
Abstract
An intronic expansion (AAGGG)exp in the RFC1 gene has recently been shown to cause recessively inherited cerebellar ataxia, neuropathy, and vestibular areflexia syndrome and, furthermore, a few patients with ataxia and parkinsonism have been reported. We investigated 569 Finnish patients with medicated parkinsonism for RFC1 and found biallelic (AAGGG)exp in three non-consanguineous patients with clinically confirmed Parkinson’s disease without ataxia suggesting that RFC1-related disorders include Parkinson’s disease as well.
Similar content being viewed by others


The replication factor C complex is a five-subunit ATPase required for DNA replication and repair. The gene encoding subunit 1 (RFC1) has been identified as a frequent cause of cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS) and late-onset ataxia1,2. The causative mutation is a biallelic pentanucleotide repeat expansion (AAGGG)exp in the intronic region of RFC1, but its functional consequences are not yet known. The expansion explains 90 % of CANVAS and up to 14 % of adult-onset ataxias3. The (AAGGG)exp is associated with a core haplotype1,2,4 and it has been estimated that the age of the ancestral haplotype is >25,000 years2.
Since the identification of (AAGGG)exp, other phenotypes, such as cerebellar and parkinsonian type multiple system atrophy (MSA)5,6,7, have been reported in association with RFC1. Involvement of dopaminergic neurons in the striatum has been described in a patient with CANVAS and features of Lewy body dementia4. Furthermore, one patient with cerebellar ataxia, vestibulopathy and levodopa responsive lower body parkinsonism and another patient with the clinical triad of CANVAS and levodopa responsive parkinsonism have been reported7,8. Here we report on screening of a population-based cohort of Finnish patients with medicated parkinsonism for RFC1 (AAGGG)exp.
We found nine subjects with the homozygous (AAGGG)exp-associated core haplotype among 569 patients with medicated parkinsonism (Supplementary Table 1) and two out of 269 controls. Neither of the controls, but three of the 9 patients, harbored biallelic (AAGGG)exp in RFC1 (Fig. 1). The number of repeated units varied from 144 to 820 (Supplementary Table 2, Supplementary Fig. 2). The three patients fulfilled the criteria of Parkinson´s disease (PD)9, none had ataxia and one did not have any of the characteristic features of CANVAS. Analysis of exome sequencing data excluded contributing mutations and copy number variants in known PD genes. Biallelic (AAGGG)exp was not found in the remaining 560 patients, who did not have the homozygous core haplotype.

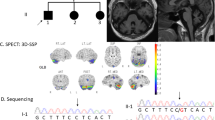
a Multiplex PCR of RFC1 and FBN1 shows no RFC1 PCR product in the region of interest in the three patients with PD (P1-P3) or CANVAS. The gel derives from the same experiment. b XL-PCR amplification of RFC1 carried out with Phire II Hot Start DNA polymerase. Lanes 1–4, healthy controls with normal fragment size variation; lanes 5–7, patients 1–3; lane 8, patient with CANVAS and biallelic (AAGGG)exp; lane 9, H2O. The gel derives from the same experiment. c Electropherogram resulting from repeat-primed PCR of patient 1 harboring the biallelic (AAGGG)exp and a control without the expansion.
An experienced neurologist (J.S.) examined patients 1 and 2 at 8 a.m. in both off and on phases, while patient 3 was not available for the clinical study but J.S. had examined her during the preceding year. The extrapyramidal signs were asymmetrical and responsive to levodopa in the three patients and beta-CIT-SPECT imaging at the time of PD diagnosis had revealed markedly asymmetrical dopamine transporter depletion in the putamina (Supplementary Fig. 1). There was some phenotypical variation (Table 1), but the clinical impression was that neither the presentation nor disease course differed from those in other PD patients.
Patient 1 (Supplementary video 1) is a 73-year-old man with onset of extrapyramidal symptoms at age 65 years. Computed tomography showed minimal frontotemporal cortical atrophy. The symptoms were quite well controlled for the first four years, after which the symptoms have deteriorated slowly. Clinical examination revealed a mild cognitive impairment with a marked ideomotor apraxia. He was independent in all activities of daily living, but his driving licence has been revoked because of cognitive difficulties. Ropinirole has been discontinued because of hallucinations. Autonomic dysfunction was deemed to be mild to moderate except for marked orthostatism. His comorbidities were arthrosis and lumbar spinal stenosis.
Patient 2 (Supplementary video 2) is a 69-year-old man with onset of extrapyramidal symptoms at age 59 years. The symptoms were easily controlled for the first nine years, but during the last year they have become more severe. Mild cognitive impairment was observed, and computed tomography showed minimal frontotemporal cortical atrophy. Pramipexole has been discontinued because of hallucinations. Patient 2 reported a short vertigo upon turning his head and vestibular areflexia was verified in a neuro-otologic examination. Autonomic dysfunction was deemed to be mild to moderate except for marked orthostatism. He has had a coronary bypass operation, an implanted cardiac pacemaker, and type 2 diabetes.
The blood pressure of patients 1 and 2 fell below 80/50 mmHg (sitting) after the administration of 250 mg of soluble levodopa. Patient 2 experienced a clinical worsening, even as his extrapyramidal signs clearly abated and a similar, although milder, effect was observed in patient 1.
Patient 3 is a 64-year-old woman with onset of extrapyramidal symptoms at age 51 years. The symptoms of PD have steadily progressed. She has been deemed indicated for deep brain stimulation, but the procedure has been deferred. Brain magnetic resonance imaging has been normal (Supplementary Fig. 1) and neuropsychological evaluation has not revealed cognitive impairment. Patient 3 has tolerated only a relatively low dose of pramipexole. She has experienced frequent falls and her autonomic dysfunction has been considered mild. Her comorbidities included hypertension and coronary heart disease.
The phenotype associated with the expansion in RFC1 is multisystemic including cerebellar, neuropathic, autonomic, extrapyramidal, cognitive and even pyramidal signs3,4,5,6,7,8. Biallelic (AAGGG)exp in RFC1 commonly manifests as CANVAS or late-onset ataxia with chronic cough3. We found that none of our patients had ataxia and that one patient (P1) was lacking all the three core features of CANVAS. The expansion in RFC1 has also been found in occasional patients with MSA, in a patient with CANVAS and levodopa responsive parkinsonism and in a patient with features of Lewy body dementia4,5,6,7,8. Bradykinesia has been reported in 26 % of ataxic RFC1 patients and it co-occurs with autonomic dysfunction yielding MSA-C phenotype in 19 % of the patients7. Only one of the previously reported MSA cases has had an unambiguous levodopa response and most of them appear to have had a more severe phenotype and a more aggressive disease course compared to our patients5,6,7. Autonomic dysfunction was manifested in two of our patients as moderate to severe orthostatic hypotension that was not present in the third patient. Autonomic dysfunction was not a central feature in any of our patients making the phenotype inconsistent with MSA.
We found that the clinical phenotype of the three patients was consistent with PD and that their extrapyramidal symptoms unambiguously responded to levodopa. Patient 3 resembled a previous case of parkinsonism with biallelic RFC1 (AAGGG)exp in levodopa response, age of onset, dopamine transporter imaging and the presence of chronic cough8. However, the disease course appears to have been considerably milder in the previous patient, as her neuropathy was restricted to sensory fibers and, most importantly, she had ataxia8, which was not present in our patients. Another patient has been reported with levodopa-responsive lower body parkinsonism and with incomplete CANVAS7. Comparison of these five cases suggests that extrapyramidal features are variable in patients with RFC1 expansion and that some patients fulfill the clinical criteria of PD.
The three patients were from the province of North Karelia. The national registry that was used to identify patients included 797 subjects with medicated parkinsonism and with a residence in North Karelia. Samples were received from 161 subjects and three patients were found with biallelic RFC1 (AAGGG)exp giving a frequency of 1.9 % (0.4–5.4 %; 95 % confidence interval). Intriguingly, the prevalence of PD is higher in North Karelia than elsewhere in Finland10, which may at least partly be attributed to factors related to geographically clustered genetic structure of the Finnish population11. Biallelic (AAGGG)exp in RFC1 may thus be one of the most common genetic causes of PD in Finland, at least in North Karelia.
The frequency of the (AAGGG)exp-associated core haplotype was 11.5 % in patients with medicated parkinsonism and that in population controls is 10.4 %12, whereas the allele frequency of the pathogenic expansion in the Finnish population is not known. However, biallelic (AAGGG)exp has shown complete penetrance by eighth decade of age7 and a minimum estimate for population frequency can be obtained based on frequencies in the patient cohorts. Such calculations give a frequency of 0.5 %, which is rather similar to that reported for non-Finnish Europeans1.
Currently, some 20 genes have been associated with monogenic PD13. Our study shows that the biallelic (AAGGG)exp in RFC1 can be found in patients with PD expanding the phenotypic spectrum of RFC1 disease. The findings, however, may be specific to the Finnish population and, therefore, other populations need to be examined in order to investigate the potential role of RFC1 as a monogenic cause of PD.
Methods
Patients and controls
Patients with medicated parkinsonism in the provinces of Northern Ostrobothnia, Kainuu and North Karelia were identified in the national medication reimbursement registry of Kela, Finland. DNA was obtained from 569 patients. The controls consisted of 269 geographically matched healthy subjects12.
Molecular genetics
Four polymorphisms defining the (AAGGG)exp-associated core haplotype (4-39364970-T-C (rs6844176), 4-39363236-T-C (rs17584703), 4-39327482-G-A (rs11096992) and 4-39317086-A-G (rs2066790) (GRCh38)) were investigated using restriction fragment length polymorphism with FastDigest® RseI, TaaI, BseJI and Eco105I (Thermo Fisher Scientific, Waltham, MA, U.S.A.). Core haplotype frequencies were estimated using Arlequin 3.5.2.2 software14. A more detailed haplotype of the three patients with PD was constructed using exome sequencing data. PCRs for large (XL-PCR) and complex amplicons were carried out using Phusion High-fidelity DNA polymerase1 with HF buffer or Phire Hot-start DNA polymerase (Thermo Fisher Scientific). Flanking multiplex PCR for FBN1 as a control and RFC1 was done using TaKaRa Ex Taq Hot Start® polymerase (Takara Bio, Kusatsu, Japan). Fluorescent-labeled repeat-primed PCR (RP-PCR) was carried out for (AAGGG)exp, (AAAAG)exp, (AAAGG)exp and (ACAGG)exp, and the products were analyzed with a GeneScan™ 600LIZ standard (Thermo Fisher Scientific) using capillary sequencer (for detailed reaction conditions, see Supplementary Table 3).
Exome sequencing was carried out as previously15 in the patients with biallelic RFC1 (AAGGG)exp to exclude contributing mutations in known PD genes13. Sequencing data were processed using GATK 4.0.6.0 with current Best PracticesTM (Broad Institute)16,17. Copy number variants were analyzed from the exome data by using XHMM18 with lenient parameters19.
The number of repeats was determined by using long-range sequencing. Unsheared, purified genomic DNA (3 µg) was used to construct sequencing libraries using the Oxford Nanopore Ligation Sequencing Kit (SQK-LSK109) (Oxford Nanopore Technologies, Oxford, UK) following the manufacturer’s instructions. The enzyme incubation times were doubled with the final AMPure purification incubation of 10 min at 37 °C. The library was loaded onto a flow cell (FLO-MIN106D) on a GridION (Oxford Nanopore Technologies). Target regions were enriched using the adaptive sampling option20 on a GridION of high accuracy mode with a bed file assigning the RFC1 locus along with 58 other loci associated with repeat expansion diseases, and their surrounding regions. Sequencing was performed for 3 days with two additional library loadings. Sequences were basecalled using guppy 4.3.4 during the run on the GridION and aligned to GRCh38 using minimap2.14 (https://github.com/lh3/minimap2) and LAST v1132 (https://gitlab.com/mcfrith/last). Tandem-genotypes v1.3.0 was used to find changes in the length of tandem repeats. When biallelic repeat expansions at the RFC1 locus were detected, the names of all reads encompassing the RFC1 locus were picked up using the tandem-genotypes –v option, and FASTA of such reads were generated from FASTQ using seqkit (https://github.com/shenwei356/seqkit). The constructed consensus sequences for both alleles were generated from FASTA files by lamassemble (https://gitlab.com/mcfrith/lamassemble). Detailed repeat analyses were performed using RepeatAnalysisTools (https://github.com/PacificBiosciences/apps-scripts/tree/master/RepeatAnalysisTools).
Ethics approval and consent to participate
The study protocol was approved by the Ethics Committee of Oulu University Hospital (EETTMK 51/2017) and by Kela (87/522/2017), and written informed consents were given by the patients or their legal caregivers. Written informed consent for the publication of identifiable material was given by patient 1 and 2 (supplementary videos 1 and 2).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Sequence data cannot be made publicly available because of restrictions imposed by the EU and Finnish General Data Protection Regulation (GDPR). Access to sequence data can be applied from the Innovation Agent of the University of Oulu (maarit.jokela@oulu.fi; innovationcentre@oulu.fi). Qualified researchers will be required to complete “Material and data transfer agreement for the transfer of human materials (personal data)”. Genetic variation data have been submitted to ClinVar (SCV002032059). Other data are available within the article or supplementary materials.
References
Cortese, A. et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 51, 649–658 (2019).
Rafehi, H. et al. Bioinformatics-based identification of expanded repeats: a non-reference intronic pentamer expansion in RFC1 causes CANVAS. Am. J. Hum. Genet. 105, 151–165 (2019).
Cortese, A. et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain 143, 480–490 (2020).
Nakamura, H. et al. Long-read sequencing identifies the pathogenic nucleotide repeat expansion in RFC1 in a Japanese case of CANVAS. J. Hum. Genet. 65, 475–480 (2020).
Wan, L. et al. Biallelic intronic AAGGG expansion of RFC1 is related to multiple system atrophy. Ann. Neurol. 88, 1132–1143 (2020).
Sullivan, R. et al. RFC1-related ataxia is a mimic of early multiple system atrophy. J. Neurol. Neurosurg. Psychiatry 92, 444–446 (2021).
Traschütz, A. et al. Natural history, phenotypic spectrum, and discriminative features of multisystemic RFC1 disease. Neurology 96, e1369–e1382 (2021).
da Silva Schmitt, G. et al. Dopa-responsive parkinsonism in a patient with homozygous RFC1 expansions. Mov. Disord. 35, 1889–1890 (2020).
Postuma, R. B. et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 30, 1591–1601 (2015).
Havulinna, A. S. et al. Geographical variation of medicated parkinsonism in Finland during 1995 to 2000. Mov. Disord. 23, 1024–1031 (2008).
Kerminen, S. et al. Changes in the fine-scale genetic structure of Finland through the 20th century. PLoS Genet. 17, e1009347 (2021).
Lipponen, J. et al. Molecular epidemiology of hereditary ataxia in Finland. BMC Neurol. 21, 382 (2021).
Blauwendraat, C., Nalls, M. A. & Singleton, A. B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 19, 170–178 (2020).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Siitonen, A. et al. Genetics of early-onset Parkinson’s disease in Finland: exome sequencing and genome-wide association study. Neurobiol. Aging 53, 195–e7 (2017).
DePristo, M. A. et al. A framework for variation discovery and genotyping using next generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011).
Van der Auwera, G. A. & O’Connor B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media, Sebastopol, 2020).
Fromer, M. & Purcell, S. M. Using XHMM software to detect copy number variation in whole‐exome sequencing data. Curr. Protoc. Hum. Genet. 81, 7–23 (2014).
Välipakka, S. et al. Improving copy number variant detection from sequencing data with a combination of programs and a predictive model. J. Mol. Diagn. 22, 40–49 (2020).
Loose, M., Malla, S. & Stout, M. Real-time selective sequencing using nanopore technology. Nat. Methods 13, 751–754 (2016).
Acknowledgements
This study was funded by grants from Sigrid Jusélius Foundation, from Yrjö Jahnsson Foundation and from Finnish Parkinson Foundation. The study received funding from Medical Research Center Oulu and state research funding from Oulu University Hospital. Part of the work was carried out with the support of Biocenter Oulu Sequencing Center, University of Oulu, Finland.
Author information
Authors and Affiliations
Contributions
LK designed the study, collected the samples, established haplotyping and XL-PCRs, analyzed the exome data and wrote the first draft of the manuscript. JS examined the patients, wrote the first draft of patient descriptions and revised the manuscript. HD and FT performed the flanking PCRs and RP-PCRs, analyzed the data and revised the manuscript. AHN participated in molecular investigations and revised the manuscript. AS analyzed the exome sequencing data and revised the manuscript. EK, SM and NM did the long-read sequencing, analyzed the data and revised the manuscript. KM designed and supervised the study and revised the manuscript. All authors have approved the submission of the manuscript in its current form.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kytövuori, L., Sipilä, J., Doi, H. et al. Biallelic expansion in RFC1 as a rare cause of Parkinson’s disease. npj Parkinsons Dis. 8, 6 (2022). https://doi.org/10.1038/s41531-021-00275-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-021-00275-7
This article is cited by
-
Investigation of RFC1 tandem nucleotide repeat locus in diverse neurodegenerative outcomes in an Indian cohort
neurogenetics (2023)
-
Screening for RFC-1 pathological expansion in late-onset ataxias: a contribution to the differential diagnosis
Journal of Neurology (2022)
-
Movement disorders and neuropathies: overlaps and mimics in clinical practice
Journal of Neurology (2022)
