
Abstract
The alteration of a four-oxide alumino-borosilicate glass close to International Simple Glass (ISG) was studied in Si saturation conditions at pH 3, pH 7, and pH 9 using solution and post mortem solid analyses. In particular, the study examined the impact of structural disorder on the glass alteration generated by swift heavy ion irradiation. It is shown that the pre-irradiated glass altered three to four times more than the non-irradiated glass, depending on the pH. This increased alteration can be attributed to two factors: an increased hydrolysis speed of the Si–O–B bonds, which control the glass dissolution under these Si saturation conditions, and a less passivating alteration layer. The latter effect suggests that the glass structural disorder is retained by the first gel formed, which means very low Si atom mobility during alteration. However, gel maturation tends to mitigate the detrimental effect of radiation damages. These results will contribute to the development of a predictive residual rate model.
Similar content being viewed by others
Introduction
The chemical durability of borosilicate glasses has been the subject of many studies ever since this type of material was chosen by certain countries as a containment matrix for long-lived radioactive waste1,2,3. However, nuclear glasses are very complex materials, and the study of the reaction mechanisms involved when the glass interacts with water requires work with model materials. Since 2013, therefore, the scientific community has used a six-oxide borosilicate glass, called ISG glass, as the reference to establish a robust knowledge base concerning the dissolution mechanisms and kinetics of nuclear glasses1,4,5. In a silica-rich solution, ISG glass dissolution is slowed down by several orders of magnitude compared to the initial dissolution rate because of an affinity reduction effect on the silicate network dissolution reaction, and by the formation of a passivating gel layer between the glass and the solution6,7. The rate which results from these processes is called the residual rate. The physical and chemical processes at microscopic, or even molecular, scale that limit the residual rate remain to be established6. Many studies have focussed on the formation and maturation of the passivating gel layers formed on nuclear glasses (for more details, see the following review articles by Frankel et al.4, Gin et al.5, and Thorpe et al.8). Diffusion properties of hydrous species in these gels depend on the composition of both the glass and the solution (Si concentration, pH), on the temperature, but also on time (maturation) and, to a certain extent, on the glass structure. Besides, short and medium structural order within the glass depend on the cooling rate and the irradiation conditions9,10. The question of whether or not there is a link between the glass structure, the gel properties and the residual rate still remains open11. Finding an answer would enable a better understanding of the passivating layer formation mechanism, and thereby open the way to developing reliable kinetic models to predict the long-term behaviour of these materials.
The work described here concerned a 4-oxide glass (CJ2 glass). It has the same elemental molar ratios as ISG and as the complex SON68 glass, a simulant for R7T7 glass produced at industrial scale in France. The study compared leaching behaviour under residual rate conditions of samples irradiated by swift heavy ions (Xe ~1 GeV, 1021 keV cm−3) with that of non-irradiated samples. The irradiation conditions corresponded to the ionic trace regime. According to the literature, this regime leads to modifications in the glass structure and macroscopic properties similar to those observed under the effect of recoil nuclei from alpha disintegrations12,13. As the residual rate is strongly dependant on the pH6,14,15, three different pH were used in these experiments: pH 3 (to maximise the residual rate and heighten the phenomena), pH 7 (to simulate the pH of the water present on the radioactive waste geological disposal site in France), and pH 9 (to simulate the pH imposed by the glass in initially pure water). The test temperature was set at 90 °C and the solutions were initially enriched in 29Si(aq), an isotope present at 4.7% in natural abundance. Following the silicon isotopes in the alteration layer by time-of-flight secondary ion mass spectrometry (ToF-SIMS) provided data concerning its formation mechanism (precipitation vs. in situ condensation without complete hydrolysis of the glass). The Si concentration in the leaching solutions was close to saturation of amorphous silica, to enable the residual rate to be reached quickly16,17. For the three tested pH, the residual rate was determined by monitoring tracer elements like B and Na in the solutions for a period of 3 months. At the end, after the leaching experiments, the two samples, which had been altered at pH 9 were soaked at room temperature in 18O labelled water (isotope present at 0.2% in natural abundance) and methylene blue (an ~1 nm size chemically inert molecule, compared to 0.27 nm for a water molecule) followed by ToF-SIMS analysis in cryogenic mode to conserve the pore water during the analysis. The diffusion and exchange properties of the gel layers were evaluated according to 18O and S (a proxy for methylene blue) depth profiling.
Results
Non-altered glasses
Figure 1 shows the Raman spectra acquired for the pre-irradiated and pristine glasses. For the pre-irradiated glass, the main point is that the R-band broadens and shifts towards 500 cm−1 indicating a change in the distributions of the Si–O–Si angle and of the various rings. A decrease in the mean Si–O–Si angle and an increase in the average ring size in the glass can explain this shift towards the highest wave numbers. A shift in the Qn band related to the connectivity between SiO2 tetrahedra (900–1300 cm−1) towards lower wave lengths can also be seen, showing a depolymerisation of the silicate network under irradiation. Finally, the three-coordinated B band (1300–1500 cm−1) is more intense on the irradiated glass, illustrating a decrease in the average coordination of B. Figure 2 gives the evolution of the R-band depending on the depth. Stabilisation of the R-band shift indicates saturation damage from the surface to a depth of at least 50 µm. The transition zone between the homogeneously irradiated glass and the pristine glass is located between 55 and a little more than 60 μm, in agreement with the SRIM calculations (Fig. 13). It can therefore be considered that the glass is completely and homogenously damaged to saturation to a depth of at least 50 µm.

Analyses from the surface of the glass samples.

Raman shift of the R-band depending on the depth, for the pre-irradiated glass.
NMR MAS spectroscopy of 11B showed a decrease of four fold coordinated B (BO4) atoms from 48 (±2) % in the non-irradiated material to 36 (±2) % in the pre-irradiated glass (Fig. 3). This variation in the B speciation lead to an increase in the number of non-bridging oxygens in the pre-irradiated glass, which should a priori make this glass less durable9.

NMR MAS spectra of 11B in the no- irradiated and pre-irradiated CJ2 glasses.
As a whole, the modifications observed on our materials are in agreement with what has been reported in the literature on borosilicate glasses, i.e., this type of irradiation decreases the coordination of B, increases the structural disorder, the stored energy and decreased the density of the glass12,13.
Solutions analyses
Table 1 and Fig. 4 give the solution data sets. Note that in the tests at pH 9 (experiments A and B), the pH remained stable throughout the experiment. In the experiments at pH 3 and pH 7, adjustments had to be made (addition of HNO3) to maintain the pH between 2.8 and 3.5 for the samples with the target of pH 3, and 6.5 and 7.5 for the pH 7 target value samples. It can be considered that the slight fluctuations observed did not affect the quality of the results.

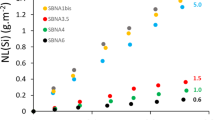
Normalised mass losses of B, Na, and Al as a function of time for the six experiments.
The Si concentrations were globally stable in all the experiments. Those which were significantly higher than the initial concentration—taking into account an uncertainty of ±10% for the measurements—were not due to a dissolution of the Si of the glass but to a slight evaporation of the solution (experiments A and B). There were several observations to support this:
-
The solutions in experiments A and B initially contained ~42 mg L−1 of Li, coming from the LiOH added to dissolve the silica. There was no addition of Li during the two experiments at pH 9. The increase in the Li concentration, about 3% at the end of experiment A and 4.4% at the end of experiment B, is the proof that there was a slight solution evaporation during the tests. This evaporation occurred particularly when samples were taken from the solution. The other solutions not containing Li, the evaporation was estimated from the mass loss measured between the samplings. Although this method is less accurate, it suggests that less than 5% of the starting water has evaporated by the end of the experiment.
-
The isotopic ratio 29Si/Sitot in solution did not vary significantly in any of the experiments. It remained at between 0.94 and 1.09 whatever the experiment and the lapsed time. Any dissolution of the silicate network would have led to a drop in this isotopic ratio by an addition of mainly 28Si to the solution.
-
Finally, the ToF-SIMS analyses that enabled chemical and isotopic data to be obtained for the alteration layer confirm the fact that the Si in the glass was not dissolved.
It can therefore be concluded that under these test conditions, the Si of the glass was not dissolved, whatever the pH. The B and the Na had overall similar behaviour, and their dissolution is close to congruence. The uncertainty for the B/Na congruence ratio was 20%, which does not allow any precise remark about the possible retention of one element rather than the other within the gel. This said, the ToF-SIMS profiles given hereafter show a depletion of these elements in the layer but with a slight retention of B, unlike Na. Overall, these elements can be considered as tracers of glass alteration. Thus, on the basis of these results, it appears that the glass was altered more when the pH was low, and that the pre-irradiated samples were systematically more altered than their non-irradiated homologues (by factors of 2.4 to 3.1, depending on the pH). Finally, Al was systematically much less dissolved than B and Na, even in an acidic medium, which is known to favour its dissolution18. It should also be mentioned that the concentrations measured in our experiments are low compared to those usually measured in the residual rate regime. For example, B concentrations 1000 times higher were measured after 3 months of alteration of CJ2 glass at 90 °C pH 9 in a previous study19. The difference comes from the S/V, which is extremely low in our study. Therefore, it can be expected to have no effect of fluid composition variations on glass behaviour.
When the normalised mass losses of B are presented as a function of the square root of time (Fig. 5), it can be seen that the curves for the pre-irradiated glasses have a fairly obvious bend after a few tens of days, unlike the non-irradiated samples. This may indicate a change in the passivation mechanism. Note that, for all cases, the residual rate was calculated between 55 and 84 days, thus beyond the inflexion point for the irradiated samples.

Exp. A, C, E = non-irradiated samples, Exp. B, D, F = irradiated samples. Despite slight variations due to B retention in gels, NL(Na) followed the same trends. The dotted lines are a visual guide to show a linear slackening as a function of the square root of time.
Solids analyses
At the end of the experiments, coupons were cross-sectionned and gel layer was observed by SEM (Fig. 6 and Supplementary Fig. S1). Thereby the two large faces along with two small sides of each altered coupons were observed. It was noticed that the gel formed on the large faces was uniform and similar on the two faces. Direct comparison between pre-irradiated and non-irradiated samples is thus made possible on this basis, as it is recalled that only the large faces of coupons B, D and F were irradiated. The characteristics of the gel formed on small sides (only two over the four were observed) depend on the state of the surface. If polished, the gel was uniform, but on unpolished sides (called ‘as cut’) a thicker and uneven gel layer developed (clearly visible on samples A, B and C). Gel thicknesses are reported on Table 2. Several observations can be made:
-
Gels formed on irradiated samples are 2.84, 3.75, and 4.05 times thicker than on reference samples at pH 9, pH 7, and pH 3, respectively.
-
As expected, gel thickness on small polished sides of pre-irradiated samples (those small side have not been irradiated) is close to that measured on large sides of non-irradiated sample. This is verified for pH 7 and pH 3. Data are not available for pH 9.
-
Gel thickness for small ‘as cut’ sides are much thicker than on polished sides (up to five times).

A pH9 non-irradiated. B pH9 irradiated. C pH7 non-irradiated. D pH7 irradiated. E pH3 non-irradiated. F pH3 Irradiated. In the first column are shown alteration layers formed on the large faces of the coupons. In the second column are shown the alteration layers formed on a small side of the corresponding coupon. For a given coupon, it is not certain that all the four small sides display the same pattern. R resin, AL alteration layer. Nearby the AL is the uncorroded glass.
The SEM data are compared to those obtained by ToF-SIMS and solution analysis in the discussion.
The ToF-SIMS analyses carried out on the altered samples are given in Figs. 7 to 9. Note that ToF-SIMS analyses were performed on one large side of each coupon. Overall, they confirm the solution data sets and provide additional information. The alteration layers are thicker for the lower pH samples, and for the pre-irradiated glasses. The results also confirm the fact that Si and Al were completely retained in the alteration layer for the tests at pH 9 and 7 (Fig. 7). Even if they are more qualitative (fewer counts) than the normalised profiles, the mappings also indicate a high retention of Si and Al in the alteration layers formed at pH 3 (Figs. 8 and 9).

Note that the mappings are recorded within the green square shown on the optical image but do not correspond to the red square.

Note that the mappings are recorded within the green square shown on the optical image but do not correspond to the red square.

Note that the mappings are recorded within the green square shown on the optical image but do not correspond to the red square.
In all the experiments conducted at pH 9 and 7, the isotopic ratio 29Si/28Si is very slightly higher in the alteration layer than for the pristine glass, but this difference is minimal (0.06 for A, B, C, and 0.1 for D, compared to 0.05 in the pristine glass). In fact, if an isotopic equilibrium with the solution had been reached, an isotopic ratio 29Si/28Si ≫ 1 would have been measured in the alteration layer. As the latter remained very close to the natural abundance (0.05), it shows that in all the experiments the alteration layer was formed by the preferential dissolution of B and Na, without the complete hydrolysis of the bonds around Si and with a passage in the aqueous form before redepositing. If the Si in the glass had passed into the solution, this would have led to a mixture of the two isotopes and the redeposit in the gel would have occurred at the same proportions as the solution. The isotopic ratio 29Si/28Si profile would then have had a gradient with a value close to that of the initial solution in the first layers of the gel, and a progressive decrease as the solution was enriched in 28Si. This was observed for the CJ1 glass, a glass, which is homologous with CJ2 glass but free of Al20. For the CJ2 glass, it is the mechanism involving preferential hydrolysis of B and Na and in situ re-condensation of Si‒OH, which dominates the formation of the alteration layer. This mechanism has already been identified for ISG glass, and opposes the interfacial dissolution re-precipitation mechanism proposed by other authors21,22,23,24.
Unlike the solution analyses, the ToF-SIMS profiles, which give better sensitivity than solution analyses, brought out differences in behaviour between B and Na. If the profiles from experiments A to D are examined carefully, it can be seen that the B was partially retained in the alteration layer, and all the more so when the glass was more altered. The Na was less retained than B in the alteration layer, and its content stabilised there at about 10% of its level in the glass. This value of 10% is approximate, as the normalisation of intensities does not provide a precise quantification. However, this low value for Na, an element, which is supposed to charge compensate tetra-coordinated Al, does not seem sufficient for a total compensation. As the glass contained 3.3 times more Na than Al, there would need to have been a retention >30% of Na (without counting the compensation of tetra-coordinated B) in the alteration layer to conserve all the 4-coodinated Al. It is therefore possible that part of the Al increased its coordination in the alteration layer, unless perhaps the Li added to the initial solution in order to adjust the pH acted as a charge compensator.
Discussion
Table 2 groups the data obtained for the gel thicknesses, from SEM observation, ToF-SIMS analyses and ICP analyses. A global consistency of the results cannot be achieved for two reasons:
-
Certain small sides of the glass coupons have not been polished prior to leaching experiment. Unexpectedly, those sides underwent severe and uneven corrosion, likely because surfaces contained small cracks that favoured alteration and triggered surface increase25. As small sides represent 31–35% of the total surface of the samples, the impact of the phenomenon is not negligible but impossible to quantify.
-
B is partly retained in the gel. This was not taken into account for normalised mass loss calculation but it can be quantified for samples A–D from ToF-SIMS profiles. B retention in gel is discussed further.
Despite these limitations, to assess the impact of irradiation on glass alteration, direct and reliable comparison can be made from SEM observations performed on large faces of the coupons, along with ToF-SIMS analyses, which were also performed on large faces. These comparisons show that whatever the pH, pre-irradiated samples were more altered than non-irradiated samples. This result was already observed at pH 911, but it was never reported in neutral and acid pHs. The difference in gel thickness between irradiated and non-irradiated samples increases with decreasing pH (from a factor 2.8 to a factor 4.0 for pH 9 to pH 3, respectively). This might be related to the capacity of the gel to reorganise, which is favoured in basic pH, as suggested by a previous work on ISG15.
The residual rate, rr, was calculated by linear regression on the normalised mass losses for B between 55 and 83 days, i.e., beyond the inflexion point for the pre-irradiated samples. The value of rr was not calculated for sample F, as alteration went beyond the irradiated area. It should be borne in mind that the value of the residual rates is indicative since the calculation is based on the geometric surface, and therefore ignore the roughness of the small sides. Nevertheless, the residual rates can be compared two by two assuming that the weight of the small sides over the whole of the alteration is similar for a given pH. The results are given in Fig. 10.

An uncertainty of 30% for the value of rr is taken. A, C, E correspond to non-irradiated samples, whereas B and D are related to pre-irradiated samples. Result for experiment F is not provided given that alteration went deeper than the irradiated depth.
For experiment A, rr was close to that determined after 1 year of alteration for CJ2 glass powder altered at pH 9 (3.2 × 10−3 g m−2 d−1 after normalisation at the specific surface of the powder). This gives ~8 × 10−3 g m−2 d−1 after normalisation at the geometric surface, a value, which can be compared to that of our study19. On the basis of the powder experiment, which lasted more than 20 years, it can be expected that the residual rate in experiment A will continue to decrease with time, and that the alteration will carry on uniformly over the entire surface without the formation of secondary minerals. Interestingly, the increase in rr for the irradiated sample (experiment B vs. A) such as observed in our experiments is aligned with residual rate measurements conducted on 238/239Pu or 244Cm-doped glasses that underwent ballistic damages due to recoil nuclei26. Here, the ratio between rr (irradiated)/rr(non-irradiated) is 2.5 and 1.5 at pH 9 and pH 7, respectively. These values are lower than the increase in gel thickness at the end after 83 days of alteration. This supports the idea proposed in a recent paper that the gels formed on irradiated samples reorganised faster than on reference samples11.
The retention of B in the alteration layer took place in the pristine glass/alteration layer interface zone. It was particularly high when the glass was greatly altered (positions of the arrows in Fig. 11). As above explained, for the experiments E and F, this quantification was not possible using the ToF-SIMS mappings. When comparing the B releases into solution for the experiments conducted at pH 9 (A and B), pH 7 (C and D), and at pH 3 (E and F), it was noted above that the latter followed a square root of time evolution for the non-irradiated samples (A, C, and E) whereas the pre-irradiated samples showed an equivalent trend for 15 to 25 days before the curve bent (Fig. 5). The glass alteration rates therefore decreased more notably for the pre-irradiated glasses than for the non-irradiated samples. These observations suggest firstly that the negative effect of pre-irradiation on glass behaviour could be transitory, and secondly that the negative effect of the solution acidity on the residual rate could be counter-balanced by an accumulation of B in the alteration layer, whose effect would be to slow down diffusion of water molecules and thus the progress of alteration (Fig. 10). These coupled phenomena, linking B and water diffusion in the gel, was demonstrated in a previous ISG glass study6. This mechanism will be discussed in more detail below.

The dotted vertical lines indicate the position of the pristine glass/alteration layer interface, and the arrows show the B content at that interface. The retention of B in the alteration layer increased with the thickness of the altered glass.
For ISG-type glasses, the residual rate resulted from the ability of B to continue dissolving after the silicate network no longer (or extremely slowly) dissolved, because Si reached the solution saturation level. In glass compositions like that studied here, there are few B‒O‒B bonds, and therefore the bonds, which do exist cannot form a percolating sub-network. However, there are as many Si‒O‒B bonds as there are for Si‒O‒Si27. This suggests that a sub-network built with Si‒O‒B linkages, a priori more fragile than the purely silicate network5 percolates within the structure and can lead to dissolution of the B atoms without necessarily breaking the Si‒O‒Si bonds nearby. Furthermore, it was calculated that B is responsible for around 90% of the pore formation in gels28. Apart from the formation of a cavity in which water can readily diffuse, the hydrolysis of a Si‒O‒B bond and the dissolution of B leads to the formation of a silanol group (Si‒OH). If two silanols are close enough to each other, they can re-form a siloxane bond (Si‒O‒Si) by expelling a water molecule29. The departures of B and Na together with the condensation of silanols create the porous structure of the gel. The pH could impact water mobility in gels through variations in hydrogen bonding interactions30. Another key process that needs to be considered, is that the gel structure evolves during the reaction, as the hydrolysis and condensation reactions continue within the gel, even though the composition of both the fluid and the gel remain constant28,30,31,32. The reactivity of the silicate network resulting from the departure of mobile elements leads to its reorganisation and to the closing of some pores31,33,34. Knowing that water diffusion coefficient through closed pores is around 10 orders of magnitude lower than in open channels31, it appears that gel maturation/reorganisation can impact the residual rate. As noted above, a previous study showed that pre-irradiated ISG samples altered in conditions similar to samples A and B (90 °C, pH 9) corroded around four times faster than non-irradiated reference samples and formed gels that reorganised faster11. The last result was obtained using TEM in cryogenic mode to preserve the gel structure during observation.
The comparison of the non-irradiated and irradiated glasses highlights significant differences among the three pH levels studied. Whether the glass was irradiated or not, the chemical elements making up the material remained the same, and it has been shown from Si isotopes that the silicate networks (Si‒O‒Si bonds) were only slightly affected by alteration in the Si saturation conditions of the solution. For a given pH, the difference in alteration rates could thus be due to the influence of the structural damages caused by irradiation (structural changes at short and medium distance and variations in the internal energy state of the system) on the hydrolysis of the Si‒O‒B bonds and/or on diffusion coefficients of aqueous species within the alteration layers. In a diluted medium, it has been shown that at pH 9 there is an increase (factor of 4.7) in the initial dissolution rate of the pre-irradiated CJ2 glass compared to a non-irradiated sample35. This value is close to that determined by Lonartz et al. under similar conditions for a Na-borosilicate glass36. Remember that the initial dissolution rate is determined based on the release of Si in solution in a highly diluted medium. These conditions enable the intrinsic reactivity of the glass to be tested in the absence of an alteration layer (resistance to hydrolysis of the silicate network). In the irradiation conditions implemented in our study, the increase in the Si‒O‒M (M = Si, B, Al) bond hydrolysis rate of the glass was established, but in a residual regime where the glass alteration was controlled by several mechanisms (hydrolysis of the Si‒O‒B bonds, transport of water and of B in the gel), it is not known exactly which of these mechanisms was impacted.
To investigate whether the transport properties of the aqueous species in the alteration layer were affected by the structural changes within the glass, an additional “tracing” experiment was carried out on samples A and B at pH 9. This involved submerging the two samples in a solution enriched in H218O and methylene blue for 24 h, and then using ToF-SIMS to analyse the penetration profiles of the isotopically labelled water and the methylene blue, a chemically inert molecule with a hydrodynamic diameter of 1 nm (compared to 0.27 nm for the water molecule). The experiment was carried out at ambient temperature to avoid generating further glass alteration, and the ToF-SIMS analyses were performed after freezing the samples in liquid nitrogen. A cooled sample holder was used in order to preserve the pore water as much as possible during the analysis. It was a tricky analysis situation, and work had to be carried out quickly, as the high vacuum in the analysis chamber can cause water sublimation in spite of the low temperatures. The results were as follows (Fig. 12):
-
S, the methylene blue label, penetrated more than 10 nm into the alteration layer of the non-irradiated glass, and 20 nm into that of the pre-irradiated glass. As the abrasion was fast (6 nm steps for experiment A and 25 nm steps for experiment B), it is not possible to give more precise penetration depths. The profiles are therefore not presented in the figure.
-
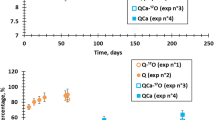
18O, the water molecule label for the solution, penetrated as far as the alteration border, marked by the departure of B atoms.
-
18O exchanged much more with the 16O in the alteration layer of the irradiated glass than in that of the non-irradiated sample.

The alteration fronts are shown by the vertical dotted lines. The isotopic ratios 18O/16O are quantitative, and given on the axis on the right. The alteration layer formed on the irradiated glass incorporated ~3 times more H218O than the non-irradiated glass.
Based on our results, the following reasoning can be followed. On the hypothesis that 18O mainly exchanged with O from water molecules present in pores of the gel31, this tracing experiment revealed the existence of open channels within the gel, which only enabled small molecules (water, but not methylene blue) to diffuse rapidly between the solution and the glass surface. An important point to note is that the quantity of exchangeable water, and therefore the proportion of channels enabling a swift circulation of water molecules, is ~3 times larger in irradiated than in non-irradiated glass (comparison between the plateau levels). This result strongly suggests that the structure of the two gels are different. The higher amount of open channels in the pre-irradiated sample could result from damages created by irradiation in the solid (fission tracks, increase in large sized-rings, and increase in NBO). Another implication of this result is that the gel former atoms (Si and Al) were not mobile during the glass alteration. Indeed, if all the Si and Al atoms were hydrolysed before precipitating within the gel layer, it is probable that the gel would have lost its glass structural characteristics. This aligns with past results showing that glasses irradiated with 252Cf beyond the formation of traces of fission keep these traces in their morphology within the alteration film37. The difference in terms of O exchange rate could also be due to a maturation process occurring at a faster rate on the pre-irradiated samples, as suggested by Tribet et al.11, and leading to bigger and more interconnected pores. Indeed, as above explained, gel reorganises over time. Because of that, the gels characterised at the end of experiments A and B have evolved after they have formed. If one considers than Si and Al are low mobile, the position along the profile can be related to time. It means that the portion of the gel near the interface with the pristine glass (inner gel) is younger than that located closer to the solution. The latter is thus more mature and reorganised than the former. Comparing young and mature gels formed on the two materials (Fig. 12), it appears than the differences are maximum for the young gels, suggesting that reorganisations within the gel plays a major role on alteration in residual rate regime.
If it is considered that the alteration took place at a constant volume, which seems to have been the case here at pH 7 and pH 9, following the method applied to ISG28, the free volume created by B and Na leaching would lead to 43% porosity. Based on this, an isotopic count for the O atoms shows that around 4.3% of the O atoms present in the alteration layer pore water were exchanged during the tracing experiment on non-irradiated glass, compared to 13% for the irradiated samples. These fractions remain quite low, showing either that the porosity in the layers is mainly closed, or that external layers of the gel (close to the interface with the solution) inhibit water diffusion, or simply that the exchange rate is slow. The fractions for the O exchanged in this study were, however, higher than those determined for ISG glass after 1 year of alteration at pH 7, 90 °C. For the latter, the fraction of pore water exchanged was 2.5%, as found by tracing identical to that used in our study31. This weaker exchange needs to be considered in relation to a faster rate decrease (factor of 1000 for ISG glass, compared to about 250 for the CJ2 glass). At this point in the investigations, no conclusion can be drawn as to the location in the gel where the diffusion is the slowest.
The experiments performed during this study did not reveal whether the gels will tend towards having the same diffusion properties because of the maturation process (pore closures and coalescence). Further work will be needed to clarify this point.
This study leads to the following concluding remarks. The CJ2 glass pre-irradiated with heavy ions altered more quickly than the non-irradiated glass under residual rate conditions in acidic, neutral, or basic media. For the three pH levels studied, the alteration rate decreased with time, because of the formation of a passivating alteration layer. This layer retained part of the hydrolysed B, which could impact water diffusion and therefore reinforce its passivating properties, as has been shown in a previous study6. In a basic medium, this work has demonstrated that the alteration layer formed on irradiated glass is less passivating, at least in the early stages, than on non-irradiated glass. We have thus established that the structure of the alteration layer initially inherited from the glass structure, and then matured, likely changing the diffusion coefficients of water and aqueous species. According to a previous study on ISG, it seems that maturation occurs at a faster rate on pre-irradiated samples11, which could explain why the residual rate, even higher at the beginning of the experiment decreased faster than for the non-irradiated material. As concerns the debate regarding the alteration layer formation mechanisms, this work has confirmed that the gel formed on this type of glass is not formed by congruent dissolution and re-precipitation, but rather by a preferential dissolution mechanism for the B atoms, followed by silanol re-condensation involving very low Si atom mobility. From these various observations, it can be concluded that the effects of pre-irradiation on glass dissolution in the residual rate regime are transient.
Following this work, it is important to clearly identify the alteration layer zone or zones contributing to diffusion inhibition. In order to explain the differences between irradiated and non-irradiated samples, the authors of the study above cited11, suggested that the driving force for glass alteration is exerted only at the interface zone between the gel and the glass. Another study performed with SON68 glass in residual rate regime proposed that water diffusion in the uncorroded glass controls the release of Na through ion exchange and B dissolution38. On the basis of our results, it would appear that the question must remain open. Overall, coherency needs to be established among all the results available in the literature. Although the effect of pre-irradiation with 1 GeV Xe on rr is close to that observed on Pu and Cm-doped glass26, the scenarios of irradiation were different (unidirectional irradiation and decoupling between irradiation and alteration in our case), so that this comparison is not sufficient to conclude about the best conditions to simulate the behaviour of nuclear glass in geological repository.
Methods
Glass preparation
The CJ2 glass has the following molar composition: 64.93 SiO2, 13.64 Na2O, 17.3 B2O3, 4.13 Al2O3. The elemental molar ratios of this glass are identical to those of ISG and SON68 glasses. It was prepared from oxide or carbonate precursors melted at 1450 °C in a Pt-Rh crucible for 3 h. After casting and cooling, the glass was ground up and then re-melted to ensure its homogeneity. Next it was poured into a graphite crucible and annealed at 550 °C for 1 h. Monolithic samples were prepared by slicing and polishing the large faces to reach the 1 µm grade diamond powder leading to a roughness of a few nm. Polishing was performed without water. Coupons were cleaned with absolute ethanol in an ultrasonic bath. Note that most of the small sides of the coupons were let as cut. The dimensions of the coupons were 5.7 × 5.4 × 1.5 mm3 and 6.9 × 6.6 ×1.5 mm3 for the non-irradiated samples and the irradiated samples, respectively. This means that the small sides represent 35% and 31% of the total surface of the coupons for the non-irradiated samples and the irradiated samples, respectively.
Irradiation scenario
The CJ2 was irradiated by a unidirectional Xe beam at 952 MeV with a fluence of 7.5 × 1012 ions cm−2 (GANIL, Caen). The damage saturation is expected to be reached for this fluence10 over the majority of the irradiated thickness. Simulation with the SRIM code39 indicated an irradiated depth of ~64 µm and a dose of 1.1 × 1021 keV cm−3, i.e., about 73 MGy. The energy deposited (Fig. 13) was mainly due to electronic interactions with high losses of electronic energy (16–18 keV.nm−1) and, to a lesser extent and in a highly localised manner at the end of the Xe ion paths, to nuclear interactions (<0.6 keV nm−1).

Deposits of electronic energy (in blue) and nuclear energy (in orange) as a function of the depth for the irradiation of the CJ2 glass by Xe ions at 952 MeV. Simulation carried out using the SRIM software.
Beyond a certain ion track threshold (around 2–3 keV nm−1)10,13, as was the case for the irradiation conditions here, the formation of ion tracks can be observed in the glass. According to the inelastic thermal spike modelling, these ion tracks induce a local glass melting along the passage of a Xe particle because of the high local energy deposit (The energy transmitted to the electrons through inelastic interactions is transferred to the glass network through an electron-phonon coupling), followed by a fast hyper-quenching, which solidifies the disordered material in a higher enthalpy state13.
Irradiation in the above threshold electronic collisions (ATEC) regime leads to evolutions in the structure and the macroscopic properties of the glass similar to those observed after nuclear interactions generated by recoil nuclei induce by alpha decay or after external irradiation involving heavy ions of a few MeV10,12,13. However, few studies concerning the leaching of irradiated glass have implemented this type of high-energy irradiation. To our knowledge, only one such study is described in the literature, and it focusses how it effects the initial dissolution rate36. The work reported an increase by a factor of 3.7 in the initial dissolution rate of a ternary glass, which underwent irradiation by Au ions at 1 GeV.
Analysis of non-altered glasses by Raman spectroscopy
The pristine and the pre-irradiated glasses were analysed by Raman spectroscopy on a Horiba Jobin Yvon XploRA Plus spectrometer with 1800 g.mm−1 at the CEA DE2D lab. The spectra were recorded at a 532 nm wavelength perpendicularly to the pristine glass sample surface and transversally, starting with a polished section, for the pre-irradiated glass sample. For each analysis, 20 accumulations of 30 s were obtained in the exact same condition and position.
Analysis of non-altered glasses by NMR spectroscopy
The speciation of B in the pristine and the irradiated glasses was studied by NMR MAS spectroscopy for 11B on a Bruker Avance II spectrometer at the CEA Saclay site. The measurements were carried out in a magnetic field at 11.72 T and at a rotation frequency of 12.5 kHz.
Leaching experiments
The six experiments were carried out under the following conditions (Table 3): The solutions were prepared by dissolving 29SiO2 (Cortecnet, 99.69% of 29Si) at 90 °C pH 10 in order to obtain 120 mg L−1 of Si. The pH 10 was obtained by adding LiOH to 18.2 MΩ cm deionized water. Part of the water was then evaporated to obtain the saturation concentration for amorphous silica at 3 pH needed for the study. The pH levels were adjusted with ultrapure HNO3 solutions. Each experiment used one 0.5 × 0.5 × 0.2 cm CJ2 glass pellet. The geometric surface of each pellet was calculated after measurement of the sides with a precision calliper. The samples were cleaned with absolute ethanol before being placed in the reactor on a support enabling the solution to be in a half-height position. The reactors used were 20 mL containers in polyfluoroalkyl (PFA). To limit evaporation, these reactors were held in an over-pack container, also made of PFA and containing a few mL of deionized water. The experiments were carried out in an oven set at 90 ± 2 °C. On each sampling date, the reactors were taken out of their containers, dried, weighed, and opened. The pH was measured at 90 °C and, if necessary, adjusted to the target value. Between 0.5 mL and 1 mL of solution were pipetted out, then diluted with ultrapure HNO3 0.5 N to obtain 2 mL of solution for the analyses. The weights were obtained using 0.1 mg precision scales.
Solution analyses
The solutions were analysed by ICP-OES and ICP-MS at the CEA LMAT laboratory, Marcoule (France). Li, B, Na and Al were analysed using an ICP/MS quadripolar ICAP_Q (ThermoFisher Scientific), on samples diluted between 10 and 1000 times based on calibration lines from certified mono-elemental standards. The total “Si” was analysed on an ICP/AES ICAP (ThermoFisher Scientific), on samples diluted 10 times based on calibration lines from certified mono-elemental standards. The 29Si was analysed on an ICP/MS quadripolar ICAP_Q (ThermoFisher Scientific), on samples diluted between 200 and 2000 times based on calibration lines from a certified enriched 29Si standard. A trueness test for the calibration was carried out by the addition of a known amount of natural isotopy standard to one of the samples, which confirmed the values measured. The ratio 29Si/total Si was calculated next, to show that virtually all the Si present in the initial solutions consisted of 29Si. The relative uncertainties associated with these concentration measurements were ±10%.
From the elemental concentrations, the normalised mass losses were calculated with the formula:
where C(i)t is the concentration of i in solution at the time t, Vt the volume of solution at time t, S the glass surface area, and xi the mass fraction of the element i in the glass. NL(i) is expressed in g m−2. The B equivalent thicknesses, which give the thickness of the glass that has been altered assuming that no B is retained in the alteration layer, were calculated as follows:
where ρ is the mass of the glass (2.41 g cm−3). Eth(B) is expressed in µm. The uncertainty for NL(i) and ETh(B) is 15%. Eth(B) could also correspond to the gel thickness under the hypothesis of isovolumicity (volume of altered glass = volume occupied by the gel).
Altered solid analyses by ToF-SIMS
The samples A–F were analysed by ToF-SIMS at the end of the experiment. These analyses were carried out by TESCAN ANALYTICS in Fuveau (France) on a TOF 5 device from IONTOF. Analyses were performed in abrasion-analysis mode for samples A–D, and in mapping mode after embedding in resin and the preparation of polished sections for samples E and F because of their thick alteration layers.
For the samples A–D, isotopic and elemental profiles were obtained by alternating the analysis and abrasion cycles according to the following sequences:
Analysis:
-
Primary ions: Bi3++, 25 keV, I ~ 0.1 pA
-
Area analysed: 50 × 50 µm², 256 × 256 pixels
-
Negative secondary ions analysed
-
Neutralisation of the surface by a pulsed flux of low energy electrons (<20 eV)
-
Trigger level: 20 mV
Abrasion:
-
Primary ions: Cs+, 2 keV, I ~ 92 nA
-
Area eroded: 200 × 200 µm²
-
Analysis: acquisition of 10 to 400 uma scan (time of max vol = 70 µs)
-
Pause between erosion and analysis: 0.5 s
The depth calibrations were carried out by measuring the size of the craters with a 1D mechanical profilometer. The average abrasion speed corresponded to the crater depth divided by the abrasion time, considering that the speed was constant for the entire profile. The values obtained were 1.11 nm s−1 for A, 1.03 nm s−1 for B, 1.05 nm s−1 for C, and 1.06 nm s−1 for D. For each species of interest point-to-point intensities were normalised to their mean value calculated in the pristine glass. The interface between the pristine glass and the alteration layer is where the B and Na profiles show a sharp drop, anticorrelated with that of H.
The ToF-SIMS images of the samples E and F were obtained under the following conditions:
-
Primary ions: Bi3++, 25 keV, I ~ 0.7 pA, 70 µs
-
HCBu Mode
-
Area analysed 100–130 µm², 512 × 512 pixels
-
Negative secondary ions analysed
-
Neutralisation of the surface by a pulsed flux of low energy electrons (<20 eV)
Following the analyses described above, samples A and B were placed in a solution enriched in H218O and methylene blue at ambient temperature for 24 h. After this « tracing » step, the samples were plunged into liquid nitrogen for 10 min, then set up on a cooled sample holder before being analysed by ToF-SIMS in abrasion-analysis mode. The analysis conditions were the same as those given above for samples A–D except for the current of primary ions for the abrasion (94 nA). Keeping the samples cold during the analysis meant that the pore water remained in the alteration layer. The analysis of the isotopic ratio for O, 18O/16O was quantitative. This enabled a population estimation to be made in order to calculate the fraction of pore water exchanged during the tracing step.
Solid analyses by SEM
Altered glass samples were embedded in resin, cross sectioned and polished for scanning electron microscopy SEM observations. Observations were performed on a SEM ZEISS MERLIN at 30 kV.
Data availability
The data that support the findings of this study are available from the corresponding authors on reasonable request.
References
Gin, S. et al. An international initiative on long-term behavior of high-level nuclear waste glass. Mater. Today 16, 243–248 (2013).
Grambow, B. Nuclear waste glasses-How durable? Elements 2, 357–364 (2006).
Ojovan, M. I., Lee, W. E. & Kalmykov, S. N. An Introduction to Nuclear Waste Immobilisation. 3rd edn (Elsevier, 2019).
Frankel, G. S. et al. Recent advances in corrosion science applicable to disposal of high-level nuclear waste. Chem. Rev. https://doi.org/10.1021/acs.chemrev.0c00990 (2021).
Gin, S., Delaye, J.-M., Angeli, F. & Schuller, S. Aqueous alteration of silicate glass: state of knowledge and perspectives. NPJ Mater. Degrad. 5, 42 (2021).
Gin, S. et al. Insights into the mechanisms controlling the residual corrosion rate of borosilicate glasses. NPJ Mater. Degrad. 4, https://doi.org/10.1038/s41529-020-00145-2 (2020).
Neeway, J., Abdelouas, A., Grambow, B. & Schumacher, S. Dissolution mechanism of the SON68 reference nuclear waste glass: New data in dynamic system in silica saturation conditions. J. Nucl. Mater. 415, 31–37 (2011).
Thorpe, C. L. et al. Forty years of durability assessment of nuclear waste glass by standard methods. NPJ Mater. Degrad. 5, https://doi.org/10.1038/s41529-021-00210-4 (2021).
Angeli, F. et al. Effect of thermally induced structural disorder on the chemical durability of International Simple Glass. NPJ Mater. Degrad. 2, 31 (2018).
Mir, A. H. & Peuget, S. Using external ion irradiations for simulating self-irradiation damage in nuclear waste glasses: State of the art, recommendations and, prospects. J. Nucl. Mater. 539, 30 (2020).
Tribet, M. et al. New Insights about the Importance of the Alteration Layer/Glass Interface. J. Phys. Chem. C. 124, 10032–10044 (2020).
Peuget, S., Delaye, J. M. & Jegou, C. Specific outcomes of the research on the radiation stability of the French nuclear glass towards alpha decay accumulation. J. Nucl. Mater. 444, 76–91 (2014).
Mir, A. H. Radiation Effects on Oxide Glasses: Importance of Energy Deposition and Relaxation Processes. (University of Caen, 2015).
Gin, S. & Mestre, J. P. SON 68 nuclear glass alteration kinetics between pH 7 and pH 11.5. J. Nucl. Mater. 295, 83–96 (2001).
Fournier, M. et al. Effect of pH on the stability of passivating gel layers formed on International Simple Glass. J. Nucl. Mater. 524, 21–38 (2019).
Vienna, J. D., Ryan, J. V., Gin, S. & Inagaki, Y. Current understanding and remaining challenges in modeling long-term degradation of borosilicate nuclear waste glasses. Int. J. Appl. Glass Sci. 4, 283–294 (2013).
Gin, S., Frugier, P., Jollivet, P., Bruguier, F. & Curti, E. New insight into the residual rate of borosilicate glasses: effect of S/V and glass composition. Int. J. Appl. Glass Sci. 4, 371–382 (2013).
Hamilton, J. P., Pantano, C. G. & Brantley, S. L. Dissolution of albite glass and crystal. Geochim. Cosmochim. Acta 64, 2603–2615 (2000).
Gin, S., Beaudoux, X., Angeli, F., Jegou, C. & Godon, N. Effect of composition on the short-term and long-term dissolution rates of ten borosilicate glasses of increasing complexity from 3 to 30 oxides. J. Non-Cryst. Solids 358, 2559–2570 (2012).
Gin, S. et al. A general mechanism for gel layer formation on borosilicate glass under aqueous corrosion. J. Phys. Chem. C. 124, 5132–5144 (2020).
Geisler, T. et al. Aqueous corrosion of borosilicate glass under acidic conditions: A new corrosion mechanism. J. Non-Cryst. Solids 356, 1458–1465 (2010).
Lenting, C. et al. Towards a unifying mechanistic model for silicate glass corrosion. NPJ Mater. Degrad. 2, 10 (2018).
Ruiz-Agudo, E. et al. Control of silicate weathering by interface-coupled dissolution-precipitation processes at the mineral-solution interface. Geology 44, 567–570 (2016).
Hellmann, R. et al. Nanometre-scale evidence for interfacial dissolution-reprecipitation control of silicate glass corrosion. Nat. Mater. 14, 307–311 (2015).
Neill, L. et al. Various effects of magnetite on international simple glass (ISG) dissolution: implications for the long-term durability of nuclear glasses. NPJ Mater. Degrad. 1, https://doi.org/10.1038/s41529-017-0001-6 (2017).
Tribet, M. et al. Alpha dose rate and decay dose impacts on the long-term alteration of HLW nuclear glasses. NPJ Mater. Degrad. 5, 36 (2021).
Lu, X. N., Ren, M. G., Deng, L., Benmore, C. J. & Du, J. C. Structural features of ISG borosilicate nuclear waste glasses revealed from high-energy X-ray diffraction and molecular dynamics simulations. J. Nucl. Mater. 515, 284–293 (2019).
Collin, M. et al. Structure of International Simple Glass and properties of passivating layer formed in circumneutral pH conditions. NPJ Mater. Degrad. 2, https://doi.org/10.1038/s41529-017-0025-y (2018).
Bunker, B. C. Molecular mechanisms for corrosion of silica and silicate-glasses. J. Non-Cryst. Solids 179, 300–308 (1994).
Ngo, D. et al. Hydrogen bonding interactions of H2O and SiOH on a boroaluminosilicate glass corroded in aqueous solution. NPJ Mater. Degrad. 4, https://doi.org/10.1038/s41529-019-0105-2 (2020).
Gin, S. et al. Dynamics of self-reorganization explains passivation of silicate glasses. Nat. Commun. 9, https://doi.org/10.1038/s41467-018-04511-2 (2018).
Gouze, B., Cambedouzou, J., Parres-Maynadie, S. & Rebiscoul, D. How hexagonal mesoporous silica evolves in water on short and long term: role of pore size and silica wall porosity. Microporous Mesoporous Mater. 183, 168–176 (2014).
Cailleteau, C. et al. Insight into silicate-glass corrosion mechanisms. Nat. Mater. 7, 978–983 (2008).
Jollivet, P. et al. Investigation of gel porosity clogging during glass leaching. J. Non-Cryst. Solids 354, 4952–4958 (2008).
Jan, A. Impact of ballistic effects on the gel layer properties of simplified nuclear glasses: a Monte Carlo simulation approach. (PhD thesis, University of Montpellier, France, 2020).
Lonartz, M. I. et al. The effect of heavy ion irradiation on the forward dissolution rate of borosilicate glasses studied in situ and real time by fluid-cell raman spectroscopy. Materials 12, https://doi.org/10.3390/ma12091480 (2019).
Dran, J. C., Petit, J. C. & Brousse, C. Mechanism of aqueous dissolution of silicate-glasses yielded by fission tracks. Nature 319, 485–487 (1986).
Strachan, D. et al. On the dissolution of a borosilicate glass with the use of isotopic tracing-Insights into the mechanism for the long-term dissolution rate. Geochim. Cosmochim. Acta 318, 213–229 (2022).
Biersack, J. P., Ziegler, J. F. & and Ziegler, M. D. SRIM, The Stopping and Range of Ions in Matter (Chester, Maryland, 2008).
Acknowledgements
This study was funded by CEA, Orano, and EDF under the agreement F25197. Authors are grateful to Tescan Analytics team for the ToF-SIMS analyses and to the CEA LMAT team for the solution analyses.
Author information
Authors and Affiliations
Contributions
S.G. supervised the study and wrote the paper. A.J. and J.M.D. prepared the irradiated samples. M.C. performed the leaching experiments. L.S. and N.B. performed the Raman study. All the authors participated to the scientific discussion and contributed to the editing of the manuscript under its present form.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gin, S., Jegou, C., Sessegolo, L. et al. Effects of irradiation on the mechanisms controlling the residual rate of an alumino-borosilicate glass. npj Mater Degrad 6, 59 (2022). https://doi.org/10.1038/s41529-022-00266-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-022-00266-w
This article is cited by
-
Influence of radiation on borosilicate glass leaching behaviors
npj Materials Degradation (2024)

{kind=link}