
Abstract
The regenerative capacity of adult human tissues and organs is limited, but recent developments have seen the advent of promising new technologies for regenerative therapy. The human heart is of particular interest for regenerative medicine, as cardiac tissue damage is repaired by the formation of rigid scar tissue, which causes inevitable structural changes and progressive functional decline leading to heart failure. Cardiac regenerative medicine aims to prevent scar formation or replace existing scars to halt or reverse adverse remodeling and therapeutic approaches include the use of biomaterials, gene therapies, delivery of growth factors, and (stem) cell therapies. Regenerative therapies, however, face significant obstacles in a hostile microenvironment. While the early immune response to a myocardial infarct is essential to ensure tissue integrity and to avoid fatal cardiac rupture, excessive activation of endogenous repair mechanisms may lead to ongoing inflammation, fibrosis, and sustained autoimmune-mediated tissue damage. Anti-cardiac autoreactivity of the adaptive immune system has been suggested to be involved in structural remodeling, functional decline, and the development of heart failure. It is, therefore, crucial to first understand the endogenous response to cardiac tissue damage and how to restore immune tolerance to cardiac tissue, before additional regenerative therapies can achieve their full potential.
Similar content being viewed by others

Introduction
Heart failure after an acute myocardial infarct (MI) remains a significant cause of mortality and a major clinical challenge. An infarct induces severe tissue damage and the immune system is crucial in orchestrating the various steps of the post MI healing process. A tightly controlled interplay between immune cell populations is responsible for removing cellular debris, restoring tissue integrity to prevent fatal cardiac rupture, and then quickly resolve inflammation. An appropriate balance between inflammatory and regenerative mechanisms is essential, as excessive early inflammation may cause collateral damage to healthy tissue and is detrimental to healing. While beneficial vs. detrimental effects of early innate immune populations, including neutrophils and macrophages, have received considerable attention in recent years,1 the role of the adaptive immune system at later stages and during cardiac remodeling is far from understood. Importantly, post MI patients present elevated levels of anti-cardiac auto-antibodies,2 consistent with the involvement of the adaptive immune system and indicating an aberrant activation by self-antigens causing anti-cardiac autoimmunity. The notion that the development and progression of heart failure is promoted by an underlying autoimmune-inflammatory condition, maintaining persistent low levels of tissue damage that sustain cardiomyocyte loss, fibrosis, and pathological ventricular remodeling is recently gaining support.
We propose that restoring cardiac immune tolerance by immunomodulatory interventions post MI is essential to pave the way for more efficient outcomes of regenerative therapies.
MI and heart failure
Recent advances in the clinical response to MI have significantly reduced acute mortality.3 However, endogenous repair mechanisms gradually replace necrotic cardiac tissue by a non-contractile scar, which may lead to ventricular remodeling. This is due to the overstretching of surviving cardiomyocytes to maintain cardiac output, leading to thinning of the ventricular wall and development of heart failure.4, 5 Current management of heart failure relies on beta-blockers, diuretics, and vasodilators such as angiotensin converting enzyme inhibitors. Surgical options are to change the shape of the left ventricle or implant assist devices such as pacemakers or defibrillators.6 However, these treatments do not completely stop progression of disease and cannot restore heart function, presenting an urgent clinical need for curative treatment options that target underlying mechanisms. Current experimental approaches to treat heart failure largely focus on cardiomyocyte deficiency and aim to replace lost cardiomyocytes in the infarcted area. Efforts to increase cardiomyocyte numbers target the limited proliferative capacity of pre-existing cardiomyocytes or aim to differentiate endogenous or delivered progenitor cells into cardiomyocytes.7 Recent advances have been reviewed extensively8, 9 and include (1) biomaterial-based approaches,10 (2) gene therapy using plasmid DNA for gene transfer11 or silencing/short hairpin RNA, microRNA, and long non-coding RNA for gene modulation,12, 13 (3) exogenous administration of growth factors such as neuregulin14 or fibroblast growth factor,15 and (4) cell therapies using mesenchymal stromal cells (MSCs),16,17,18 cardiac and bone marrow-derived c-kit+ cells,19, 20 cardiosphere cells,21, 22 and pluripotent stem cell-derived cardiac cells.23 While promising results have been obtained in experimental pre-clinical models, many of these approaches still face severe obstacles concerning efficacy of production and delivery, as well as immunological rejection. In particular though, they all share the common hurdle of a hostile inflammatory microenvironment and so far benefits observed in clinical trials are disappointing or inconsistent.9, 23
Post MI autoimmunity
The immune system is a sophisticated system of cells and effector molecules essential to protect the host from pathogens and ensure tissue integrity. Two fundamental features to ensure these roles are the ability to discriminate between the body’s own (self) and foreign (non-self) agents and to react quickly if there are signs of “danger” usually associated with tissue damage, including the release of intracellular molecules (danger-associated molecular patterns, DAMPs) such as high-mobility group box-1, heat shock proteins, nucleic acids, and mitochondrial molecules.24 Tight regulatory networks are in place to restrain immune responses to self-antigens and to downregulate a response once an initial insult is cleared to minimize inflammatory damage. If these regulatory networks fail, chronic inflammatory conditions may develop, including allergies and autoimmunity. Autoimmune disease is a highly diverse group of conditions, classified according to main target organs and manifestations. While a certain level of immunological autoreactivity is normal and generally kept in check, an environmental trigger in a genetically susceptible individual can overwhelm regulatory mechanisms and result in immune-mediated tissue destruction.
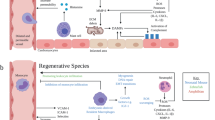
Physical trauma and the associated release of DAMPs from dying cells causes local and systemic inflammation and can be a potent trigger for the induction of autoimmunity.25 The release of previously sequestered antigens from necrotic cells progressively diversifies the autoreactive lymphocyte repertoire in a process termed epitope spreading, which is well established as a crucial contributor to chronic autoimmune tissue destruction.26 An infarct causes severe tissue trauma, with necrotic heart tissue releasing vast amounts of DAMPs together with cardiac proteins. In these highly inflammatory circumstances, the ectopically encountered cardiac antigens are recognized by autoreactive lymphocyte clones and can induce autoimmunity. These antigens include α-myosin heavy chain, a prominent example of an abundant cardiac protein that is commonly targeted by post MI anti-cardiac autoreactivity.27 Subsequent immune-mediated tissue injury provides an ongoing supply of the same auto-antigens, culminating in persistent immune autoreactivity (Fig. 1).

The role of the innate and adaptive immune system post MI. A MI causes severe tissue damage and the release of DAMPs and cardiac self-antigens such as myosin and troponin. Immediately after injury, DAMPs lead to an acute inflammatory response, characterized by the influx of a vast number of innate immune cells, which initiate and orchestrate wound repair. Negative feedback mechanisms activated immediately and the declining availability of DAMPs eventually resolves early inflammation. However, in parallel, the release of massive amounts of self-antigens in an inflammatory environment breaks tolerance mechanisms and induces long-lived antigen-specific adaptive immune cells, which cause ongoing autoimmune tissue damage and continuous supply of self-antigen. IS; immune system
Auto-antibodies and B-lymphocytes
An abundance of clinical literature, spanning over three decades, documents the presence of auto-antibodies specific to cardiac antigens in patients with a wide range of heart diseases.2, 28 Antibody-mediated cardiac damage results from binding to and influencing signaling of surface receptors including β-adrenergic and muscarinic receptors29, 30 or from immune complex-mediated direct cellular toxicity.31 However, assigning a causative pathogenic role to specific anti-cardiac antibodies is technically challenging and most reports rely on their presence and theoretical ability to cause damage. Importantly, low titers of auto-antibodies to various antigens can also be detected in healthy subjects, where they do not cause any clinically evident pathology.32 A lack of inflammatory stimulation presumably allows control mechanisms to work appropriately, and under physiological conditions many intracellularly localized cardiac antigens such as myosin and troponin are not easily accessible to the immune system.
Information on other B-lymphocyte functions in MI, such as the secretion of cytokines and growth factors, and antigen presentation, is missing to date. B-lymphocytes infiltrate the mouse myocardium after experimental MI33 and one B-lymphocyte depletion study shows improved post MI recovery of heart function.34 Moreover, activated B-lymphocytes produce cytokines depending on their environment35 and excessive levels of pro-inflammatory cytokines such as TNF-α directly contribute to myocardial dysfunction by depressing contractility, inducing myocyte apoptosis,36 and inducing fibroblast differentiation into myofibroblasts, a crucial step toward myocardial remodeling.37
T-lymphocytes
The involvement of T-lymphocytes in post MI repair and regeneration has been investigated only recently. However, the presence of mature B-lymphocytes producing isotype-switched antibodies, which are dependent on antigen-specific T helper cells, has long provided indirect proof of their involvement. Knowledge about the exact role of T-lymphocytes post MI is still very limited, but considering the diversity of the T-lymphocyte population and the rapidly changing environmental conditions in the heart and the periphery after an infarct, it is likely that there will be tightly regulated changes in subpopulations, effector functions, and beneficial or detrimental effects depending on time point and exact type of damage. Results to date show that post MI effector T-lymphocytes are antigen-specific, primed in the heart-draining lymph nodes and infiltrate the heart.33, 38 Their infiltration of the tissue after activation in the lymph node suggests roles beyond B-lymphocyte activation, but corresponding data are scarce and highly dependent on model system and exact analytical timepoint. Immediately post MI, regulatory T-lymphocytes appear to play a beneficial role.39, 40 Conversely, CD4+ effector T-lymphocytes produce inflammatory cytokines such as IFN-γ and IL-17 in the post MI myocardium38 and pro-inflammatory cytokines are known to increase cardiomyocyte death and enhance fibroblast proliferation and pro-fibrotic gene expression.41 Direct cytotoxic effects of infiltrating CD8+ T-lymphocytes have also been suggested.42 In experimental autoimmune myocarditis, CD4+ helper T-lymphocytes producing IFN-γ and autoreactive cardiac myosin-specific cytotoxic CD8+ T-lymphocytes are major mediators of cardiac damage,43, 44 which suggests similar roles for these two major broad T-lymphocyte subpopulations in post MI cardiac autoreactivity.
Dendritic cells (DCs)
Although not traditionally viewed as part of the adaptive immune system, DCs are crucial in the activation of adaptive antigen-specific T-lymphocytes. Through their activation state when presenting antigen, they direct T-lymphocytes toward either an effector or regulatory phenotype. Under homeostatic conditions, presentation of self-antigen induces a regulatory/tolerogenic phenotype, while in an activated context inflammation-mediated activation of DC and antigen presentation may induce autoreactive effector lymphocytes. We and others have demonstrated that DC numbers indeed increase in infarcted hearts in experimental models45 and decreased numbers of DC in human infarcted myocardial tissue are associated with impaired reparative fibrosis and the development of cardiac rupture soon post MI.46 This early benefit and role in fibrotic repair, however, may well present a threat at later stages, causing excessive fibrosis and subsequent remodeling. The role of endogenous DC populations in inducing and shaping post MI cardiac autoreactivity and their impact on cardiac remodeling toward heart failure is currently under investigation.
How does adaptive autoreactivity impede regenerative therapies?
It is accepted that impediments to therapy exist immediately after an infarct, when the innate immune system responds with astounding speed and force. However, after the initial clearance of acute post MI inflammation, adaptive autoreactivity may proceed insidiously with potentially grave effects. An autoimmune response against major cardiac proteins is very likely to confound a full regenerative outcome in heart disease: it may even destroy tissue in previously unaffected regions of the heart and increase the strain on remaining healthy cardiomyocytes to compensate for the loss. This will exacerbate the remodeling process and accelerate maladaptive left ventricular dilation and heart failure.
Regenerative therapies therefore not only have to deal with the status quo of damage at the time of administration, but also face ongoing autoimmune tissue destruction. Some therapies rely on local delivery of cells and factors into the infarct zone; an approach that seems most efficient considering retention of therapeutic agents where they seem to be needed most, but fails to address ongoing remote damage.
In addition, regenerative therapies, particularly those involving allogeneic cells, must withstand an immunologically distorted environment both in the periphery and in the local microenvironment of the tissue.47 Increased levels of inflammatory cytokines may aggravate immune rejection and prevent engraftment, or even change therapeutic cell phenotypes and impair appropriate function of exogenously delivered stem cells.48
From immunosuppression to immunomodulation
The scenarios described above may give the impression that the immune system in its entirety is adverse to efficient post MI healing and regeneration. On the contrary, it is fundamental in orchestrating the early healing response. Attempts to improve post MI cardiac regeneration by broad immunosuppression have resulted in poor healing, scar formation, and heart failure in animal models and clinical studies. General immunosuppression with corticosteroids and non-steroidal anti-inflammatory drugs disrupts the healing process by impairing collagen deposition and scar formation, leading to an increased chance of left ventricle rupture.49,50,51,52 Similar to full blown immunosuppression, clinical trials using intravenous immunoglobulin, which broadly blocks the full antibody repertoire as well as several other innate and adaptive immune pathways, have yielded limited and inconsistent benefits,53, 54 likely because they do not target the balance between regulatory/regenerative and inflammatory subpopulations of lymphocytes.
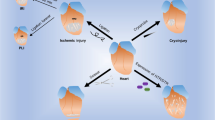
Thus, the way forward is immunomodulation, aiming to restore cardiac tolerance and achieve appropriate balance between inflammatory and regulatory immune cell populations and factors (Fig. 2).55 Combination therapies of regenerative and immunomodulatory treatments have so far largely focused on preventing immune-mediated rejection of transplanted cells or materials by blocking of pro-inflammatory pathways or co-delivery of regulatory immune cells.56 However, similar principles could be applied to the restoration of endogenous cardiac tolerance. Notably, beneficial effects of some post MI therapies have in fact been attributed to a modulating effect on the immune response. For example, statins are in routine clinical use in cardiovascular disease and after an acute MI, with the primary goal of reducing cholesterol synthesis. However, statins are now known to modulate the immune response by enhancing regulatory T-lymphocyte numbers and function while inhibiting pro-inflammatory T-lymphocyte subpopulations.57 A prominent example of immunomodulatory cell therapy is the use of MSCs, which in themselves combine cell therapy with immunomodulation due to their stem cell-like and immunosuppressive characteristics.58

Regenerative therapies are hampered by direct and indirect immunological mechanisms. Immunological rejection and a hostile inflammatory environment may prevent stem cell engraftment or change their beneficial properties. At the same time, anti-cardiac autoreactivity causes further and additional tissue damage. Immunomodulatory therapies may prevent anti-cardiac autoimmunity as well as immunological rejection and excessive inflammation
There are a variety of approaches to boost immune-regulatory mechanisms, but due to their central and unique role in defining the T-lymphocyte phenotype, DC are likely to be an ideal intervention point to skew the post MI anti-cardiac lymphocyte population toward tolerance. Notably, immunomodulatory cell therapies using DC have shown promising results in experimental pre-clinical settings as well as in clinical trials in autoimmune patients with type 1 diabetes or rheumatoid arthritis.59, 60 Indeed, induction of antigen-specific immune tolerance to cardiac antigens has been achieved in a rat model of cardiac damage, which resulted in a milder inflammatory infiltrate, less collagen deposition, and improved cardiac performance.61 Most relevant however, two very recent studies reported beneficial effects of cardiomyocyte antigen-specific tolerogenic DC on post MI function and remodeling in mice.62, 63 Importantly, priming DC with post MI serum was as protective as using cardiac tissue lysate, providing a first proof of concept that post MI tolerogenic DC therapy may be translatable into clinical use.63 Due to their migratory ability and systemic mode of action, DC can be injected subcutaneously, which will avoid common efficacy problems associated with intravenous or intramyocardial cell delivery. Tolerogenic DC can be generated from a patient’s own blood and treatment could be performed within a few days after infarct together with or preferably even before other regenerative therapies. It is therefore both crucial and feasible to design therapeutic strategies for cardiac regeneration targeting ongoing anti-cardiac autoreactivity.
Open questions
Research aimed at unraveling the vast complexity of the endogenous immune response to injury is making rapid progress, but many unanswered questions remain.
How is post MI cardiac autoreactivity induced and which immune cell types are involved at what stage?
Answers to this questions would allow specific targeting of detrimental vs. beneficial cell types, effector molecules or molecular pathways at the most effective window in time. Identification of the targeted auto-antigens might even allow for “counter-vaccination” to induce immune tolerance.
What is the immunological role of cardiac resident cell types in exacerbating post MI immune responses and autoreactivity?
Cardiac cells including cardiomyocytes, endothelial cells, pericytes, fibroblasts, and tissue-resident macrophages are far from mere victims of an immunological attack but crucial active players in shaping the post MI immune response. A better understanding of their immunological characteristics and the cross-talk between cardiac cells and immunological factors may help to protect them from immune-mediated damage.
What is the contribution of genetic diversity?
The exact immune response and the balance between cell populations and factors varies dramatically between individuals. Autoimmune prone (and known autoimmune) patients have a worse outcome post MI. Knowing which factors predispose a post MI patient to detrimental autoreactivity and rapid progression to heart failure may allow earlier and more successful intervention.
Conclusion
Although the field is still in its infancy, there is accumulating evidence that cardiac regeneration is hampered by excessive activation of the immune system and that the progression of ischemic damage to adverse remodeling and heart failure in particular is exacerbated by an ongoing adaptive immune response. Current experimental therapies rely on the assumption that the damaging incident—the infarct—has passed, and it is now time to start repairing the myocardium. Unfortunately, this might be a flawed assumption. While acute early inflammation may be resolved, adaptive autoreactivity will linger, may cause persistent low-level tissue damage, even in previously healthy areas, and may counteract any beneficial effects of regenerative therapies.
The ideal post MI treatment to prevent progression to heart failure should therefore aim to induce immune tolerance to cardiac antigens as a priority. This will prevent the development of pathological adaptive immune autoreactivity, and thus provide other therapies with an environment truly permissive of regeneration.
Data availability
Data sharing not applicable to this article, as no data sets were generated or analyzed during the current study.
References
Epelman, S., Liu, P. P. & Mann, D. L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 15, 117–129 (2015).
Kaya, Z., Leib, C. & Katus, H. A. Autoantibodies in heart failure and cardiac dysfunction. Circ. Res. 110, 145–158 (2012).
Roger, V. L. et al. Heart disease and stroke statistics—2012 update: a report from the American Heart Association. Circulation 125, e2–e220 (2012).
Dargie, H. Heart failure post-myocardial infarction: a review of the issues. Heart 91, ii3–ii6 (2005). discussion ii31, ii43–ii38.
Stewart, S., MacIntyre, K., Hole, D. J., Capewell, S. & McMurray, J. J. More “malignant” than cancer? Five-year survival following a first admission for heart failure. Eur. J. Heart Fail. 3, 315–322 (2001).
Janaswamy, P., Walters, T. E., Nazer, B. & Lee, R. J. Current treatment strategies for heart failure: role of device therapy and LV reconstruction. Curr. Treat. Options Cardiovasc. Med. 18, 57 (2016).
Malliaras, K. et al. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol. Med. 5, 191–209 (2013).
Lin, Z. & Pu, W. T. Strategies for cardiac regeneration and repair. Sci. Transl. Med. 6, 239rv231 (2014).
Doppler, S. A., Deutsch, M. A., Lange, R. & Krane, M. Cardiac regeneration: current therapies-future concepts. J. Thorac. Dis. 5, 683–697 (2013).
Huyer, L. D. et al. Biomaterial based cardiac tissue engineering and its applications. Biomed. Mater. 10, 034004 (2015).
Collesi, C. & Giacca, M. Gene transfer to promote cardiac regeneration. Crit. Rev. Clin. Lab. Sci. 53, 359–369 (2016).
Hodgkinson, C. P., Kang, M. H., Dal-Pra, S., Mirotsou, M. & Dzau, V. J. MicroRNAs and cardiac regeneration. Circ. Res. 116, 1700–1711 (2015).
Ounzain, S. & Pedrazzini, T. The promise of enhancer-associated long noncoding RNAs in cardiac regeneration. Trends Cardiovasc. Med. 25, 592–602 (2015).
Bersell, K., Arab, S., Haring, B. & Kuhn, B. Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138, 257–270 (2009).
Zhang, Y. H. et al. Exogenous basic fibroblast growth factor promotes cardiac stem cell-mediated myocardial regeneration after miniswine acute myocardial infarction. Coron. Artery Dis. 22, 279–285 (2011).
Dong, F. et al. Myocardial CXCR4 expression is required for mesenchymal stem cell mediated repair following acute myocardial infarction. Circulation 126, 314–324 (2012).
Hatzistergos, K. E. et al. Bone marrow mesenchymal stem cells stimulate cardiac stem cell proliferation and differentiation. Circ. Res. 107, 913–922 (2010).
Lee, R. H. et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 5, 54–63 (2009).
Loffredo, F. S., Steinhauser, M. L., Gannon, J. & Lee, R. T. Bone marrow-derived cell therapy stimulates endogenous cardiomyocyte progenitors and promotes cardiac repair. Cell Stem Cell 8, 389–398 (2011).
Tang, X. L. et al. Long-term outcome of administration of c-kit(POS) cardiac progenitor cells after acute myocardial infarction: transplanted cells do not become cardiomyocytes, but structural and functional improvement and proliferation of endogenous cells persist for at least one year. Circ. Res. 118, 1091–1105 (2016).
Malliaras, K. et al. Stimulation of endogenous cardioblasts by exogenous cell therapy after myocardial infarction. EMBO Mol. Med. 6, 760–777 (2014).
Malliaras, K. et al. Intracoronary cardiosphere-derived cells after myocardial infarction: evidence of therapeutic regeneration in the final 1-year results of the CADUCEUS trial (cardiosphere-derived autologous stem cells to reverse ventricular dysfunction). J. Am. Coll. Cardiol. 63, 110–122 (2014).
Gouadon, E. et al. Concise review: pluripotent stem cell-derived cardiac cells, a promising cell source for therapy of heart failure: where do we stand? Stem Cells 34, 34–43 (2016).
Land, W. G. The role of damage-associated molecular patterns (DAMPs) in human diseases: part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ. Med. J. 15, e157–e170 (2015).
Kono, H. & Rock, K. L. How dying cells alert the immune system to danger. Nat. Rev. Immunol. 8, 279–289 (2008).
Vanderlugt, C. L. & Miller, S. D. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol. 2, 85–95 (2002).
Lv, H. & Lipes, M. A. Role of impaired central tolerance to alpha-myosin in inflammatory heart disease. Trends Cardiovasc. Med. 22, 113–117 (2012).
De Scheerder, I. et al. Post-cardiac injury syndrome and an increased humoral immune response against the major contractile proteins (actin and myosin). Am. J. Cardiol. 56, 631–633 (1985).
Yu, X. et al. Development of cardiomyopathy and atrial tachyarrhythmias associated with activating autoantibodies to beta-adrenergic and muscarinic receptors. J. Am. Soc. Hypertens. 3, 133–140 (2009).
Bornholz, B., Roggenbuck, D., Jahns, R. & Boege, F. Diagnostic and therapeutic aspects of beta1-adrenergic receptor autoantibodies in human heart disease. Autoimmun. Rev. 13, 954–962 (2014).
Fleming, S. D. & Tsokos, G. C. Complement, natural antibodies, autoantibodies and tissue injury. Autoimmun. Rev. 5, 89–92 (2006).
Rose, N. R. Infection, mimics, and autoimmune disease. J. Clin. Invest. 107, 943–944 (2001).
Yan, X. et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 62, 24–35 (2013).
Zouggari, Y. et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 19, 1273–1280 (2013).
Lund, F. E. Cytokine-producing B lymphocytes-key regulators of immunity. Curr. Opin. Immunol. 20, 332–338 (2008).
Nian, M., Lee, P., Khaper, N. & Liu, P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ. Res. 94, 1543–1553 (2004).
Porter, K. E., Turner, N. A., O’Regan, D. J. & Ball, S. G. Tumor necrosis factor alpha induces human atrial myofibroblast proliferation, invasion and MMP-9 secretion: inhibition by simvastatin. Cardiovasc. Res. 64, 507–515 (2004).
Hofmann, U. et al. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125, 1652–1663 (2012).
Matsumoto, K. et al. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J. 52, 382–387 (2011).
Tang, T. T. et al. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 107, 232 (2012).
Yan, X. et al. Deleterious effect of the IL-23/IL-17A axis and gammadeltaT cells on left ventricular remodeling after myocardial infarction. J. Am. Heart Assoc. 1, e004408 (2012).
Tae Yu, H. et al. Characterization of CD8(+)CD57(+) T cells in patients with acute myocardial infarction. Cell. Mol. Immunol. 12, 466–473 (2015).
Huber, S. A., Graveline, D., Born, W. K. & O’Brien, R. L. Cytokine production by Vgamma(+)-T-cell subsets is an important factor determining CD4(+)-Th-cell phenotype and susceptibility of BALB/c mice to coxsackievirus B3-induced myocarditis. J. Virol. 75, 5860–5869 (2001).
Huber, S. A., Sartini, D. & Exley, M. Vgamma4(+) T cells promote autoimmune CD8(+) cytolytic T-lymphocyte activation in coxsackievirus B3-induced myocarditis in mice: role for CD4(+) Th1 cells. J. Virol. 76, 10785–10790 (2002).
Gallego-Colon, E. et al. Cardiac-restricted IGF-1Ea overexpression reduces the early accumulation of inflammatory myeloid cells and mediates expression of extracellular matrix remodelling genes after myocardial infarction. Mediators Inflamm. 2015, 484357 (2015).
Nagai, T. et al. Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J. Am. Heart Assoc. 3, e000839 (2014).
Lyon, A. & Harding, S. The potential of cardiac stem cell therapy for heart failure. Curr. Opin. Pharmacol. 7, 164–170 (2007).
Rameshwar, P., Qiu, H. & Vatner, S. F. Stem cells in cardiac repair in an inflammatory microenvironment. Minerva Cardioangiol. 58, 127–146 (2010).
Timmers, L. et al. Cyclooxygenase-2 inhibition increases mortality, enhances left ventricular remodeling, and impairs systolic function after myocardial infarction in the pig. Circulation 115, 326–332 (2007).
Giugliano, G. R., Giugliano, R. P., Gibson, C. M. & Kuntz, R. E. Meta-analysis of corticosteroid treatment in acute myocardial infarction. Am. J. Cardiol. 91, 1055–1059 (2003).
Brown, E. J. Jr. et al. Scar thinning due to ibuprofen administration after experimental myocardial infarction. Am. J. Cardiol. 51, 877–883 (1983).
Gislason, G. H. et al. Risk of death or reinfarction associated with the use of selective cyclooxygenase-2 inhibitors and nonselective nonsteroidal antiinflammatory drugs after acute myocardial infarction. Circulation 113, 2906–2913 (2006).
Gullestad, L. et al. Intravenous immunoglobulin does not reduce left ventricular remodeling in patients with myocardial dysfunction during hospitalization after acute myocardial infarction. Int. J. Cardiol. 168, 212–218 (2013).
McNamara, D. M. et al. Controlled trial of intravenous immune globulin in recent-onset dilated cardiomyopathy. Circulation 103, 2254–2259 (2001).
van den Akker, F., Deddens, J. C., Doevendans, P. A. & Sluijter, J. P. Cardiac stem cell therapy to modulate inflammation upon myocardial infarction. Biochim. Biophys. Acta 1830, 2449–2458 (2013).
Zakrzewski, J. L., van den Brink, M. R. & Hubbell, J. A. Overcoming immunological barriers in regenerative medicine. Nat. Biotechnol. 32, 786–794 (2014).
Forero-Pena, D. A. & Gutierrez, F. R. Statins as modulators of regulatory T-cell biology. Mediators Inflamm. 2013, 167086 (2013).
Wood, K. J., Bushell, A. & Hester, J. Regulatory immune cells in transplantation. Nat. Rev. Immunol. 12, 417–430 (2012).
Creusot, R. J., Giannoukakis, N., Trucco, M., Clare-Salzler, M. J. & Fathman, C. G. It’s time to bring dendritic cell therapy to type 1 diabetes. Diabetes 63, 20–30 (2014).
Bell, G. M. et al. Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Ann. Rheum. Dis. 76, 227–234 (2017).
Ramos, G. C. et al. The autoimmune nature of post-infarct myocardial healing: oral tolerance to cardiac antigens as a novel strategy to improve cardiac healing. Autoimmunity 45, 233–244 (2012).
Zhu, R. et al. Interleukin-37 and dendritic cells treated with interleukin-37 plus troponin I ameliorate cardiac remodeling after myocardial infarction. J. Am. Heart Assoc. 5, e004406 (2016).
Choo, E. H. et al. Infarcted myocardium-primed dendritic cells improve remodeling and cardiac function after myocardial infarction by modulating the Treg and macrophage polarization. Circulation, doi:10.1161/CIRCULATIONAHA.116.023106. (2017).
Acknowledgements
This work was supported by the Medical Research Council UKRMP Immunomodulation Hub [MR/L022699/1] and the BHF Cardiovascular Regenerative Medicine Centre [RM/13/1/30157 to S.E.H.]. We thank Dr. Hanneke de Kort, LUMC, Leiden University, for critical reading of the manuscript.
Author information
Authors and Affiliations
Contributions
S.S. and S.E.H. conceived and drafted the manuscript; N.R., P.F., and F.M.W. critically revised the work. All authors were responsible for final approval of the completed version, and are accountable for all aspects of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sattler, S., Fairchild, P., Watt, F.M. et al. The adaptive immune response to cardiac injury—the true roadblock to effective regenerative therapies?. npj Regen Med 2, 19 (2017). https://doi.org/10.1038/s41536-017-0022-3
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41536-017-0022-3
This article is cited by
-
Elevated resting heart rate as a predictor of inflammation and cardiovascular risk in healthy obese individuals
Scientific Reports (2021)
-
Immunomodulation for optimal cardiac regeneration: insights from comparative analyses
npj Regenerative Medicine (2021)
-
Immunomodulation in Heart Failure with Preserved Ejection Fraction: Current State and Future Perspectives
Journal of Cardiovascular Translational Research (2021)
-
Merging microarray studies to identify a common gene expression signature to several structural heart diseases
BioData Mining (2020)
-
Zebrafish cardiac regeneration—looking beyond cardiomyocytes to a complex microenvironment
Histochemistry and Cell Biology (2020)
