
Abstract
Naval vessels regularly mix fuel and seawater as ballast, a practice that might exacerbate fuel biodegradation and metal biocorrosion. To investigate, a metagenomic characterization and metabolite profiling of ballast from U.S. Navy vessels with residence times of 1-, ~20-, and 31 weeks was conducted and compared with the seawater used to fill the tanks. Aerobic Gammaproteobacteria differentially proliferated in the youngest ballast tank and aerobic-specific hydrocarbon degradation genes were quantitatively more important compared to seawater or the other ballast tanks. In contrast, the anaerobic Deltaproteobacteria dominated in the eldest ballast fluid with anaerobic-specific hydrocarbon activation genes being far more prominent. Gene activity was corroborated by detection of diagnostic metabolites and corrosion was evident by elevated levels of Fe, Mn, Ni and Cu in all ballast samples relative to seawater. The findings argue that marine microbial communities rapidly shift from aerobic to anaerobic hydrocarbonoclastic-dominated assemblages that accelerate fuel and infrastructure deterioration.
Similar content being viewed by others


Introduction
The impact of humans on planetary systems led to the proposal that mankind has entered a new geologic epoch, the Anthropocene1,2. A distinguishing feature is the ‘Great Acceleration’ phase wherein a host of physical, chemical, and biological rate processes increased dramatically over the second half of the twentieth century. For example, from 1950 to 2015, worldwide oil production increased >8.7 fold from just over 500 to 4362 million tons3. There is a nearly equivalent increase (>8.5 fold) in world steel production from 189 to 1,621 million tons over the same time period4. The importance of steel to modern societies prompted one scholar to argue that man is still very much in the Iron Age5. Herein, we suggest that the relationship between fuel and steel is intricately linked, particularly in marine environments where metal corrosion and hydrocarbon biodegradation occur throughout the carbon-steel infrastructure. The complex and rapidly changing nature of such problems tend to resist simple control measures. Arguably, the ballast tanks of military ships, where diesel fuel and seawater intentionally come into contact, are near ideal microcosms in efforts to understand the underlying patterns of such deterioration processes.
In the 1960s, the U.S. Navy recognized shipboard corrosion problems in seawater-displaced fuel ballast tanks6,7 and in tanks storing various fuels8,9. Fuel souring and corrosion of ballast tanks were linked to marine sulfate-reducing bacteria (SRB)8,9. Similar corrosion-based ship ‘wasting’ in ballast tanks was observed on submerged horizontal surfaces and tank bottoms in a cargo vessel10. Huang and colleagues11, also reported corrosion of surfaces and uncoated tank bottoms in oil cargo holds of oil tanker ships and enumerated relatively large populations of SRBs.
The capacity of traditional fuels to biodegrade and potentially impact steel corrosion has recently been explored to establish a baseline to which various formulations can be compared. To that end, a standardized protocol was developed12 to assess, (i) removal of organosulfur components13, (ii) the inclusion of fatty acid methyl esters14,15, (iii) the impact of different feedstocks used to make biofuel16, (iv) the method of biofuel processing16, (v) subtle differences in hydrocarbon composition and structure17, and (vi) the impact of next-generation biofuel formulations18.
The in vitro studies demonstrated the susceptibility of fuel components to biodegradation under anaerobic conditions and the link between hydrocarbon oxidation and the production of corrosive sulfides. The assumption is that information on microbial successional patterns and metabolic processes could be extrapolated to in situ environments where fuel and metal come into contact. To test this prospect, we investigated the marine microflora and associated metabolic activities in the ballast tanks of several Navy ships and compared the findings to harbor water and to seawater-only ballast from another ship. The goal was to identify the predominant mechanisms impacting the fate of diesel fuel once inside a darkened ballast tank for various lengths of time and how it might be related to shipboard infrastructure corrosion.
Results
Ship ballast and seawater
Prior to sampling, interviews with navy personnel revealed that crews retained ballast fluids on board for varying lengths of time. Since the ships were of the same class (Arleigh Burke destroyers), possessed seawater-compensated carbon steel ballast tanks, were stationed in the same harbor, and used the same seawater and fuel as ballast, comparison between ships as a function of ballast residence time seemed pragmatic. Nevertheless, variations in vessel operating practices, distances traveled, fuel use patterns, and other between-vessel differences were also evident. Thus, the harbor water and a research vessel that possessed carbon steel ballast tanks but did not mix fuel and seawater (Ship NF—no fuel) for ballast served as comparative controls.
Characterization of fluid samples
Harbor water and ballast tank fluids of varying shipboard residence time were obtained from three naval vessels as well as from ship NF. The samples were analyzed for potential electron acceptors and selected dissolved metals (Table 1). The shortest residence time ballast water (Ship #1) contained ~50% less dissolved oxygen than the San Diego harbor, but its sulfate concentration was comparable to both the seawater and Ship #2. In contrast, the ship with the longest ballast residence time (Ship #3) exhibited 4 mM sulfate depletion in 8 mo. Dissolved Mn, Cu, Fe, and Ni concentrations in the ballast waters were substantially higher than in the harbor water. After 1 week of residence (Ship #1), these metals increased ~4.5–15-fold (Table 1). Dissolved Cu and Ni levels were much higher than Mn in Ships #1 and #2, but Ship #3 had the opposite trend with Mn being 10-fold higher than the other elements. In all ships, particles were mostly copper sulfides, with a smaller percentage of nickel sulfides, and an even smaller fraction of iron sulfides. In a few cases, both copper and nickel sulfides were present in the same particle but manganese sulfides were not detected (Table 1). Comparison of the metals in the ballast tanks relative to the harbor water suggests that the intake fluids accounted for a negligible fraction of the shipboard dissolved metal status and implicates metal dissolution through corrosion as the more likely explanation. The ballast of a comparator ship that did not mix fuel and seawater (Ship NF) contained higher levels of Cu and Ni than the harbor water, but less dissolved and particulate metal than the other vessels.
Marine populations and community composition
Microbial populations in the harbor and samples from the Navy ships were quantified by direct microscopy, domain-specific qPCR, and single-copy marker gene quantification based on the metagenomic analysis (Table 2). Insufficient DNA was recovered from Ship NF for metagenomic sequencing without amplification which would have biased the findings and negated comparability. Direct counting revealed that the microbial population size was comparable in all Navy ships (105 cells mL−1), independent of ballast residence time and between 2 and 10-fold less than the harbor community. Quantitative PCR enumeration of bacterial and archaeal 16S rRNA genes ranged from 105–107 and 102–103 gene copies mL−1, respectively. By this measure, the Bacteria bloomed after 1 wk aboard Ship #1 relative to the harbor community but returned to near comparable levels with increased ballast residence time as in Ships #2 and #3 (Table 2). Archaea were several orders of magnitude less abundant than Bacteria in all samples. The largest picoeukaryote populations were found in the harbor water, whereas sharply decreased numbers were found in all ballast samples.
Microbial diversity indices, including the Shannon index, observed OTUs, and Simpson’s evenness metric (Table 3) indicate that all communities showed a bias towards some taxa (e.g. more uneven distribution of species) and a trend of increasing dominance of particular microbial types with increasing ballast tank residence time. There is also a limited change in the range of species richness (e.g. observed species) of only 8% between Ship #1 and Ship #2.
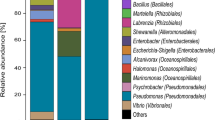
The composition of microbial populations within each sample was determined by comparison of each 16S rRNA gene library extracted from the metagenome samples against the Silva SSU reference database (Fig. 1a). Taxa belonging to the Gammaproteobacteria (19.7%, 64.6%, 32.3%, 23.1%), Alphaproteobacteria (24.2%, 10.8%, 11.4%, 7.6%), Flavobacteria (20.5%, 7.1%, 12.1%, 1.8%), and the Deltaproteobacteria (3.1%, 4.5%, 21.1%, 43.3%) accounted for substantive proportions of the communities within the harbor water and Ships #1, #2 and #3, respectively.

Class-level depiction (a) and Order-level (b) comparison for the Delta- and Gammaproteobacteria.
A decrease in number of OTUs was observed from the harbor to Ship NF to Ships # 1, 2, and 3 (Supplementary Table 1, analysis of 16S amplicon libraries). All ballast tank samples had much lower Chao1 richness scores than did the harbor sample (Supplementary Table 1). Data from 16S rRNA amplicon libraries gave similar trends in relative abundance for most taxa (Supplementary Fig. 1) as those derived from metagenome sequencing for samples from the harbor and ships containing fuel. The sample from a ship whose ballast tank did not contain fuel (Ship NF) was most similar to the harbor sample but with approximately half the relative abundance of Flavobacteria, higher relative abundance of Alphaproteobacteria and Gammaproteobacteria but no increase in the relative abundance of Deltaproteobacteria.
Based on analysis of the metagenomes, Rhodobacterales (8.9%) and members of the SAR 11 clade (7.7%) were the Alphaproteobacteria accounting for greatest proportion of the total harbor community. Within the harbor water Gammaproteobacteria, sequences were predominantly affiliated with Oceanospirillales and Alteromonadales at 8.9% and 4.6% total relative abundance, respectively (Fig. 1b). The harbor contained several groups of marine Eukaryota and were dominated by Alveolata (11.0%) and Stramenopiles (4.0%). Despite accounting for ~20% of the total marine community in the harbor, the Eukaryota were substantially reduced in the ballast tank assemblages (Table 2). The decline after only a week of residence time was also reflected in the reduced level of eukaryotic rRNA gene sequences detected in the other ships (Fig. 1). Sequences classified as Archaea were relatively minor and never exceeded 0.3% in any shipboard community.
The Gammaproteobacteria were enriched in the Ship #1 ballast tank assemblage to become the dominant taxonomic Class, with the concomitant decline of the Alphaproteobacteria and the Flavobacteria. Alteromonadales, Oceanospirillales, and Thiotrichales increased to account for 42%, 35%, and 19% of all observed Gammaproteobacteria, respectively (Fig. 1b). Epsilonproteobacteria were also enriched 10-fold (2.8%) after one week in the ballast tank of Ship #1 and were represented almost exclusively (97%) by members of the Campylobacterales.
Deltaproteobacteria were the dominant Class observed in the Ship #2 community at 21.0%, a ~3.5-fold increase relative to Ship #1. Members of the Desulfuromonadales and Desulfobacterales accounted for the majority of the Deltaproteobacteria at 80 and 10%, respectively (Fig. 1b). Though the total proportion of Gammaproteobacteria was smaller in Ship #2 vs. Ship #1, the Alteromonodales were enriched to represent 71% of this taxon (Fig. 1b). Taxa affiliated with the Clostridia (7.2%) and Mollicutes (4.6%) accounted for substantive portions of the total ballast tank community in Ship #2, with increases of ~3-fold and ~5-fold relative to Ship #1. The Alphaproteobacteria, Flavobacteria, and Epsilonproteobacteria were all slightly higher in relative abundance in the Ship #2 community relative to Ship #1.
The ballast tank community of Ship #3, with the longest residence time, was the most different from the harbor water assemblage (Fig. 1a). Deltaproteobacteria (43.3%) was the most abundant taxon, with members belonging to the Desulfobacterales, Desulfarculales, and Desulfovibrionales at 66%, 27%, and 6%, respectively (Fig. 1b). This community also contained the largest population of Bacteroidetes (8.2%), represented predominantly by unclassified members of the Bacteroidales (57% of the Bacteroidetes), and the smallest segment of Flavobacteria observed in any sample. Gammaproteobacteria, Alphaproteobacteria, and Epsilonproteobacteria also accounted for smaller community abundance in Ship #3 relative to Ship #2.
The trends in relative proportions seen in the metagenome data were supported by the proportions of aerobic alkane degraders, Marinobacter in particular (Supplementary Table 2) as well as of Deltaproteobacteria (Supplementary Table 3) found in the 16S rRNA amplicon libraries. In particular, relative abundances in Ship NF of Marinobacter and Deltaproteobacteria were similar to those in the harbor and less than in the three ships that used fuel in ballast tanks.
Functional genes associated with the utilization of common electron donors and acceptors were characterized by translated Blast analysis (Fig. 2). The nitrification genes, ammonia monooxygenase (amoA) and hydroxylamine dehydrogenase (hao) did not exceed 0.3% normalized abundance within any sample library. Genes associated with the oxidation of reduced inorganic sulfur species were present within all of the samples analyzed. Sulfur-oxidizing system (SOX) protein B (soxB) declined from 5.9% in the harbor water metagenome to ~4.2% of the functional capacity to Ships #1 and #2 and to barely detectable levels in Ship #3 (0.5%) (Fig. 2). Nitrogen fixation nitrogenase gene (nifH) generally increased with ballast water residence time from a low in the harbor water (0.9%) to 5.5%, 7.1%, and 8.8% of the libraries for Ships #1, #2, and #3, respectively.

Gene abundances are normalized to single-copy marker gene (rpoB) abundance for each sample. Colors are scaled based on values in discrete rows. Gene abbreviations: amoA, ammonia monooxygenase; hao, hydroxylamine oxidoreductase; soxB, sulfur oxidation gene; sqr, sulfide:quinone oxidoreductase; ccoN cbb3-type cytochrome C oxidase; coxA, aa3-type cytochrome C oxidase; aprA, adenylylsulfate reductase; phsA, thiosulfate reductase; dsrA, dissimilatory sulfite reductase.
Genes affiliated with the reduction of oxygen and nitrate exhibited a marked increase between the harbor water and Ships #1 and #2. The aerobic respiration genes cytochrome c oxidase subunit I (ccoN and coxA) had the highest levels of any inorganic redox genes assayed in the harbor and ballast tank waters of Ships #1 and #2. However, there was a 50% decrease in cytochrome c oxidase subunit I gene abundance after 8 months in Ship #3. Nitrate reductase genes (cytoplasmic narG and periplasmic napA) had a combined abundance that markedly increased in ballast tank waters of Ships #1 (22.1%) and #2 (38.0%) relative to the harbor water (9.0%) and then declined to 11.3% in the Ship #3 library. The potential for sulfate and thiosulfate reduction was investigated through an assessment of the adenylylsulfate reductase (aprA) and thiosulfate reductase (phsA/prsA) genes, respectively. Total sulfidogenesis potential was marked by the abundance of the dissimilatory sulfite reductase gene, dsrA. All genes associated with the reduction of sulfoxyanions remained at relatively low levels in all samples excluding Ship #3. Between Ships #2 and #3, there were increases of 4-fold in aprA, 5-fold in phsA, and 5-fold in dsrA genes. These were the highest levels observed for each of these genes within this study and represented increases relative to the harbor water library ranging between 3.6-fold for adenylylsulfate reductase to 30-fold for thiosulfate reductase.
Evidence for hydrocarbon biodegradation
Normalized gene abundances (Fig. 3) and observed metabolic intermediates (Fig. 4) associated with the activation and biodegradation of diesel fuel components were readily evident in the shipboard ballast tank systems. Oxygen-dependent mechanisms for the activation of diesel hydrocarbons were assayed by quantifying the proportion of genes annotated as alkane 1-monooxygenase (alkB) for aliphatic substrates, ethylbenzene dioxygenase (etbAa), toluene monooxygenase (tmoA), benzene/toluene dioxygenase (todC1), and xylene monooxygenase (xylM) for BTEX hydrocarbons, and naphthalene 1,2-dioxygenase (nahAc) and 1-methylnaphthalene hydroxylase for polyaromatic hydrocarbons. Phenol hydroxylase (dmpL/poxC), phenol 2-monooxygenase, catechol 1,2-dioxygenase (catA) and catechol 2,3-dioxygenase (catE), along with salicylaldehyde dehydrogenase and salicylate hydroxylase were used as metrics for the genetic potential for the aerobic microbial metabolism of mono- and polyaromatic hydrocarbons. Alkyl-, benzyl-, and naphthylmethyl-succinate synthases (assA, bssA, nmsA) served as indicator genes for anaerobic activation of aliphatic, mono-, and polyaromatic hydrocarbons by addition to fumarate. The ethylbenzene dehydrogenase gene ebdA served as a marker for the potential of anaerobic activation of ethylbenzene by dehydrogenation and subsequent hydroxylation of the ethyl-substituent.

Gene abundances are normalized to single copy marker gene (rpoB) abundance for each sample. Numbers correspond to genes associated with metabolic reactions indicated in Fig. 4.

Blue boxes indicate compounds identified in Ship #1 only, red in Ship #3 only, and black were observed in both samples. Numbers above arrows correspond to transformations catalyzed by metabolic genes listed in Fig. 3.
Alkane monooxygenase (12.4%) was the only hydrocarbon activation gene that constituted >2.0% of the total harbor water community potential. Genes associated with the anaerobic activation of hydrocarbons were not detected. Similarly, none of the aerobic or anaerobic signature hydrocarbon biodegradation metabolites (Fig. 4) were detected in the harbor seawater.
Aerobic BTEX metabolizing capability in the Ship #1 community increased significantly relative to the harbor community with ethylbenzene dioxygenase, xylene monooxygenase, and phenol hydroxylase rising by 366-, 92.6-, and 8.0-fold respectively. The ring-cleaving enzyme catechol 2,3-dioxygenase increased significantly in Ship #1 sample by 11.4-fold to 25.3% of the total community, while catechol 1,2-dioxygenase increased by 3-fold to 2.5% of the assemblage. Metabolite analyses identified the presence of phenylethanol, 3-methylcatechol, 4-methyl catechol, and methylnaphthalenediol, indicative of the oxygen-dependent biological activation of mono- and polyaromatic hydrocarbons. There was an increase of 12% in the proportion of the flora possessing alkB and genes related to the aerobic activation of naphthalenes were <1%. Genes for the alkyl- (2.1%), monoaromatic- (0.3%), and naphthylmethyl- (1.2%) succinate synthases all showed slight increases in Ship #1 samples relative to the harbor water metagenome, but these values were perhaps biased by the presence of other common glycyl-radical enzymes (e.g. pyruvate-formate lyase) that share substantive protein sequence homology. Despite these slight gene enrichments, no succinate-derivative metabolites were detected in Ship #1 samples.
Analysis of the community in Ship #2 revealed a further 11.6% enrichment of alkB possessing taxa relative to Ship #1. In contrast, the genes for ethylbenzene dioxygenase, xylene monooxygenase, and phenol hydroxylase declined sharply and each accounted for less than 2.0% of the total community metagenome. The abundance of the nmsA gene, coding for the anaerobic activation of methylnaphthalene, represented 7.7% of the total gene pool in Ship #2. This was an increase of the same gene abundance relative to Ship #1 of 440.1%. Overall, the total proportion of hydrocarbonoclastic taxa in Ship #2 decreased compared to Ship #1, though there was an enrichment in oxygen-independent activation genes (Fig. 5).

Pie chart area is proportional to the total abundance of aerobic and anaerobic hydrocarbon activation genes normalized to rpoB for each library. Blue represents the aerobic, and orange represents anaerobic hydrocarbon activation gene subpopulations.
The Ship #3 ballast tank contained the largest hydrocarbon-activating community of any sample investigated. Alkane 1-monooxygenase (6.1%) and catechol dioxygenase (catA/catE) genes (0.4%, 2.0%) decreased to their lowest relative proportion observed in any sample. In contrast, the community possessing assA (5.7%), bssA/hbsA/ibsA (4.7%) and nmsA (20.5%) were found in the highest respective abundances (Fig. 5). Mono- and poly-aromatic succinate metabolites were present corresponding to alkylated aromatic hydrocarbon substrates (Fig. 4). No catechols or aromatic dihydrodiol intermediates were measured in the Ship #3 water sample. Naphthoic and alkanoic acids, aliphatic alcohols, and phenylethanol were also putatively identified in Ship #3 and Ship #1 waters (Fig. 4).
Discussion
The study of seawater-compensated fuel ballast tanks holding fluids for varying lengths of time provides insight into the temporal dynamics of microbial community composition changes and associated metabolic activity shifts through metagenomic and metabolite profiling analyses coupled with geochemical assessments. Ribosomal RNA gene analysis revealed the surface waters of San Diego Harbor to be primarily colonized by Bacteria belonging to the Alphaproteobacteria, Flavobacteria, and Gammaproteobacteria and marine eukaryotes (Fig. 1a). The harbor receives terrestrial discharges from urban, industrial, and military instillations in the surrounding metropolitan area. The bacterioplankton community in San Diego harbor resembles the northern Gulf of Mexico profiles that were dominated by Alphaproteobacteria and Bacteriodetes, with ratios of Alpha- to Gammaproteobacteria of <1.719. The comparable ratio in the San Diego Bay of 1.4 no doubt reflected the complex natural and anthropogenic inputs enriching for marine heterotrophic taxa that differ substantively from open ocean surveys.
The microbial community composition was significantly altered after a one-week residence time in a Navy ballast tank (Ship #1) relative to the harbor assemblage. Despite the total cell density decrease of ~100-fold (Table 2), there was only a slight decrease in community diversity and an increase in the observed taxa representing the OTU (Table 3). Gammaproteobacteria were the dominant community members, predominantly driven by relative increases in the Orders Altermonodales, Oceanospiralles, and Thiotrichales (Fig. 1b). These bacterial Orders contain numerous well-documented hydrocarbonoclastic aerobic taxa belonging to the genera Marinobacter, Alcanivorax, and Cycloclasticus, respectively. Previous studies noted the enrichment of these groups in marine waters impacted by oil spills across the globe20,21,22,23. Marinobacter hydrocarbonoclasticus preferentially metabolizes short to moderate chain length (C8-C11) n-alkanes and selected monoaromatic hydrocarbons24. Pure cultures of M. vinifirmus and M. hydrocarbonoclasticus were able to catalyze the complete oxidation of BTEX hydrocarbons under aerobic conditions25. Marine Alcanivorax spp. are known to aerobically biodegrade a wide range of aliphatic petroleum constituents26,27,28, but are thought incapable of aromatic hydrocarbon decay29,30. However, recent work suggests that Alcanivorax may also contribute to the biodegradation of PAHs31. Cycloclasticus pugetii is an obligate aerobe with the capacity to metabolize mono- and polynuclear aromatic hydrocarbons (PAHs)32. Cycloclasticus strains isolated from marine coastal waters20,21 were also associated with the aerobic biodegradation of a variety of alkylated PAHs and dibenzothiophenes. Genetic studies revealed the presence of distinct naphthalene and biphenyl cluster dioxygenases in several Cycloclasticus isolates20. The increased proportions of Alteromonodales and Thiotrichales likely account for the enrichment of aromatic hydrocarbon degradation genes and the presence of signature aerobic metabolites (e.g. alkylcatechols and dihydromethylnaphthalene) observed in Ship #1 (Figs. 3, 4). Phylogenetic, genomic, and metabolic evidence suggests that the ballast tank community of Ship #1 was enriched in taxa responsible for the aerobic oxidation of the more water-soluble aromatic hydrocarbon components of diesel fuel.
Ship #2 was predominantly colonized by members of the Alteromonadales, but also had an increase in Deltaproteobacteria, specifically the Desulfuromonadales (Fig. 1b). In concert with the decreased relative abundance of the Gammaproteobacteria, there was also a decrease in the aerobic hydrocarbon-activation genes relative to Ship #1 (Fig. 3). Despite the bloom in Desulfuromonadales, there was surprisingly little enrichment in genes associated with sulfidogenesis (Fig. 2). Anaerobic hydrocarbon-activation genes exhibited only a slight increase in nmsA abundance relative to Ship #1 (Fig. 3). The Desulfuromonadaceae are strictly anaerobic heterotrophs, with some aerotolerance capacity, that typically respire sulfur compounds to sulfide, with the notable exception of several genera that are almost exclusively fermentative33. Members of this Family typically utilize fatty acids and alcohols as growth substrates, but hydrocarbon metabolism is not widely distributed34. However, several species of Desulfuromonas are known to reduce Fe(III) and Mn(IV) as electron acceptors33,35,36. The oxygen tolerance and ability to utilize metals as electron acceptors, suggests that these taxa bloomed in response to increasing scarcity of oxygen in Ship #2 ballast tank. In the absence of metabolite or genomic evidence suggesting direct fuel biodegradation, it is likely that aerobic catabolites produced by the cohabitating Alteromonadales helped provide the requisite electron donors and carbon sources.
After 32 weeks aboard ship, the ballast tank community of Ship #3 was predominantly anaerobic taxa affiliated with Deltaproteobacteria (42%), specifically the Desulfobacteraceae (27%) and Desulfarculaceae (11%) (Fig. 1a). There was also a concomitant increase in sulfuroxyanion reductive respiration genes to the highest levels observed in this study (Fig. 2). Conversely, the Ship #3 community possessed the smallest population of c-type cytochrome oxidase containing taxa (Fig. 2). Metagenome analysis estimated that 27.0% of the genomes encoded an adenylylsulfate reductase and 24.8% a thiosulfate reductase (Fig. 2). This community also possessed the highest levels of anaerobic hydrocarbon activation genes, including alkylsuccinate synthase (5.7%), monoaromatic succinate synthases (4.7%), and naphthylmethylsuccinate synthases (20.5%). Pure cultures and enrichments containing Deltaproteobacteria able to anaerobically metabolize a variety of normal, iso- and cyclic alkanes, as well as mono- and polycyclic aromatic hydrocarbons were recently reviewed37. The cultures were mostly derived from marine environments and could couple the oxidation of their parent hydrocarbon with the reduction of sulfate and production of sulfide. Indeed, the sulfate concentration in Ship #3 was noticeably lower than in the harbor or any of the other ballast tanks (Table 1).
The mechanisms of hydrocarbon activation by Deltaproteobacteria are fundamentally different from hydrocarbonoclastic aerobes in that molecular oxygen is not a co-reactant. There are at least four biochemical strategies governing the anaerobic attack on hydrocarbons37,38. The most commonly reported pathway is the activation of the parent substrates via fumarate addition reactions39. There was an increase in genes encoding this pathway in Ship #3 and many of the succinyl derivatives associated with such bioconversions were found as signature metabolites in this sample (Figs. 3, 4).
From an ecological perspective, fuel-compensated ballast tanks are microcosms of other hydrocarbon-laden marine ecosystems. After a short exposure (1 week) to petroleum in the absence of light, the coastal marine community based on phototrophic primary production shifted to one supported by aerobic heterotrophic respiration of diesel hydrocarbons and an almost complete loss of the eukaryotic life forms. Ship NF did not show an increase in the relative abundance of hydrocarbonclastic aerobes. The resulting hydrocarbonoclastic community of Ship #1 is reminiscent of the successional patterns observed during the response of open-ocean surface waters to the influx of petroleum from the Deep Water Horizon oil spill40. Here too, surface water assemblages underwent shifts within the first week and experienced strong enrichments in hydrocarbonoclastic aerobes22. The shift in community proportions encoding hydrocarbon activation genes, as well as the relative abundance of genes from oxygen-dependent and independent pathways also changed as a function of time (Fig. 5). After one week, the hydrocarbonoclastic community containing aerobic biodegradation genes was enriched 2.4 times that observed in the harbor. At 20 weeks and beyond, the abundance of the same genes decreased to only about 1.5 times that of the harbor assemblage. Within Ship #1, 95% of the activation genes were associated with aerobic mechanisms associated with well-documented aerobic hydrocarbonoclastic taxa, aerobic respiration genes, signature aerobic catabolic intermediates, and a decreased dissolved oxygen content (Figs. 1, 2, 4, and 5; Table 1). As ballast residence time increased, the fuel-degrading activation gene subset was increasingly affiliated with anaerobic biodegradation mechanisms. At 32 weeks, the hydrocarbon-degrading community predominantly employed oxygen-independent mechanisms (68%) with key metabolites associated with the fumarate addition-mediated degradation of BTEX and PAH fuel components (Figs. 4,5).
Biodegradation activity was associated with the corrosion of the ballast tanks. Dissolved metal concentrations within each Navy ballast tank indicate corrosion of the carbon steel and/or the copper-nickel sluice pipes as the values are considerably greater than ship NF or the surrounding harbor waters (Table 1). Dissolved manganese, copper, and nickel were positively correlated with the increase in sulfidogenic taxa between Ships #1 and #2 (Fig. 5). There was also a strong increase in the abundance of sulfide-bearing particulates of each metal species associated with the clearly sulfidogenic community of Ship #3 (Supplementary Fig. 2). The influence of biogenic sulfide in Ship #3 on the dissolved metal concentrations is demonstrated by the inverse correlation between the relative frequency of metals observed in metal-sulfide particles (Cu > Ni > Fe > Mn) and the solubility equilibrium constant (Ks) for the reaction:
where X = Cu2+, Ni2+, Fe2+, and Mn2+, with log Ks values ranging from −15.2 for CuS, 1.5 for α−NiS, 3.0 for FeS, and 10.0 and 7.0 for MnS rambergite and alabandite minerals, respectively41. Localized corrosion in low carbon steels in marine environments shows that pit initiation begins at the immediate surroundings of MnS inclusions where strained interfaces act as anodes and the surrounding unstrained iron as cathodes. Hydrolysis in the surrounding region acidifies the local region releasing Mn ions into solution42. The increasing dissolved manganese concentration with increasing residence time illustrates ongoing corrosion of carbon steel. Despite increased sulfidic conditions in Ship #3, the predicted solubilities of alabandite and rambergite phase MnS compounds were not exceeded (12.97 mg per liter—alabandite phase; 499.40 mg per liter—rambergite phase) explaining the lack of MnS particles in the samples. Corrosion of carbon steel in Ships #2 and #3 is further demonstrated by the presence of dissolved iron. In Ship #3, the measured dissolved iron concentration (603.0 µg per liter) is nearly equivalent to the calculated equilibrium concentration (441.20 µg per liter) presuming the simultaneous precipitation of FeCO3 and FeS minerals. Copper-nickel alloys are commonly utilized as marine infrastructure, but are highly susceptible to corrosion and leaching by sulfides, organic acids and other products of microbial metabolism43,44. Previous studies have shown strong correlations between hydrocarbon biodegradation-driven sulfidogenesis and both generalized and pitting corrosion for carbon steel and copper-nickel alloys in incubations of San Diego Bay seawater and F-76 diesel fuel15,45. Analysis of metal concentrations and particulates suggests that the CuNi-alloy sluice pipes transporting water and fuel between ballast tanks were arguably the primary sites of active biocorrosion in Ships #1 and #2 and that aggressive pitting corrosion of the ballast tank carbon steel was occurring in Ship #3. The concentrations of dissolved copper and nickel in Ship #1 and Ship #2 are greater than the equilibrium solubility of covellite (CuS) (3.88 × 10−7 µg per liter) and α-NiS (19.4 µg per liter), suggesting that in these ships, the copper-nickel slice pipes initially had an oxidized coating of copper hydroxides and/or carbonates (solubility 9–85 µg per liter) and nickel oxide and/or carbonate (solubility 30–1033 mg per liter). Microbial biofilm formation on copper and copper-nickel alloys can facilitate loss of the passivating layer of copper and nickel oxides and carbonates46,47,48,49 thereby providing a second mechanism for release of dissolved copper and nickel. Additionally, a thermodynamic model based on the premise of microbial sulfidogenesis in the presence of various metals/alloys predicts that sulfides will react with metal oxides to produce metallosulfide particles, thus exacerbating the depletion of the protective oxide layers50. Regardless of the mechanisms for corrosion, as sulfidogenic activity increases from Ship #2 to Ship #3, a decrease in the concentration of dissolved copper and nickel occurs due to the formation and precipitation of sulfide particulates.
This study has important implications for the operation and maintenance of fuel compensated ballast tanks of the U.S. Navy. Petroleum-derived diesel fuels are susceptible to biodegradation when in contact with seawater, regardless of oxygenation status or residence time and shipboard infrastructure is negatively impacted by microbiologically influenced corrosion minimally driven by the production of sulfides. Studies conducted on simulated ballast tank systems highlighted the inherent stratified nature of these habitats with zones of variable oxygenation51,52. The deleterious impacts of microbial activity were not homogenously distributed within the ballast systems and tank bottoms tended to be sites of aggressive general and localized corrosion10,15,53,54. Ship operators have estimated that a vessel on standard operations consumes ~1000 gallons of fuel per day, necessitating the introduction of fresh seawater into ballast tanks on a regular basis (personal communication). Cyclic oxygenation/deoxygenation has repeatedly been observed to promote the highest rates of generalized and pitting corrosion55. Furthermore, this condition promotes the persistence of biofilms harboring both aerobic and anaerobic taxa with the capacity for sulfur cycling and the formation of corrosive sulfur granules10. As treatment of incoming seawater for the removal of oxygen and sulfate is not feasible, a critical consideration might be to eliminate the introduction of sediments (and associated microbial load) into ballast tanks and to ensure all internal shipboard surfaces are coated. Sediments tend to settle onto horizontal surfaces and promote further stratification within ballast tanks and likely serve as a source of sulfide-producing microbes. Given the stratified nature of these systems, further research is warranted to determine how well the sampling of expansion tanks represents the various ballast tank zones. Ultimately, the successional trends in the ecology of ballast tank ecosystems were identified that are critical for understanding the impact of microorganisms on infrastructure and will serve to inform future studies to lessen the biological wastage of naval resources.
Methods
Sample collection
Ballast tank fluid samples were collected in sterile polypropylene bottles from valves in the expansion tanks (Fig. 6) on board the Navy vessels (Ship #1, 2, and 3). The ballast tank systems on the ships consist of carbon steel tanks with sacrificial anodes and protective coatings connected via copper-nickel alloy sluice pipes. Unlike the Navy ships, the ballast tank system on the Ship NF was not designed to mingle fuel and seawater. The sample from Ship NF was pumped from the ballast tank underlying the mess area through an access hatch, preserved with DNAzol® (Molecular Research Center Inc.; Cincinnati, OH) as indicated below, and shipped by crew members to the laboratory for analysis. For each Navy ship, one 250 mL aliquot of ballast water was preserved with 5 mL of DNAzol for DNA extraction. Another 1 L aliquot was acidified with 6 N HCl (pH < 2) for metabolite profiling. Finally, a separate bottle was filled to capacity (>250 mL) without preservative for chemical and metals analyses. In addition, several liters of seawater were collected from the San Diego Harbor where each ship was docked for comparative analyses. All samples were shipped overnight on ice to the laboratory and stored at 4 °C until use.

In reality, a ballast tank group is formed by interconnected chambers of varying shapes and sizes. The expansion tanks are where seawater enters and exits the ballast system and where samples were obtained. Fuel enters the first tank and pushes seawater through the rest of the tanks via the sluice pipes.
Chemical analyses
Dissolved oxygen was measured in the field using a Firesting-O2 oxygen meter (Pyro Science GmbH; Aachen, Germany) operated according to manufacturer instructions. Nitrate and sulfate were quantified by ion-chromatography as previously described12. For the determination of dissolved metals, each sample was filtered with a 0.45 µm polyethersulfone filter (Whatman Puradisc) and diluted to a metal concentration range between 0 to 40 mg per liter using 0.1% (v/v) nitric acid (Fluka, Ultrapure). Dissolved metal concentrations were measured using a Varian AA 240FS Atomic Absorption Spectrometer with a graphite tube furnace (Varian GTA120). Sample volume injected into the graphite furnace was 10 µL with argon as purging gas. Temperature gradient started at 85 °C and was held for 5 s, then ramped to 95 °C and held for 40 s, then ramped to 700 °C for 7 s before going to 2400 °C for 4.8 s. At this final temperature, absorption was measured using a background correction and a spectral bandwidth/slit width of 0.2 A six-point calibration curve ranging from 0 to 40 mg per liter was used for each metal examined. All samples were measured in triplicate, and each time after measuring five samples, two known calibration standards and the blank were reanalyzed with recalibration if necessary.
Suspended particles in ballast tank samples were collected by filtration on a 0.45-micron filter while in an anaerobic chamber. Individual particles were analyzed with a scanning field emission electron microscope (SUPRA 55VP, Carl Zeiss Microimaging, GmpH, Gottingen) equipped with energy dispersive X-ray analysis (EDX) (Princeton Gamma-Tech, Inc. - PGT). The EDX spectra were collected using a 20-keV primary beam and analyzed using the Spirit V. 1.07.05 software of PGT.
Microbial enumeration
Microbial cells were enumerated from unpreserved Navy ballast fluids by direct epifluorescence microscopy after fixing the samples with 1/10 volume of 37% formaldehyde and staining with 4,6-diamidino-2-phenylindole. Fixed cells were then filtered onto 0.22 µm, 25 mm diameter black polycarbonate membrane filters (Sterlitech, Kent WA). At least 30 fields were counted per sample and results were averaged.
DNA extraction and qPCR analyses
Samples preserved for DNA extraction were filtered (250 mL; 0.45 µm pore size PES filter) upon receipt, stored at −80 °C until use, and extracted from the filters as previously described56. The same DNA extracts from the harbor sample and from Ships #1, 2, and 3 were used for metagenome, qPCR, and 16S rRNA amplicon libraries. Quantitative PCR was used to enumerate population densities of bacteria, archaea and picoeukaryotes within each sample. Bacterial and archaeal 16S rRNA genes were amplified with Eubac8F/338R and A8F/344R primer pairs57. Marine picoeucaryote 18S rRNA genes were amplified using the 345F/499R primer pair as previously described58. Dissimilatory sulfate reduction genes, ade nosine-5’-phosphosulfate reductase A gene (aprA) and dissimilatory sulfite reductase A (dsrA), were assessed with the RH1apsF/RH2apsR and RH1-dsr-F/RH3-dsr-R primer pairs, respectively59.
Metagenome analysis
Shotgun metagenome libraries were sequenced on an Illumina MiSeq platform (Oklahoma Medical Research Foundation, Oklahoma City, OK). Sequencing adapters were trimmed using Cutadapt60 to a minimum average PHRED score of 30 and mate paired with Trimmomatic61. Unpaired reads were not retained for analyses. Community composition was assessed from unassembled mate-paired reads with significant homology to known small subunit (SSU) rRNA genes. Reads were searched against the Silva SSU Reference database release 10862 pre-clustered at 97% sequence similarity using USEARCH63. Hits with a minimum of 70% homology to reference SSU gene sequences were extracted for further analysis. Putative SSU reads were analyzed using QIIME 1.9.164 through closed-reference OTU clustering (97% similarity) and taxonomic assignment against the Silva 111 SSU reference sequence and taxonomy databases. Alpha diversity metrics were computed for each sample based upon metagenome reads containing SSU fragments rarefied to 1800 sequences for each library.
Functional gene annotation was performed by translated homology searching of unassembled mate-paired reads against the KEGG database65,66 using DIAMOND51 in the sensitive mode retaining only the top hit (E < 10–5) for each query. Putative anaerobic hydrocarbon activation genes were identified by a translated homology search in USEARCH against the AnHyDeg database52. Acceptance criteria were 60% identity over alignments with at least 35 amino acids. Functional gene abundances were calculated by dividing the number of hits to specific reference gene by the number of hits to the single-copy marker gene, rpoB, to normalize for differential library population size67. A modified approach was used to estimate the total hydrocarbon-activating proportion of each community from that previously used68 by a summation for selected aerobic and anaerobic specific activation genes of the following: (hits to functional genei /length of functional genei)/hits to rpoB. Aerobic activation genes included: alkB, etbAa, tmoA, todC1, dmpB, nahAc, and 1-methylnaphthalene hydroxylase-like genes. Anaerobic activation genes were ebdA, assA, bssA, nmsA.
16S rRNA amplicon libraries for high-throughput sequencing
An aliquot of the DNA extracted from filters was used to generate 16S amplicon libraries with distinctive barcodes as previously described69. Briefly, the M13-519F/785R primer set70 was used to amplify the V3-V5 region of the 16S rRNA gene, and a 12 base pair sequence conjugated to M1371 was used to barcode each sample (Supplementary Table 4). Amplicon libraries were sequenced as described above and analyzed as previously described69. Paired-end reads were trimmed and stitched together, and default parameters in QIIME were used to demultiplex, cluster, and remove chimeras.
Metabolite analyses
Mass spectral analysis of diagnostic metabolites associated with oxygen-dependent and independent hydrocarbon activation of diesel fuel components was performed on acidified ballast tank samples. Methods used for the targeted (GC-MS) and global survey of organic intermediates were as reported previously72.
Thermodynamic equilibrium solubility calculations
The equilibrium solubility of the carbonate and sulfide mineral compounds of manganese, copper and nickel in seawater were calculated using MINEQL 5.0, with the following chemical parameters: total carbonates (2 mM), ionic strength (0.5 M), and a temperature of 25 oC. Since the San Diego Harbor seawater and ballast water ranged in pH from 7.65 to 8.2873 and between 7.71 and 8.3 (data not shown), respectively, a constant pH of 8.2 was used in the MINEQL 5.0 calculations. The small pH differences from the constant value resulted in a <1% change in the metal sulfide equilibrium solubility values. A closed system was modeled for each metal carbonate and sulfide compound contained in the MINEQL 5.0 thermodynamic database, and CO2 (gas) and H2S (gas) were removed as fixed entities.
Data availability
Metagenome and 16S amplicon sequence files are available at the NCBI Sequence Read Archive under accession numbers SRR14067720 for Ship #1, SRR14067719 for Ship #2, SRR14067718 for Ship #3, SRR14067717 for Harbor SD, and SRR14121797 for the 16S amplicon library.
References
Gillings, M. R. & Paulsen, I. T. Microbiology of the anthropocene. Anthropocene 5, 1–8 (2014).
Crutzen, P. J. Geology of mankind. Nature 415, 23 (2002).
Economist P. World Energy 2016-2050: Annual Report. Peak Oil Barrel [Internet]. Available at: http://peakoilbarrel.com/world-energy-2016-2050-annual-report/ (2016).
Smil, V. Still the Iron Age: Iron and Steel in the Modern World (Butterworth-Heinemann, 2016).
Hazlett, R. N. Examination of Corrosion Deposits from USS IWO JIMA Aviation Gasoline Storage Tanks Memorandum Report 1779 (U.S. Naval Research Laboratory, Washington, DC, 1967).
Hazlett, R. N. Control of Sulfate-reducing Bacteria in Navy Fuel Systems Memorandum Report 1737 (U.S. Naval Research Laboratory, Washington, DC, 1966).
Klemme. D. E. & Leonard J. M. Inhibitors for Marine Sulfate-reducing Bacteria in Shipboard Fuel Storage Tanks Memorandum Report 2324 (U.S. Naval Research Laboratory, Washington, DC, 1971).
Klemme, D. E. & Neihof R. A. Control of Marine Sulfate-reducing Bacteria in Water-displaced Shipboard Fuel Storage Tanks Memorandum Report 2069 (U.S. Naval Research Laboratory, Washington, DC, 1969).
Cleland, J. H. Corrosion risks in ships’ ballast tanks and the IMO pathogen guidelines. Eng. Fail. Anal. 2, 79–84 (1995).
Huang, R. T., McFarland, B. L. & Hodgman R. Z. Microbial influenced corrosion in cargo oil tanks of crude oil tankers. In: Paper NACE-97535 Presented at the Corrosion97, New Orleans, Louisiana, March (NACE International, 1997).
Liang, R. & Suflita, J. M. in Hydrocarbon and Lipid Microbiology Protocols. Springer Protocols Handbooks. (eds McGenity, T., Timmis, K. & Nogales, B.) 211–226 (Springer, Berlin, Heidelberg, 2015). https://doi.org/10.1007/8623_2015_76.
Lyles, C. N. et al. Impact of organosulfur content on diesel fuel stability and implications for carbon steel corrosion. Environ. Sci. Technol. 7, 6052–6062 (2013).
Aktas, D. F. et al. Anaerobic metabolism of biodiesel and its impact on metal corrosion. Energy Fuels 24, 2924–2928 (2010).
Lee, J. S. et al. Sulphide production and corrosion in seawaters during exposure to FAME diesel. Biofouling 28, 465–478 (2012).
Liang, R. et al. Anaerobic biodegradation of alternative fuels and associated biocorrosion of carbon steel in marine environments. Environ. Sci. Technol. 50, 4844–4853 (2016).
Aktas, D. F. et al. Anaerobic hydrocarbon biodegradation and biocorrosion of carbon steel in marine environments: The impact of different ultra low sulfur diesels and bioaugmentation. Int. Biodeterior. Biodegrad. 118, 45–56 (2017).
Liang, R., Harvey, B. G., Quintana, R. L. & Suflita, J. M. Assessing the biological stability of a terpene-based advanced biofuel and its relationship to the corrosion of carbon steel. Energy Fuels. 29, 5164–5170 (2015).
King, G. M., Smith, C. B., Tolar, B. & Hollibaugh, J. T. Analysis of composition and structure of coastal to mesopelagic bacterioplankton communities in the northern Gulf of Mexico. Front. Microbiol. 3, 1–14 (2013). article 438.
Geiselbrecht, A. D., Hedlund, B. P., Tichi, M. A. & Staley, J. T. Isolation of marine polycyclic aromatic hydrocarbon (PAH)-degrading Cycloclasticus strains from the Gulf of Mexico and comparison of their PAH degradation ability with that of puget sound Cycloclasticus strains. Appl. Environ. Microbiol. 64, 4703–4710 (1998).
Kasai, Y., Kishira, H. & Harayama, S. Bacteria belonging to the genus Cycloclasticus play a primary role in the degradation of aromatic hydrocarbons released in a marine environment. Appl. Environ. Microbiol. 68, 5625–5633 (2002).
Dubinsky, E. A. et al. Succession of hydrocarbon-degrading bacteria in the aftermath of the Deepwater Horizon oil spill in the Gulf of Mexico. Environ. Sci. Technol. 47, 10860–10867 (2013).
King, G. M., Kostka, J. E., Hazen, C. & Sobecky, P. A. Microbial responses to the Deepwater Horizon oil spill: From coastal wetlands to the deep sea. Ann. Rev. Mar. Sci. 7, 377–401 (2015).
Striebich, R. C. et al. Characterization of the F-76 diesel and Jet-A aviation fuel hydrocarbon degradation profiles of Pseudomonas aeruginosa and Marinobacter hydrocarbonoclasticus. Int. Biodeterior. Biodegrad. 93, 33–43 (2014).
Berlendis, S. et al. First evidence of aerobic biodegradation of BTEX compounds by pure cultures of Marinobacter. Appl. Biochem. Biotechnol. 160, 1992–1999 (2010).
Yakimov, M. M. et al. Alcanivorax borkumensis gen. nov., sp. nov., a new, hydrocarbon-degrading and surfactant-producing marine bacterium. Int. J. Syst. Evol. Microbiol. 48, 339–348 (1998).
Harayama, S., Kasai, Y. & Hara, A. Microbial communities in oil-contaminated seawater. Curr. Opin. Biotechnol. 15, 205–214 (2004).
Liu, C. & Shao, Z. Alcanivorax dieselolei sp. nov., a novel alkane-degrading bacterium isolated from sea water and deep-sea sediment. Int. J. Syst. Evol. Microbiol. 55, 1181–1186 (2005).
Schneiker, S. et al. Genome sequence of the ubiquitous hydrocarbon-degrading marine bacterium Alcanivorax borkumensis. Nat. Biotechnol. 24, 997–1004 (2006).
Lai, Q., Li, W. & Shao, Z. Complete genome sequence of Alcanivorax dieselolei type strain B5. J. Bacteriol. 194, 6674 (2012).
Dombrowski, N. et al. Reconstructing metabolic pathways of hydrocarbon-degrading bacteria from the Deepwater Horizon oil spill. Nat. Microbiol. 1, 16057 (2016).
Dykesterhouse, S. E., Gray, J. P., Herwig, R. P., Cano Lara, J. & Staley, J. T. Cycloclasticus pugetii gen. nov., sp. nov., an aromatic hydrocarbon-degrading bacterium from marine sediments. Int. J. Syst. Evol. Microbiol. 45, 116–123 (1995).
Finster, K. & Bak, F. & Pfennig, N. Desulfuromonas acetexigens sp. nov., a dissimilatory sulfur-reducing eubacterium from anoxic freshwater sediments. Arch. Microbiol. 161, 328–332 (1994).
Greene, A. C. in The Prokaryotes 143–155 (Springer, 2014).
Roden, E. E. & Lovley, D. R. Dissimilatory Fe(III) reduction by the marine microorganism Desulfuromonas acetoxidans. Appl. Environ. Microbiol. 59, 734–742 (1993).
Coates, J. D., Lonergan, D. J., Philips, E. J., Jenter, H. & Lovley, D. R. Desulfuromonas palmitatis sp. nov., a marine dissimilatory Fe(III) reducer that can oxidize long-chain fatty acids. Arch. Microbiol. 164, 406–413 (1995).
Davidova I. A., Marks C. R., & Suflita J. M. in Handbook of Hydrocarbon and Lipid Microbiology 207–243 (Springer, 2019).
Callaghan, A. V. Enzymes involved in the anaerobic oxidation of n-alkanes: from methane to long-chain paraffins. Front. Microbiol. 4, 89 (2013).
Callaghan, A. V. Metabolomic investigations of anaerobic hydrocarbon-impacted environments. Curr. Opin. Biotechnol. 24, 506–155 (2013).
Yang, T. et al. Pulsed blooms and persistent oil-degrading bacterial populations in the water column during and after the Deepwater Horizon blowout. Deep Sea Res. II: Top. Stud. Oceanogr. 129, 282 (2016).
NIST Standard Reference Data, NIST Critically Selected Stability Constants of Metal Complexes: Version 8.0. https://www.nist.gov/srd/nist46 (2004).
Rieders, N., Nandasiri, M., Mogk, D. & Avci, R. New insights into sulfide inclusions in 1018 carbon steels. Metals 11, 428 (2021).
Pope, D. H. Microbial Corrosion in Fossil-fired Power Plants: A Study of Microbiologically Influenced Corrosion and A Practical Guide For Its Treatment And Prevention Final Report. United States https://www.osti.gov/biblio/5623575 (1987).
Wagner, P. & Little, B. Impact of alloying on microbiologically influenced corrosion. A review. Mater. Perfor. 32, 65–68 (1993).
Liang, R. et al. Anaerobic biodegradation of biofuels and their impact on the corrosion of a Cu-Ni alloy in marine environments. Chemosphere 195, 427–436 (2018).
Guillaume, I., Constant, H., Lemaire, J.-C., Valensi, G. & Brisou, J. Role de certaines bacteries dans la corrosion du cuivre, du nickel et du zinc en milieu doux et marin. Electrochim. Acta 15, 1445–1454 (1970).
Bautistt, B. E. T. et al. Influence of extracellular polymeric substances (EPS) from Pseudomonas NCIMB 2021 on the corrosion behavior of 70Cu-30Ni alloy in seawater. J. Electroanal. Chem. 737, 184–197 (2015).
Zhang, Y. et al. Metagenomic resolution of functional diversity in copper surface-associated marine biofilms. Front. Microbiol. 10, 2863 (2019).
Aissaoui, N. et al. Unravelling surface changes on Cu-Ni alloy upon immersion in aqueous media simulating catalytic activity of aerobic biofilms. Appl. Surf. Sci. 503, 144081 (2020).
McNeil, M., & Odom, A. in Microbiologically Influenced Corrosion Testing (eds. Kearns, J. & Little B) 173–179 (ASTM International, 1994).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Callaghan, A. V. & Wawrik, B. AnHyDeg: A curated database of anaerobic hydrocarbon degradation genes Available from: https://github.com/AnaerobesRock/AnHyDeg (2016).
Lee, J. S., Ray, R. I., Lemieux, E. J., Falster, A. U. & Little, B. J. An evaluation of carbon steel corrosion under stagnant seawater conditions. Biofouling 20, 237–247 (2004).
Heyer, A. et al. Ship ballast tanks a review from microbial corrosion and electrochemical point of view. Ocean Eng. 70, 188–200 (2013).
Lee, J. S., Ray, R. I., Little, B. J. & Lemieux, E. J. Evaluation of deoxygenation as a corrosion control measure for ballast tanks. Corrosion 61, 1173–1188 (2005).
Oldham, A. L., Drilling, H. S., Stamps, B. W., Stevenson, B. S. & Duncan, K. E. Automated DNA extraction platforms offer solutions to challenges of assessing microbial biofouling in oil production facilities. AMB Express 2, 60 (2012).
Stevenson, B. S. et al. Microbial communities in bulk fluids and biofilms of an oil facility have similar composition but different structure. Environ. Microbiol. 13, 1078–1090 (2011).
Zhu, F., Massana, R., Not, F., Marie, D. & Vaulot, D. Mapping of picoeucaryotes in marine ecosystems with quantitative PCR of the 18S rRNA gene. FEMS Microbiol. Ecol. 52, 79–92 (2005).
Ben-Dov, E., Brenner, A. & Kushmaro, A. Quantification of sulfate-reducing bacteria in industrial wastewater, by real-time polymerase chain reaction (PCR) using dsrA and apsA genes. Microb. Ecol. 54, 439–451 (2007).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Pruesse, E., Quast, C., Knittel, K., Fuchs, B. & Ludwig, W. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196 (2007).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M. & Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114 (2012).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Howard, E. C., Sun, S., Biers, E. J. & Moran, M. A. Abundant and diverse bacteria involved in DMSP degradation in marine surface waters. Environ. Microbiol. 10, 2397–2410 (2008).
Biers, E. J., Sun, S. & Howard, E. C. Prokaryotic genomes and diversity in surface ocean waters: Interrogating the global ocean sampling metagenome. Appl. Environ. Microbiol. 75, 2221–2229 (2009).
Oldham, A. L., Sandifer, V. & Duncan, K. E. Effects of sample preservation on marine microbial diversity analysis. J. Microbiol. Methods 158, 6–13 (2019).
Klindworth, A. et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41, e1 (2013).
Hamady, M., Walker, J., Harris, J., Gold, N. J. & Knight, R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nat. Methods 5, 235–237 (2008).
Bonifay, V., Aydin, E., Aktas, D. F., Sunner, J., & Suflita, J. M. in Hydrocarbon and Lipid Microbiology Protocols (eds McGenity, T. J., Timmis, K. N. & Nogales, B.) 111–161 (Springer, 2017).
Stransky, C. et al., 2018 San Diego Regional Harbor Monitoring Program Final Report. Submitted to, Port of San Diego, City of San Diego, City of Oceanside, and County of Orange. Wood Project No.: 1715100804 https://pantheonstorage.blob.core.windows.net/environment/Regional-Harbor-Monitoring-Program-2018-Final-Report.pdf (2020).
Acknowledgements
This study was financially supported by a grant (Award N000141010946) from the Office of Naval Research. We appreciated the help of Commander Bill Partington and ONR Science Advisor Dr. Heidi M. Boose for access to US Navy ships and assistance with the sampling effort. We also thank the officers and crews serving aboard each of the vessels sampled in this study. We thank Dr. Deniz Aktas for the metabolite profiling effort, Dr. Renxing Liang for pH and sulfate measurements as well as filtering samples prior to EDX analysis and Mr. Vince Sandifer for facilitating the revision of Fig. 1. We also acknowledge grant NSF/NNCI (ECCS1542210) for supporting the ICAL facility at Montana State University.
Author information
Authors and Affiliations
Contributions
C.R.M. conducted the field sampling, bioinformatic analyses, and drafted the manuscript. K.E.D. conducted the DNA extractions and amplification methods. A.L.O. analyzed the 16S amplicon libraries and submitted the sequences to Genbank. M.A.N. carried out the dissolved and particulate metals analyses. B.H.H. conducted the cell counts. R.A. responsible for the characterization of solids recovered from the ballast tanks. J.M.S. was the principal investigator supervising this project and participated in field sampling, manuscript preparation and revision. All co-authors reviewed and edited the manuscript prior to submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Marks, C.R., Duncan, K.E., Nanny, M.A. et al. An integrated metagenomic and metabolite profiling study of hydrocarbon biodegradation and corrosion in navy ships. npj Mater Degrad 5, 60 (2021). https://doi.org/10.1038/s41529-021-00207-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-021-00207-z
This article is cited by
-
Biodegradation of materials: building bridges between scientific disciplines
npj Materials Degradation (2023)
-
Steel rust layers immersed in the South China Sea with a highly corrosive Desulfovibrio strain
npj Materials Degradation (2022)
