often make a single cell type, whereas in vivo hepatocytes, for instance, are in close proximity to non-parenchymal cells such as stellate cells, Kupffer cells and endothelial cells, facilitating crosstalk between parenchymal and non-parenchymal cells. This heterogeneous population, in addition to nutrients, growth factors and vasculature all work together to support the genetic expression and viability of the hepatocytes86. However, there are steps to overcome this limitation with the use of co-cultures – for instance, co-culturing hepatocytes with endothelial and stromal cells aids maturation of hepatocytes87. In the context of UCDs, urea cycle activity and ammonia metabolism are confined to zone 1 located hepatocytes nearest the portal vein, as part of the regulated compartmentalisation of the hepatic lobule88. However, at present, only partial zonation is able to be achieved, which may influence how well ammonia metabolism can be modelled89. Nevertheless, these co-cultures cannot completely recapture the complexity of the in vivo environment.
A safety issue for clinical translation is the risk of teratoma formation90. This is particularly a consideration for UCDs as these diseases have occasionally been associated with hepatocellular carcinoma4,91,92. There is a risk of potential tumorigenicity and malignant tumours if transplanted cells are contaminated with undifferentiated iPSCs. However, there are treatments that could potentially treat these teratomas if they occur93. There are also purification methods such as flow cytometry sorting and small chemical molecules that initiate death of undifferentiated iPSCs which could help lower the risk of tumorigenicity. However, the effectiveness of such methods has yet to be fully elucidated94. Autologous iPSC therapies effectively eliminate the potential for immune rejection. The generation of clinical grade autologous iPSC-derived cells entails high production costs associated with a rigorous quality control system. As the generation of the patient-specific tissue of interest can take several months, this timeframe might not be suitable for neonatal-onset UCD patients, which require an urgent liver replacement strategy95. Therefore, an alternative approach is to use allogeneic iPSC-derived cell sources which would enable biobanking a limited number of approved iPSCs from assorted human leukocyte antigen (HLA)-homozygous donors that would match the bulk of the population96. These cells would have all undergone regulatory clearance and have been thoroughly tested meaning that the cells could be more readily used to treat patients who require timely intervention. However, patients would be required to undergo lifelong immunosuppression treatment.
For clinical trial applications, ethical consent should be obtained freely. For children, eligibility should be considered only if there is minimal or low risk with potential for direct benefit, and the procedure is as favourable as available alternatives97. Regulatory guidelines such as the US Food and Drug Administration (FDA)98, the European Medicines Agency (EMA) (https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-human-cell-based-medicinal-products_en.pdf) and the International Society for Stem Cell Research are in place to ensure that patient safety is at the forefront99,100,101. Adhering to these guidelines, alongside potential intellectual property and patent considerations, can add substantial time and effort to the setting up and delivery of iPSCs studies in accordance with Good Clinical Practice. Additionally, scalable technology to meet regulatory standards remains a challenge for generating genetically modified iPSCs for cell therapy, as it requires special equipment alongside skilled operators102,103,104. Thus, these limitations may prevent potential therapeutic treatments from reaching patients due to prohibitive cost and time requirements.
Future directions
iPSC differentiation provides a unique opportunity to study a genetic disease in a specific genetic and epigenetic background in vitro and screen for therapeutic candidates. iPSCs have improved our knowledge of the pathophysiological mechanisms of several diseases, alongside accelerating progress in our understanding of human physiology at the cellular level. In targeting UCDs, iPSC differentiation into hepatocytes has been the primary focus as the urea cycle is only expressed in totality in this cell type. Optimisation of the hepatocyte differentiation is key in reproducing a defective ureagenesis. Differentiation in additional cell subtypes has enabled to study novel mechanistic approaches and pathophysiology, especially in endothelial cells67 and osteoblasts68 in ASLD. In the future, iPSC differentiation towards other cell types, particularly involved in the specific pathogenesis of the central nervous system, will be of utmost interest.
iPSCs can be used as a platform for drug discovery, testing different pathways of UCDs for potential therapeutic treatment. For example, induced autophagy has recently been shown to increase ureagenesis, via the cell-penetrating autophagy-inducing Tat-Beclin-1 (TB-1) peptide in OTC and ASL deficiencies when tested in an animal model105,106. This approach could be translated to drug screening using iPSCs for other compounds. Drug screening via this approach has been tried in different contexts, including using Sendai virus, and would be amenable to discover novel therapies to restore ureagenesis in UCD or to protect the brain in the context of hyperammonaemia107,108.
Advancements in both iPSCs differentiation protocols and CRISPR/Cas9 techniques are continually emerging. For instance, recently researchers have managed to cultivate four organs in a microbioreactor from iPSCs from a single donor. The organ models were positioned in separate compartments of the bioreactor and connected by a microfluidic network109. In addition, another study has generated an in vitro whole-organ “Bioreactor grown Artificial Liver Model” (BALM) which enables the long-term 3D culture of iPSC-derived hepatocyte-like cells. This model also allows the transduction of adeno-associated viral and lentiviral vectors, meaning this model could provide a more representative preclinical therapy testing environment110. There are also numerous techniques to help identify off-target effects associated with CRISPR/Cas9 editing, such as in vitro and cell-based assays83,111,112. These allow off-target effects to be more readily assessed and thus can be taken into account when performing differentiation experiments. Alongside this, the rapid evolution of next-generation sequencing techniques provides means to analyse transcriptomics and chromatin accessibility of both iPSCs and differentiated cell populations. Utilising different multi-omics approaches allows refinement of in-depth analysis and the discovery of novel pathophysiological mechanisms113.
Conclusion
UCD-derived iPSCs are progressively expanding their applications in several areas, from supporting a genetic diagnosis in the genomic era, studying pathophysiology to drug development and validation. Differentiated iPSCs creating patients’ avatars are of particular interest in proof-of-concept studies where they recapitulate key aspects of the disease in a specific disease-enabling genetic and epigenetic background, allowing novel pathophysiological insights. By providing this invaluable opportunity to analyse a genotype of interest within the genetic background of the patient, this allows functional studies in a physiologically relevant model. iPSCs offer the unique opportunity to implement personalised medicine and perform drug screens. The field still faces several challenges, particularly with optimising iPSCs differentiation and successfully translating proof-of-concept studies to effective long-term treatments whilst minimising the risk of adverse effects. While considerable efforts are in place to overcome these challenges, iPSCs present a promising avenue for UCD research and therapy development.
References
Gebhardt, R. & Matz-Soja, M. Liver zonation: Novel aspects of its regulation and its impact on homeostasis. World J. Gastroenterol. 20, 8491–8504 (2014).
Summar, M. L. et al. The incidence of urea cycle disorders. Mol. Genet Metab. 110, 179–180 (2013).
Matsumoto, S. et al. Urea cycle disorders-update. J. Hum. Genet. 64, 833–847 (2019).
Baruteau, J. et al. Expanding the phenotype in argininosuccinic aciduria: need for new therapies. J. Inherit. Metab. Dis. 40, 357–368 (2017).
Savy, N. et al. Acute pediatric hyperammonemia: current diagnosis and management strategies. Hepat. Med. 10, 105–115 (2018).
Nagamani, S. C., Erez, A. & Lee, B. Argininosuccinate lyase deficiency. Genet. Med. 14, 501–507 (2012).
Darwish, A. A., McKiernan, P. & Chardot, C. Paediatric liver transplantation for metabolic disorders. Part 1: Liver-based metabolic disorders without liver lesions. Clin. Res. Hepatol. Gastroenterol. 35, 194–203 (2011).
Darwish, A. A., McKiernan, P. & Chardot, C. Paediatric liver transplantation for metabolic disorders. Part 2: Metabolic disorders with liver lesions. Clin. Res. Hepatol. Gastroenterol. 35, 271–280 (2011).
Raper, S. E. et al. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 80, 148–158 (2003).
Inc, U. P. Safety and Dose-Finding Study of DTX301 (scAAV8OTC) in Adults With Late-Onset OTC Deficiency (CAPtivate), https://clinicaltrials.gov/ct2/show/NCT02991144 (2016).
Arcturus Therapeutics, I. Safety, Tolerability, and Pharmacokinetics of ARCT-810 in Stable Adult Subjects With Ornithine Transcarbamylase Deficiency, https://clinicaltrials.gov/ct2/show/NCT04442347 (2020).
Bernard Kok, X. L., David Ebeid, Xianghong Li, Ryan Weiss, Nairika Meshgin, Arturo Barcenas, Naha Parayath, Jivan Yewle, Brian Truong, Blair Madison, Bruce F. Scharschmidt, Julian D. Down, Devon J. Shedlock, Jack Rychak, Eric M. Ostertag and Jingjing Jiang. in 14TH INTERNATIONAL CONGRESS OF INBORN ERRORS OF METABOLISM (Sydney, Australia (held online), 2021).
Haberle, J. et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J. Rare Dis. 7, 32 (2012).
Mauhin, W. et al. Update on lysinuric protein intolerance, a multi-faceted disease retrospective cohort analysis from birth to adulthood. Orphanet J. Rare Dis. 12, 3 (2017).
Lee, J. S. et al. Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell 174, 1559–1570 e1522 (2018).
Waisbren, S. E., Stefanatos, A. K., Kok, T. M. Y. & Ozturk-Hismi, B. Neuropsychological attributes of urea cycle disorders: a systematic review of the literature. J. Inherit. Metab. Dis. 42, 1176–1191 (2019).
Braissant, O., McLin, V. A. & Cudalbu, C. Ammonia toxicity to the brain. J. Inherit. Metab. Dis. 36, 595–612 (2013).
Redant, S. et al. Management of late onset urea cycle disorders-a remaining challenge for the intensivist? Ann. Intensive Care 11, 2 (2021).
Gropman, A. L., Summar, M. & Leonard, J. V. Neurological implications of urea cycle disorders. J. Inherit. Metab. Dis. 30, 865–879 (2007).
Liu, X. B. et al. Hepatic arginase deficiency fosters dysmyelination during postnatal CNS development. JCI Insight 4, https://doi.org/10.1172/jci.insight.130260 (2019).
Baruteau, J. et al. Argininosuccinic aciduria: recent pathophysiological insights and therapeutic prospects. J. Inherit. Metab. Dis. 42, 1147–1161 (2019).
Diez-Fernandez, C. et al. Argininosuccinate neurotoxicity and prevention by creatine in argininosuccinate lyase deficiency: An in vitro study in rat three-dimensional organotypic brain cell cultures. J. Inherit. Metab. Dis. 42, 1077–1087 (2019).
Lerner, S. et al. ASL metabolically regulates tyrosine hydroxylase in the nucleus locus coeruleus. Cell Rep. 29, 2144–2153 e2147 (2019).
Lerner, S. et al. ASL expression in ALDH1A1(+) neurons in the substantia nigra metabolically contributes to neurodegenerative phenotype. Hum. Genet. 140, 1471–1485 (2021).
Barilli, A. et al. In Lysinuric Protein Intolerance system y+L activity is defective in monocytes and in GM-CSF-differentiated macrophages. Orphanet J. Rare Dis. 5, 32 (2010).
Rivera-Barahona, A. et al. Functional characterization of the spf/ash splicing variation in OTC deficiency of mice and man. PLoS ONE 10, e0122966 (2015).
Allegri, G. et al. Comprehensive characterization of ureagenesis in the spf(ash) mouse, a model of human ornithine transcarbamylase deficiency, reveals age-dependency of ammonia detoxification. J. Inherit. Metab. Dis. 42, 1064–1076 (2019).
Deignan, J. L., Cederbaum, S. D. & Grody, W. W. Contrasting features of urea cycle disorders in human patients and knockout mouse models. Mol. Genet. Metab. 93, 7–14 (2008).
Takahashi, K. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 (2007).
Evans, M. J. & Kaufman, M. H. Establishment in culture of pluripotential cells from mouse embryos. Nature 292, 154–156 (1981).
Martin, G. R. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc. Natl Acad. Sci. USA 78, 7634–7638 (1981).
Liu, H., Dong, H., Robertson, K. & Liu, C. DNA methylation suppresses expression of the urea cycle enzyme carbamoyl phosphate synthetase 1 (CPS1) in human hepatocellular carcinoma. Am. J. Pathol. 178, 652–661 (2011).
Cheng, P. N. et al. Pegylated recombinant human arginase (rhArg-peg5,000mw) inhibits the in vitro and in vivo proliferation of human hepatocellular carcinoma through arginine depletion. Cancer Res. 67, 309–317 (2007).
Westerink, W. M. & Schoonen, W. G. Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol. Vitr. 21, 1581–1591 (2007).
Corbett, J. L. & Duncan, S. A. iPSC-derived hepatocytes as a platform for disease modeling and drug discovery. Front Med (Lausanne) 6, 265 (2019).
Guo, L. et al. Similarities and differences in the expression of drug-metabolizing enzymes between human hepatic cell lines and primary human hepatocytes. Drug Metab. Dispos. 39, 528–538 (2011).
Gramignoli, R. et al. Rapid and sensitive assessment of human hepatocyte functions. Cell Transpl. 23, 1545–1556 (2014).
Heslop, J. A. et al. Mechanistic evaluation of primary human hepatocyte culture using global proteomic analysis reveals a selective dedifferentiation profile. Arch. Toxicol. 91, 439–452 (2017).
Richert, L. et al. Gene expression in human hepatocytes in suspension after isolation is similar to the liver of origin, is not affected by hepatocyte cold storage and cryopreservation, but is strongly changed after hepatocyte plating. Drug Metab. Dispos. 34, 870–879 (2006).
LeCluyse, E. L., Witek, R. P., Andersen, M. E. & Powers, M. J. Organotypic liver culture models: meeting current challenges in toxicity testing. Crit. Rev. Toxicol. 42, 501–548 (2012).
Yamaguchi, T. et al. Generation of functional human hepatocytes in vitro: current status and future prospects. Inflamm. Regen. 39, 13 (2019).
Giancotti, A. et al. Functions and the emerging role of the foetal liver into regenerative medicine. Cells 8, https://doi.org/10.3390/cells8080914 (2019).
Larijani, B. et al. Stem cell therapy in treatment of different diseases. Acta Med Iran. 50, 79–96 (2012).
Ma, C., Peng, Y., Li, H. & Chen, W. Organ-on-a-chip: a new paradigm for drug development. Trends Pharm. Sci. 42, 119–133 (2021).
Wu, Q. et al. Organ-on-a-chip: recent breakthroughs and future prospects. Biomed. Eng. Online 19, 9 (2020).
Deng, J. et al. Engineered liver-on-a-chip platform to mimic liver functions and its biomedical applications: a review. Micromachines (Basel) 10, https://doi.org/10.3390/mi10100676 (2019).
van den Berg, A., Mummery, C. L., Passier, R. & van der Meer, A. D. Personalised organs-on-chips: functional testing for precision medicine. Lab Chip 19, 198–205 (2019).
Liu, X. et al. Tumor-on-a-chip: from bioinspired design to biomedical application. Microsyst. Nanoeng. 7, 50 (2021).
Esch, E. W., Bahinski, A. & Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Disco. 14, 248–260 (2015).
Rajan, S. A. P. et al. Probing prodrug metabolism and reciprocal toxicity with an integrated and humanized multi-tissue organ-on-a-chip platform. Acta Biomater. 106, 124–135 (2020).
Robinson, N. B. et al. The current state of animal models in research: A review. Int J. Surg. 72, 9–13 (2019).
Roberts, S. F., Fischhoff, M. A., Sakowski, S. A. & Feldman, E. L. Perspective: Transforming science into medicine: how clinician-scientists can build bridges across research’s “valley of death”. Acad. Med 87, 266–270 (2012).
Zabulica, M. et al. Gene Editing Correction of a Urea Cycle Defect in Organoid Stem Cell Derived Hepatocyte-like Cells. Int J Mol Sci 22, https://doi.org/10.3390/ijms22031217 (2021).
Nitzahn, M. et al. CRISPR-Mediated Genomic Addition to CPS1 Deficient iPSCs is Insufficient to Restore Nitrogen Homeostasis. Yale J. Biol. Med 94, 545–557 (2021).
Guan, J. et al. Generation of a human induced pluripotent stem cell line (SDQLCHi036-A) from a patient with ornithine transcarbamylase deficiency carrying a deletion involving 3-9 exons of OTC gene. Stem Cell Res 52, 102220 (2021).
Yang, X. et al. Generation of an induced pluripotent stem cell line (SDQLCHi009-A) from a patient with 47,XXY and ornithine transcarbamylase deficiency carrying a hemizygote mutation in OTC. Stem Cell Res 43, 101704 (2020).
Laemmle, A. et al. Aquaporin 9 induction in human iPSC-derived hepatocytes facilitates modeling of ornithine transcarbamylase deficiency. Hepatology https://doi.org/10.1002/hep.32247 (2021).
Kimani, J. K. et al. Functional analysis of novel splicing and missense mutations identified in the ASS1 gene in classical citrullinemia patients. Clin. Chim. Acta 438, 323–329 (2015).
Lin, Y. et al. Citrullinemia type I is associated with a novel splicing variant, c.773 + 4A > C, in ASS1: a case report and literature review. BMC Med Genet 20, 110 (2019).
Diez-Fernandez, C., Rufenacht, V. & Haberle, J. Mutations in the Human Argininosuccinate Synthetase (ASS1) Gene, Impact on Patients, Common Changes, and Structural Considerations. Hum. Mutat. 38, 471–484 (2017).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 (2015).
Yuan, F. et al. Generation of an ASS1 heterozygous knockout human embryonic stem cell line, WAe001-A-13, using CRISPR/Cas9. Stem Cell Res. 26, 67–71 (2018).
Yoshitoshi-Uebayashi, E. Y. et al. Modelling urea-cycle disorder citrullinemia type 1 with disease-specific iPSCs. Biochem. Biophys. Res. Commun. 486, 613–619 (2017).
Akbari, S. et al. Robust, long-term culture of endoderm-derived hepatic organoids for disease modeling. Stem Cell Rep. 13, 627–641 (2019).
Baruteau, J. et al. Argininosuccinic aciduria fosters neuronal nitrosative stress reversed by Asl gene transfer. Nat. Commun. 9, 3505 (2018).
Nagamani, S. C. et al. Nitric-oxide supplementation for treatment of long-term complications in argininosuccinic aciduria. Am. J. Hum. Genet. 90, 836–846 (2012).
Kho, J. et al. Argininosuccinate lyase deficiency causes an endothelial-dependent form of hypertension. Am. J. Hum. Genet. 103, 276–287 (2018).
Jin, Z. et al. Nitric oxide modulates bone anabolism through regulation of osteoblast glycolysis and differentiation. J. Clin. Investig. 131, https://doi.org/10.1172/JCI138935 (2021).
Lee, P. C. et al. Restoring ureagenesis in hepatocytes by CRISPR/Cas9-mediated genomic addition to arginase-deficient induced pluripotent stem cells. Mol. Ther. Nucleic Acids 5, e394 (2016).
Sin, Y. Y. et al. Inducible arginase 1 deficiency in mice leads to hyperargininemia and altered amino acid metabolism. PLoS ONE 8, e80001 (2013).
Sin, Y. Y., Price, P. R., Ballantyne, L. L. & Funk, C. D. Proof-of-concept gene editing for the murine model of inducible arginase-1 deficiency. Sci. Rep. 7, 2585 (2017).
Gomez-Lechon, M. J. & Tolosa, L. Human hepatocytes derived from pluripotent stem cells: a promising cell model for drug hepatotoxicity screening. Arch. Toxicol. 90, 2049–2061 (2016).
Zhu, S. et al. Mouse liver repopulation with hepatocytes generated from human fibroblasts. Nature 508, 93–97 (2014).
Sin, Y. Y., Ballantyne, L. L., Richmond, C. R. & Funk, C. D. Transplantation of gene-edited hepatocyte-like cells modestly improves survival of arginase-1-deficient mice. Mol. Ther. Nucleic Acids 10, 122–130 (2018).
Kim, Y. et al. Malfunction in mitochondrial beta-oxidation contributes to lipid accumulation in hepatocyte-like cells derived from citrin deficiency-induced pluripotent stem cells. Stem Cells Dev. 25, 636–647 (2016).
Sampaziotis, F., Segeritz, C. P. & Vallier, L. Potential of human induced pluripotent stem cells in studies of liver disease. Hepatology 62, 303–311 (2015).
Kajiwara, M. et al. Donor-dependent variations in hepatic differentiation from human-induced pluripotent stem cells. Proc. Natl Acad. Sci. USA 109, 12538–12543 (2012).
Rouhani, F. et al. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 10, e1004432 (2014).
Kim, K. et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 29, 1117–1119 (2011).
Merkle, F. T. et al. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545, 229–233 (2017).
Herai, R. H. Avoiding the off-target effects of CRISPR/cas9 system is still a challenging accomplishment for genetic transformation. Gene 700, 176–178 (2019).
Tsai, S. Q. et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 14, 607–614 (2017).
Cameron, P. et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat. Methods 14, 600–606 (2017).
Doench, J. G. et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 34, 184–191 (2016).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Roy-Chowdhury, N., Wang, X., Guha, C. & Roy-Chowdhury, J. Hepatocyte-like cells derived from induced pluripotent stem cells. Hepatol. Int 11, 54–69 (2017).
Stevens, K. R. et al. InVERT molding for scalable control of tissue microarchitecture. Nat. Commun. 4, 1847 (2013).
Ma, R., Martinez-Ramirez, A. S., Borders, T. L., Gao, F. & Sosa-Pineda, B. Metabolic and non-metabolic liver zonation is established non-synchronously and requires sinusoidal Wnts. Elife 9, https://doi.org/10.7554/eLife.46206 (2020).
Danoy, M. et al. Characterization of liver zonation-like transcriptomic patterns in HLCs derived from hiPSCs in a microfluidic biochip environment. Biotechnol. Prog. 36, e3013 (2020).
Lee, A. S., Tang, C., Rao, M. S., Weissman, I. L. & Wu, J. C. Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat. Med. 19, 998–1004 (2013).
Bigot, A., Tchan, M. C., Thoreau, B., Blasco, H. & Maillot, F. Liver involvement in urea cycle disorders: a review of the literature. J. Inherit. Metab. Dis. 40, 757–769 (2017).
Wilson, J. M., Shchelochkov, O. A., Gallagher, R. C. & Batshaw, M. L. Hepatocellular carcinoma in a research subject with ornithine transcarbamylase deficiency. Mol. Genet Metab. 105, 263–265 (2012).
Wuputra, K. et al. Prevention of tumor risk associated with the reprogramming of human pluripotent stem cells. J. Exp. Clin. Cancer Res. 39, 100 (2020).
Doss, M. X. & Sachinidis, A. Current challenges of iPSC-based disease modeling and therapeutic implications. Cells 8, https://doi.org/10.3390/cells8050403 (2019).
Ohnuki, M. & Takahashi, K. Present and future challenges of induced pluripotent stem cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, 20140367 (2015).
Jang, Y. et al. Development of immunocompatible pluripotent stem cells via CRISPR-based human leukocyte antigen engineering. Exp. Mol. Med 51, 1–11 (2019).
CIOMS. International Ethical Guidelines for Health-related Research Involving Humans. (2016).
Health, U. S. D. o., Human Services, F., Drug Administration, C. f. B. E. & Research. Guidance for human somatic cell therapy and gene therapy. Hum. Gene Ther. 12, 303–314 (2001).
Daley, G. Q. et al. Setting global standards for stem cell research and clinical translation: the 2016 ISSCR guidelines. Stem Cell Rep. 6, 787–797 (2016).
Caulfield, T. et al. Stem cell research policy and iPS cells. Nat. Methods 7, 28–33 (2010).
Krackhardt, A. M. et al. Clinical translation and regulatory aspects of CAR/TCR-based adoptive cell therapies-the German Cancer Consortium approach. Cancer Immunol. Immunother. 67, 513–523 (2018).
van der Loo, J. C. & Wright, J. F. Progress and challenges in viral vector manufacturing. Hum. Mol. Genet. 25, R42–R52 (2016).
Ausubel, L. J. et al. Production of CGMP-grade lentiviral vectors. Bioprocess Int. 10, 32–43 (2012).
Clement, N. & Grieger, J. C. Manufacturing of recombinant adeno-associated viral vectors for clinical trials. Mol. Ther. Methods Clin. Dev. 3, 16002 (2016).
Soria, L. R. et al. Beclin-1-mediated activation of autophagy improves proximal and distal urea cycle disorders. EMBO Mol. Med. 13, e13158 (2021).
Soria, L. R. et al. Enhancement of hepatic autophagy increases ureagenesis and protects against hyperammonemia. Proc. Natl Acad. Sci. USA 115, 391–396 (2018).
Elitt, M. S., Barbar, L. & Tesar, P. J. Drug screening for human genetic diseases using iPSC models. Hum. Mol. Genet. 27, R89–R98 (2018).
Imamura, K. et al. iPSC screening for drug repurposing identifies anti-RNA virus agents modulating host cell susceptibility. FEBS Open Bio 11, 1452–1464 (2021).
Ramme, A. P. et al. Autologous induced pluripotent stem cell-derived four-organ-chip. Future Sci. OA 5, FSO413 (2019).
Lorvellec, M. et al. An In Vitro Whole-Organ Liver Engineering for Testing of Genetic Therapies. iScience 23, 101808 (2020).
Kim, D. et al. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 12, 237–243 (2015). 231 p following 243.
Yan, W. X. et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 8, 15058 (2017).
Hasin, Y., Seldin, M. & Lusis, A. Multi-omics approaches to disease. Genome Biol. 18, 83 (2017).
van Karnebeek, C. D. et al. Mitochondrial carbonic anhydrase VA deficiency resulting from CA5A alterations presents with hyperammonemia in early childhood. Am. J. Hum. Genet 94, 453–461 (2014).
Kenneson, A. & Singh, R. H. Presentation and management of N-acetylglutamate synthase deficiency: a review of the literature. Orphanet J. Rare Dis. 15, 279 (2020).
Diez-Fernandez, C. & Haberle, J. Targeting CPS1 in the treatment of Carbamoyl phosphate synthetase 1 (CPS1) deficiency, a urea cycle disorder. Expert Opin. Ther. Targets 21, 391–399 (2017).
Brassier, A. et al. Long-term outcomes in Ornithine Transcarbamylase deficiency: a series of 90 patients. Orphanet J. Rare Dis. 10, 58 (2015).
Sin, Y. Y., Baron, G., Schulze, A. & Funk, C. D. Arginase-1 deficiency. J. Mol. Med (Berl.) 93, 1287–1296 (2015).
Martinelli, D. et al. The hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Orphanet J. Rare Dis. 10, 29 (2015).
Hayasaka, K. Metabolic basis and treatment of citrin deficiency. J. Inherit. Metab. Dis. 44, 110–117 (2021).
Godoy, P. et al. Gene network activity in cultivated primary hepatocytes is highly similar to diseased mammalian liver tissue. Arch. Toxicol. 90, 2513–2529 (2016).
Gupta, R. et al. Comparing in vitro human liver models to in vivo human liver using RNA-Seq. Arch. Toxicol. 95, 573–589 (2021).
Hart, S. N. et al. A comparison of whole genome gene expression profiles of HepaRG cells and HepG2 cells to primary human hepatocytes and human liver tissues. Drug Metab. Dispos. 38, 988–994 (2010).
Zeilinger, K., Freyer, N., Damm, G., Seehofer, D. & Knospel, F. Cell sources for in vitro human liver cell culture models. Exp. Biol. Med. (Maywood) 241, 1684–1698 (2016).
Bruno, S. et al. Human liver stem cells: a liver-derived mesenchymal stromal cell-like population with pro-regenerative properties. Front Cell Dev. Biol. 9, 644088 (2021).
Si-Tayeb, K. et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology 51, 297–305 (2010).
Shi, Y., Kirwan, P. & Livesey, F. J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 7, 1836–1846 (2012).
Matsunari, H. et al. Modeling lethal X-linked genetic disorders in pigs with ensured fertility. Proc. Natl Acad. Sci. USA 115, 708–713 (2018).
Barre-Sinoussi, F. & Montagutelli, X. Animal models are essential to biological research: issues and perspectives. Future Sci. OA 1, FSO63 (2015).
Sugahara, G. et al. Humanized liver mouse model with transplanted human hepatocytes from patients with ornithine transcarbamylase deficiency. J. Inherit. Metab. Dis. 44, 618–628 (2021).
Ginn, S. L. et al. Efficient in vivo editing of OTC-deficient patient-derived primary human hepatocytes. JHEP Rep. 2, 100065 (2020).
Funding
This work was supported by funding from the United Kingdom Medical Research Council Clinician Scientist Fellowship MR/T008024/1 and NIHR Great Ormond Street Hospital Biomedical Research Centre to JB. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Contributions
C.D. and J.B. designed the study. C.D. wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Duff, C., Baruteau, J. Modelling urea cycle disorders using iPSCs. npj Regen Med 7, 56 (2022). https://doi.org/10.1038/s41536-022-00252-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41536-022-00252-5
