
Abstract
One dimensional (1D) electronic system is a versatile platform hosting novel physics, such as charge density wave, Su-Schrieffer-Heeger (SSH) topological state and solitons, Tomonaga-Luttinger Liquid etc. Here, we systematically study the surface electronic properties on layered composition-tunable compounds Nb2n+1SinTe4n+2 (n = 1–5), which is predicted to be a nodal-line semimetal when n = 1 (Nb3SiTe6). Via scanning tunneling microscopy/spectroscopy, we observe 1D chains formed on the surface of the compounds. We uncover that with the increasing of n, the distance between the chains becomes larger, and the 1D electronic state is developed in the compounds with n ≥ 3. Our first-principle calculations reveal that the nodal-line in Nb3SiTe6 and the 1D electronic state in the crystals with higher n in fact arise from the same bands, which are protected by the same nonsymmorphic symmetry. Furthermore, we can understand the evolution of the electronic states on these series of compounds with such complicated structures and compositions based on a simple SSH type picture. Our experiment demonstrates a tunable and unidirectional 1D electronic system, which offers a concrete platform for the exploration of intriguing 1D electron physics and will enrich the opportunity for future condensed matter physics, material science and nanotechnology researches.
Similar content being viewed by others
Introduction
When electrons are confined into a one-dimensional (1D) wire, many fundamentally important phenomena may emerge. For example, the electron-phonon interaction can induce Peierls phase transition. Dimerization of the atomic lattice could lead to a topologically protected gap in the entire wire with a gapless solitonic excitation at the ends. Electron correlation in 1D metal may be understood by Bosonization method, which gives rise to a Tomonaga-Luttinger Liquid behavior. Despite the research interests, the experimental realization of a 1D system in real materials appears very difficult. Self-assembly growth of metal wires on semiconductor surface, e.g., In wires on Si(111), Au wires on Ge(111), have exhibited several significant progresses1,2,3,4,5,6,7,8. However, it requires in situ growth environment and elaborate parameters control. Another practically successful material system is the 1D defects, such as step edges on quantum spin hall insulator or domain boundaries on 2D transition metal dichalcogenides9,10,11,12,13,14,15,16,17,18. The main drawback of these systems is the directions of the wires are not uniformly distributed on the sample, which complicates their macroscopic transport property and hinders future device applications. IrTe2 features a strip like structures, and undertakes interesting phase transitions19,20,21, but its structure is only stable in narrow temperature range. Therefore, an air and thermal stable, unidirectional and tunable 1D system is still yet to be achieved.
Composition-tunable compound is a class of thermally stable materials with a series of possible stoichiometry ratio. A famous example is the Ruddlesden–Popper phase, An+1BnO3n+1, where A and B are metallic elements, and O is oxygen22. When n = ∞, the Ruddlesden–Popper phase becomes the well-known perovskite structure ABO3. In the Ruddlesden-Popper series of strontium iridates, scientists found that the electron correlation and Mott transition evolves with the component ratio n23. In other families of composition-tunable compounds, e. g. Srn+1CunO2n+1+δ or (BA)2(MA)n−1PbnI3n+1, it has been found that the superconducting transition temperatures and the solar cell power conversion efficiencies are also remarkably influenced by n24,25. The discovery of new family of composition-tunable compounds is always accompanied by a comprehensive understanding of substantial issues in physics, chemistry or material science.
Here, we reveal that the family of composition-tunable compounds Nb2n+1SinTe4n+2 offers an ideal platform for studying 1D electrons. We show that the low-energy electrons in these materials are confined in 1D chain-like structures and are gapless ensured by a nonsymmorphic crystal symmetry. Because these chains are intrinsic component of the crystal lattice, they share a fixed orientation. In addition, the 1D confinement can be readily tuned by changing n, which plays the role of spacing between neighboring chains in this context. As such, it overcomes the long-standing uniformity and tunability issues in realizing 1D electronic systems. Furthermore, the thermal and air stability natures of these materials will facilitate the future applications in 1D electronics.
Results
Overview of the crystals
Nb3SiTe6 was synthesized about thirty years ago26. Its isostructural phases, e.g., Ta3SiTe6, Nb3GeTe6 and Ta3GeTe6, were also prepared27,28. Scientists also recognized that this material in fact belongs to a family of composition-tunable compounds Nb2n+1SinTe4n+2, where n = 1 gives Nb3SiTe6 and n = ∞ makes Nb2SiTe426,27,28,29. Unfortunately, not much attention has been paid on their physical properties until very recently. It was reported that enhanced electron coherence was achieved in atomically thin Nb3SiTe630. It was also predicted to possess nonsymmorphic-symmetry-protected topological nodal-line in the bulk or monolayer forms31, which was later probed by angle-resolved photoemission spectroscopy and magneto-transport experiments32,33. Since then, more and more researchers start to notice these compounds30,31,32,33,34.
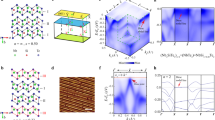
The entire class of the Nb2n+1SinTe4n+2 compounds all features layered structure, with relatively weak van der Waals type bonding between the layers. In each layer, the Nb and Si atoms sit in same plane, which are sandwiched by two layers of Te atoms. The compound can be effectively viewed as Si-doped NbTe2, because the value of n increases with the concentration of Si. The atomic structure of the compounds become complicated when n grows larger. Fortunately, the monolayer of the series can be constructed by three basic building blocks, which are named as a, b and c chains (Fig. 1a). a and b chains are both doped with Si, whose formula unit is NbSi1/2Te2, while the c chain is free of Si, i.e., it is simply NbTe2. a and b chains are glide mirror partner to each and always stick together. Moreover, all chains possess the similar width of about 0.39 nm. The class of composition-tunable compounds Nb2n+1SinTe4n+2 can thus be represented in this picture as (ab)nc, or the chemical formula (Nb2SiTe4)n(NbTe2). Remarkably, the glide mirror symmetry \(\tilde M_y = \{ M_y|0\}\) is preserved in all monolayers regardless of n, whose significance will be discussed later.

a Sketches of the monolayer Nb2n+1SinTe4n+2 compounds, which can be constructed by three building blocks. Blue, yellow and green dots stand for Te, Nb, and Si atoms respectively. Based on the chain construction method, the compounds can be expressed as (ab)nc, where a, b and c chains are the building blocks. b Scanning tunneling microscopy (STM) images of the samples at indicated compositions. All images were taken at 300 mV and 1 nA. Scale bar represents 3 nm. All samples feature bright 1D chain morphologies. The distance between chains increase with elevated n. c Typical dI/dV spectra measured on top of the bright chain of the samples with indicated n. All spectra are obtained at set point of 0.2 V and 1 nA. Arrows point to the main peaks.
We systematically measure a series of Nb2n+1SinTe4n+2 compounds. Scanning tunneling microscopy (STM) images in Fig. 1b display obvious 1D chains morphologies on all samples. (The voltage dependent STM images can be found in Supplementary Fig. 1). The distances between adjacent chains are 1.21 nm, 1.96 nm, 2.77 nm, 3.57 nm, 4.39 nm, which are consistent with the theoretical lattice period (normal to the chains) of abc, (ab)2c, (ab)3c, (ab)4c and (ab)5c, whose width should be (2n + 1) × 0.39 nm. Furthermore, based on the observations, we uncover that the low-energy electronic states (the bright lines in the figure) mainly come from the c chains, which are the Si-free NbTe2 chains. We will show that these are effectively 1D electrons for compositions with n ≥ 3.
After identifying the compositions of the samples, we pay attention to the electronic structures. We take differential conductance (dI/dV) spectra on top of the bright chains on each sample to reveal the local density of states (Fig. 1c). On the sample of n = 1, i.e., Nb3SiTe6, we observe a broad peak with peak position at 80 meV on its dI/dV spectrum. On the sample of n = 2, there are several peaks, but the main peak is located at the energy of 110 meV. On the samples with n ≥ 3, the three spectra present similar shapes, single pronounced sharp peaks located at 150 meV, 140 meV and 140 meV for n = 3, 4, 5, respectively. The origination of the peaks will be discussed latter.
Characterization of 1D electronic states
Impurities on a crystal might induce quasiparticle interferences (QPI), which has become a powerful method in modern condensed matter physics research9,35,36,37,38,39,40. Here, we apply this method to study the development of 1D electronic state on the Nb2n+1SinTe4n+2 compounds. On Nb3SiTe6 (n = 1), we focus on a segment on a bright chain (c chain) between two point defects, which manifest themselves as dark points on the chain, as shown in Fig. 2a. We measure a series of position-dependent dI/dV spectra, i.e., dI/dV (x,V) map, along the target area. For this n = 1 case, the most obvious feature is the defects induced electronic states, and we don’t find clear 1D QPI signal in the region between the defects (Fig. 2b, c). Similar observations are made on the n = 2 composition. However, one notices that a weak 1D QPI pattern on the c chain starts to emerge on the dI/dV (x,V) and dI/dV (x,y) maps (see Supplementary Fig. 2).

a STM image showing an area of Nb3SiTe6 containing 4 chains, where one of them possess two point defects (marked by arrows). Image size is 5 nm × 25 nm. b Position-dependent dI/dV spectra, which taken along the dotted line in a. c dI/dV maps measured on the same chain with two defects. Voltages are indicated. The set point is 200 mV, 1 nA. No standing waves can be observed on the surface of Nb3SiTe6.
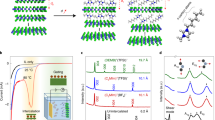
Clear signals for 1D electronic state have been found for samples with n ≥ 3. In Figs. 3 and 4, one observes clear 1D QPI patterns in the dI/dV (x,V) maps for regions on the c chain between two point defects. By a fast Fourier transform (Fig. 3c), we resolve the scattering vector-energy dispersion, which indicates a hole-like band structure. Furthermore, in Fig. 3d, our dI/dV (x,y) maps taken at various voltages clearly show that the standing waves propagate along the NbTe2 chain and thus prove that these are confined 1D electronic states. The 1D character becomes more and more pronounced with increasing n. In Fig. 4 for n = 5, we discern a typical “particle in a box” like 1D QPI pattern. This trend of enhanced 1D character with n can be intuitively understood. Because the 1D states come only from the NbTe2 chains, the a and b chains which are inactive can be viewed as spacers between the NbTe2 chains (see details in Supplementary Fig. 3). When spacer is narrow, the electrons can hop around the NbTe2 chains hence still have a 2D character (although highly anisotropy). With increasing spacer width, the coupling is suppressed and the electrons become more and more confined into the 1D chain. Moreover, on our samples, we have not found any domains with different orientations of the chains, in contrast to the previously reported pseudo-1D material systems. This is simply because that the 1D (NbTe2) chain is an intrinsic component of the crystal structure, in contrast to the artificially constructed atomic wires or extrinsic defect lines/boundaries.

a STM image showing an area of Nb7Si3Te14 containing 3 chains. Middle chain possesses two point-defects (marked by arrows). Image size is 10 nm × 23 nm. b Position-dependent dI/dV spectra, which taken along the dotted line in a. Voltage dependent patterns are clearly discerned in the region between two defects. c Fast Fourier transform of b, which directly exhibits the dispersion of the quasiparticle interference. The dotted line guides the eyes to the hole-like QPI feature. d dI/dV maps measured on another area with clear standing waves. Voltages are indicated. The set point is 200 mV, 1 nA. 1D standing waves are revealed on segment between defects of the chain.

a STM image showing an area of Nb11Si5Te22 containing 3 chains. The middle chain possesses two point-defects (marked by arrows). Image size is 12 nm × 20 nm. b Position-dependent dI/dV spectra, which taken along the dotted line in a. “Particle in a box” type patterns are clearly discerned in the region between two defects. c dI/dV maps measured on the same area as a. Voltages are indicated. The set point is 200 mV, 1 nA. The electronic waves are all propagated along 1D chains.
Discussions
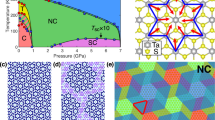
We performed first-principle simulations on the monolayers of the Nb2n+1SinTe4n+2 compounds with n = 1, 2 and 3. We find many of the observations can be qualitatively understood. Furthermore, we also uncover that the spin-orbit coupling (SOC) is not significant in our case (Supplementary Fig. 4) and thus neglect it in the calculation. In Fig. 5a–c, we compare the calculated band structure. One observes that there is a twofold band degeneracy (nodal line) at the Brillouin zone boundary along the X-M line. This degeneracy is connected with the nonsymmorphic \(\tilde M_y\) symmetry, because its combination with time reversal symmetry T satisfies \(\left( {T\tilde M_y} \right)^2 = - 1\) on X-M, leading to a protected double degeneracy on this path. The low-energy states are dominated by the two bands that cross on X-M. By analyzing the charge density distribution for these states (see Fig. 5d–f), we confirm that they are mainly distributed on the c (NbTe2) chains, and mostly from the Nb sites. Furthermore, the dispersion of the band along X-M (and also Γ-Y) is quite small, and it gets flattened out with increasing n. For n = 3, the dispersion along X-M becomes almost completely flat. This confirms that the low-energy states become localized in the x direction, forming 1D states that can only propagate along the chain direction. We have also identified that the β bands are also originated from c chains, while γ and δ bands are located on the b and c chains (see details in the Supplementary Figs. 5–7). Our simulated STM images also prove that the states close to the Fermi level are located on NbTe2 chains (Supplementary Fig. 8). We agree that the simulation of the 3D crystal will generate an exact result. However, when n in the material Nb2n+1SinTe4n+2 increases, the number of atoms in one unit-cell becomes very large. The simulation turns out to be very time consuming. Fortunately, we find the simulation of 2D layer gives rise to a good approximation, especially when we are more concerned about the issue of intra-chain hopping and inter-chain hopping. On the other hand, STM is only sensitive to the surface of a material (at most the first few atomic layers). The configuration for the surface atoms (the coordination, environment, etc.) is drastically different from atoms in the bulk, but is more similar to that in the 2D setup. It is rationalized by the simulated STM images (in Supplementary Fig. 8) based on 2D model which corroborates our experimental data.

a–c Calculated band structures of the monolayer samples with n = 1, 2, 3 respectively. Inset of panel a is the first Brillouin zone with high symmetry points marked. d–f The real space plots of the charge densities at the α points in n = 1, 2, 3 respectively. We know the bands at Fermi levels on samples come from the c chains in real spaces. (see more charge distribution plots in Supplementary Figs. 5–7).
The 1D electrons confined in the NbTe2 chain can be captured by a simple SSH like model. As mentioned, the low-energy states are mainly from the Nb atoms in the NbTe2 chain, which forms a zigzag chain with two sites per unit cell (labeled as Nb1 and Nb2), which just resembles the case for the SSH model41. Hence, we can write down the 1D model
where i labels the unit cell, t and t’ denote the intracell and intercell hopping amplitude respectively, and c† (c) are electron creation (annihilation) operator. Here, the \(\tilde M_y\) dictates that the two sites Nb1 and Nb2 are equivalent such that t = t’ (see Supplementary Fig. 9). Therefore, the spectrum is gapless with \(E_ \pm \left( k \right) = \pm 2t\cos \left( {\frac{k}{2}} \right)\), which captures the band dispersion observed in Fig. 3. Near zero energy, the dispersion is linear, of 1D Dirac type. It would also be interesting if \(\tilde M_y\) can be broken with dimerization formed along the chain, such as if we are able to use a STM tip to manipulate ad-atoms periodically on top of different bonds between Nb atoms, such that t and t’ no longer equal. Then a topological gap of \(2|t - t{^\prime}|\) can be opened in the spectrum, and there may appear topological zero energy mode at the end of the chain. These can be interesting topics to pursue in future studies.
Having the theoretical analysis in mind, we are able to understand the QPI and dI/dV features. Figure 3c shows a hole-type quasiparticle dispersion, which dispersion maximum is located at 150 meV above the Fermi level. We attribute the QPI feature to the interband scatterings in the hole branch of the 1D SSH type energy band. Therefore, the dispersion of the QPI resembles the dispersion of the energy band, which is also consistent with the simulation on monolayer Nb7Si3Te14, e.g., the hole branch along Γ-X line in the Fig. 5c. Moreover, the band maximum gives rise to a high local density of state and generates the peaks at the energies in the dI/dV spectra being measured on the c chains, just as shown in Fig. 1cn ≥ 3. Furthermore, we now know that electronic state of the complicated Nb2n+1SinTe4n+2 compounds in fact can be understood by a very simple picture. The monolayer of the crystals is constructed by a buddle of 1D chains with Dirac type dispersions, which Dirac point is protected by the glide mirror symmetry. If the chains are far from each other, e. g. on the compounds with larger n, the hopping between adjacent chains are neglectable, the sample will exhibit real 1D behavior. When the chains are closer, the interchain hopping will be turned on. However, the glide mirror symmetry still forces the bands to be doubly degenerate on the zone boundary, the Dirac nodes will change to a dispersed nodal line on the X-M line.
In summary, we experimentally discover a tunable and unidirectional 1D electronic system on the family of Nb2n+1SinTe4n+2 compounds. Based on theoretical analysis, we predict that the monolayers of Nb2n+1SinTe4n+2 compounds host various novel physics, e.g., topological nodal line semimetal state and 1D Dirac electrons. It provides ample opportunities for the scientists working in condensed matter physics, material sciences and nanotechnologies.
Methods
Experiment
Single crystals were grown by using Te as flux. Starting materials Nb (Powder, 99.99%, Alfa Aesar), Si (Lump, 99.9999%, Alfa Aesar) and Te (Lump, 99.999%, Alfa Aesar) were mixed in an Ar-filled glove box at a molar ratio of Nb: Si: Te = 3: 1: 30. The mixture was placed in an alumina crucible, which was then sealed in an evacuated quartz tube. The tube was heated to 1100 °C over 10 h and dwelt for 20 h. Then, the tube was slowly cooled down to 800 °C at a rate of 2 °C h−1 followed by separating the crystals from the Te flux by centrifuging. Shiny crystals with the size of 2 × 2 mm2 were obtained on the bottom of the crucible.
We carry out our measurements in an Unisoku 1600 scanning tunneling microscopy system. The Nb2n+1SinTe4n+2 compounds were cleaved in ultra-high vacuum at room temperature and transferred in situ to the STM head whose temperature is ~ 4.8 K. dI/dV grids were obtained by measuring one complete dI/dV spectrum on each point of a map. Lock-in amplifier, with modulation voltage of 5 mV, is used to get the dI/dV signals.
Theory
We performed first-principles calculations using Vienna ab initio simulation package42,43 with the projector augmented wave method44. The Perdew-Burke-Ernzerhof-type45 generalized gradient approximation was used for the exchange-correlation functional. The cutoff energy was set to 400 eV, and a 12 × 8 × 1 Γ-centered k-point mesh was used for the Brillouin zone sampling. The convergence criteria for the energy and force were set to be 10–5 eV and 0.01 eV Å−1, respectively. For the monolayer materials, a vacuum layer with a thickness of 20 Å was taken to avoid artificial interactions between periodic images. We don’t consider spin-orbit coupling in our simulations.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
Snijders, P. C. & Weitering, H. H. Colloquium: electronic instabilities in self-assembled atom wires. Rev. Mod. Phys. 82, 307–329 (2010).
Yeom, H. W. et al. Instability and charge density wave of metallic quantum chains on a silicon surface. Phys. Rev. Lett. 82, 4898–4901 (1999).
Crain, J. N. & Pierce, D. T. End states in one-dimensional atom chains. Science 307, 703–706 (2005).
Zeng, C. et al. Charge-order fluctuations in one-dimensional silicides. Nat. Matter 7, 539–542 (2008).
Blumenstein, C. et al. Atomically controlled quantum chains hosting a Tomonaga–Luttinger liquid. Nat. Phys. 7, 776–780 (2011).
Cheon, S., Kim, T.-H., Lee, S.-H. & Yeom, H. W. Chiral solitons in a coupled double Peierls chain. Science 350, 182–185 (2015).
Do, E. H. & Yeom, H. W. Electron quantization in broken atomic wires. Phys. Rev. Lett. 115, 266803 (2015).
Lee, G., Shim, H., Hyun, J.-M. & Kim, H. Intertwined solitons and impurities in a quasi-one-dimensional charge-density-wave system: In/Si(111). Phys. Rev. Lett. 122, 016102 (2019).
Drozdov, I. K. et al. One-dimensional topological edge states of bismuth bilayers. Nat. Phys. 10, 664–669 (2014).
Pauly, C. et al. Subnanometre-wide electron channels protected by topology. Nat. Phys. 11, 338–342 (2015).
Sessi, P. et al. Robust spin-polarized midgap states at step edges of topological crystalline insulators. Science 354, 1269–1273 (2016).
Tang, S. et al. Quantum spin Hall state in monolayer 1T’-WTe2. Nat. Phys. 13, 683–687 (2017).
Reis, F. et al. Bismuthene on a SiC substrate: a candidate for a high-temperature quantum spin Hall material. Science 357, 287–290 (2017).
Liu, H. et al. Dense network of one-dimensional midgap metallic modes in monolayer MoSe2 and their spatial undulations. Phys. Rev. Lett. 113, 066105 (2014).
Barja, S. et al. Charge density wave order in 1D mirror twin boundaries of single-layer MoSe2. Nat. Phys. 12, 785–756 (2016).
Jolie, W. et al. Tomonaga-Luttinger liquid in a box: Electrons confined within MoS2 mirror-twin boundaries. Phys. Rev. X 9, 011055 (2019).
Chen, P. et al. Large quantum-spin-Hall gap in single-layer 1T′ WSe2. Nat. Commun. 9, 2003 (2018).
Ugeda, M. M. et al. Observation of topologically protected states at crystalline phase boundaries in single-layer WSe2. Nat. Commun. 9, 3401 (2018).
Hsu, P. J. et al. Hysteretic melting transition of a soliton lattice in a commensurate charge modulation. Phys. Rev. Lett. 111, 266401 (2013).
Li, Q. et al. Bond competition and phase evolution on the IrTe2 surface. Nat. Commun. 5, 5358 (2014).
Chen, C. et al. Surface phases of the transition-metal dichalcogenide IrTe2. Phys. Rev. Lett. 95, 094118 (2017).
Ruddlesden, S. N. & Popper, P. The compound Sr3Ti2O7 and its structure. Acta Crystallogr. 11, 54–55 (1958).
Moon, S. J. et al. Dimensionality-controlled insulator-metal transition and correlated metallic state in 5d transition metal oxides Srn+1IrnO3n+1 (n = 1, 2, and∞). Phys. Rev. Lett. 101, 226402 (2008).
Hiroi, Z., Takano, M., Azuma, M. & Takeda, Y. A new family of copper oxide superconductors Srn+1CunO2n+1+δ stabilized at high pressure,. Nature 364, 315–317 (1993).
Tsai, H. et al. High-efficiency two-dimensional Ruddlesden–Popper perovskite solar cells. Nature 536, 312–316 (2016).
L, J., Badding, M. E. & DiSalvo, F. J. Synthesis and structure of Nb3SiTe6, a new layered ternary niobium telluride compound. J. Alloy Compd. 184, 257–263 (1992).
Evain, M., Van der Lee, A., Monconduit, L. & Petricek, V. Modulated structure of TaSi0.414Te2: sandwich stacking in the MAxTe2 (M = Nb, Ta; A = Si, Ge; 1/3 < x < 1/2) Series. Chem. Mater. 6, 1776–1783 (1994).
Van der Lee, A. et al. A superspace approach to the modulated structures of MAxTe2 (M = Nb, Ta; A = Si, Ge; 1/2< x < 1/3), exemplified by NbGe3/7Te2. J. Phys. Condens. Matter 6, 933–944 (1994).
Monconduit, L. et al. Synthesis, crystal and electronic structure of a new ternary layered compound: Nb2SiTe4. Comptes Rendus Acad. Sci. Ser. II 316, 25–34 (1993).
Hu, J. et al. Enhanced electron coherence in atomically thin Nb3SiTe6. Nat. Phys. 11, 471–476 (2015).
Li, S. et al. Nonsymmorphic-symmetry-protected hourglass Dirac loop, nodal line, and Dirac point in bulk and monolayer X3SiTe6 (X = Ta, Nb). Phys. Rev. B 97, 045131 (2018).
Sato, T. et al. Observation of band crossings protected by nonsymmorphic symmetry in the layered ternary telluride Ta3SiTe6. Phys. Rev. B 98, 121111(R) (2018).
An, L. et al. Magnetoresistance and Shubnikov–de Haas oscillations in layered Nb3SiTe6 thin flakes. Phys. Rev. B 97, 235133 (2018).
Naveed, M. et al. Magneto-transport and Shubnikov-de Haas oscillations in the layered ternary telluride topological semimetal candidate Ta3SiTe6. Appl. Phys. Lett. 116, 092402 (2020).
Zheng, H. et al. Atomic-Scale visualization of quantum interference on a Weyl semimetal surface by scanning tunneling microscopy. ACS Nano 10, 1378–1385 (2016).
Zheng, H. & Hasan, M. Z. Quasiparticle interference on type-I and type-II Weyl semimetal surfaces: a review. Adv. Phys. X 3, 1466661 (2018).
Zhu, Z. et al. Quasiparticle interference and nonsymmorphic effect on a floating band surface state of ZrSiSe. Nat. Commun. 9, 4153 (2018).
Nie, X.-A. et al. Robust hot electron and multiple topological insulator states in PtBi2. ACS Nano 14, 2366–2372 (2020).
Schirone, S. et al. A. Spin-flip and element-sensitive electron scattering in the BiAg2 surface alloy. Phys. Rev. Lett. 114, 166801 (2015).
Chang, K. et al. Standing waves induced by valley-mismatched domains in ferroelectric SnTe monolayers. Phys. Rev. Lett. 122, 206402 (2019).
Su, W. P., Schrieffer, J. R. & Heeger, A. J. Solitons in polyaeetylene. Phys. Rev. Lett. 42, 1698–1701 (1979).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Acknowledgements
We thank J. Zhang for the helpful discussion. We acknowledge the financial supports from NSFC (Grants No.11674226, No. 11790313, No. 11574202, No. 11874256, No. 11521404, No. 11874258, No. U1632102, No. 11861161003, and No. 11674226) and from National Basic Research Program of China (Grants No. 2019YFA0308601, No. 2016YFA0300403 and No. 2016YFA0301003), Singapore Ministry of Education AcRF Tier 2 (MOE2017-T2-2-108), additional support from a Shanghai talent program and the project supported by Shanghai Municipal Science and Technology Major Project (Grant No. 2019SHZDZX01)
Author information
Authors and Affiliations
Contributions
H.Z. and J.-F.J. supervised the project. Z.Z. did the STM measurement with the help of X.-A.N., H.-K.X., X.Y., D.-D.G, S.W., Y.-Y.L. and C.L.S.L., S.A.Y. and Y.Y. did the simulations. M.Y. grow the crystals with the help of Y.S., Z.-Q.M. and N.X. All authors discussed the result and contributed to the paper writing. Z.Z. and S.L. contributed equally to this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhu, Z., Li, S., Yang, M. et al. A tunable and unidirectional one-dimensional electronic system Nb2n+1SinTe4n+2. npj Quantum Mater. 5, 35 (2020). https://doi.org/10.1038/s41535-020-0238-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41535-020-0238-0
This article is cited by
-
Transport measurement of fractional charges in topological models
npj Quantum Materials (2023)
-
Bulk-interface correspondence from quantum distance in flat band systems
Communications Physics (2022)
-
Observation of dimension-crossover of a tunable 1D Dirac fermion in topological semimetal NbSixTe2
npj Quantum Materials (2022)
