
Abstract
Cu-doped martensitic steels (Fe–(13, 16)Cr–3W–2Cu–1C) (mass%) with multiple carbide precipitates were prepared at different quenching temperatures, and their corrosion behaviours were examined by measuring the weight loss during immersion in a 0.5 M H2SO4 solution. Lower weight losses and corrosion rates were obtained for the alloy samples prepared at higher quenching temperatures. Surface Cu enrichment was observed for all specimens with a large fraction of dissolved Cr species. Moreover, quenching from higher temperatures not only reduced the amount of M23C6 carbide but also decreased the local electrochemical potential difference between the carbide phase and the martensitic matrix via enhanced surface Cu accumulation, thus increasing corrosion resistance by suppressing microgalvanic corrosion between the constituent phases. The corrosion behaviour of the studied steels was remarkably different from those of the Cu-doped stainless and low-alloy steels with passive oxide surface films, suggesting the strong effect of multiple carbide precipitates on their corrosion behaviour.
Similar content being viewed by others

Introduction
High-speed steels (HSS) with high carbon content (0.65–1.60 mass% according to the ASTM A600 standard1) and alloying elements such as Cr, W, Mo, V, and Co2 are widely used for manufacturing cutting tools (e.g., taps and dies) because of their high hardness and wear resistance. These steels comprise martensitic matrices containing fine carbide particles with the formulae MC, M2C, M6C, and M7C3, depending on the chemical composition and steel processing method3,4,5,6,7. HSS are often manufactured by powder metallurgy techniques to obtain a more uniform distribution of fine carbide precipitates8,9. HSS also possess outstanding mechanical and tribological properties (at room and elevated temperatures), which have attracted considerable attention from researchers working in the automotive, aerospace, energy, and plastic manufacturing industries. Furthermore, HSS are often used as substitutes for more expensive cemented carbides such as WC–Co.
However, the corrosion resistance of HSS is often insufficient for practical applications because of their low Cr content (<5 mass%1). Accordingly, the corrosion behaviour of these materials has not been elucidated10,11. Various surface modification techniques, such as laser treatment12,13, coating14,15,16, and nitriding17, have been applied to increase the corrosion resistance of HSS. In contrast, martensitic stainless steels possess a higher Cr concentration (11.5–18 mass%18) and exhibit favourable combinations of high wear and corrosion resistances, although the hardness values of these steel grades are generally lower than those of HSS. However, the presence of carbide precipitates in these steels accompanied by the local decrease in the Cr content of their neighbouring regions decreases the corrosion resistance of these materials19,20,21. This phenomenon is known as sensitisation22. Lu et al.20 reported that the precipitation of large amounts of nanosized Cr-rich M23C6 carbides in tempered 13.6Cr–0.3V–0.38C (mass%) martensitic stainless steels produces numerous carbide/matrix interfaces for pitting initiation, thereby hindering the formation of passive films on their surfaces. Moreover, Anantha et al.21 conducted scanning Kelvin probe force microscopy (SKPFM) observations of the same steel grade. The obtained Volta potential map revealed that the formed carbides exhibited higher electrochemical nobilities than those of the martensitic matrix. The resulting electrochemical potential difference caused selective dissolution along the carbide boundaries.
Recently, we designed a Cu-doped carbide-reinforced martensitic steel, Fe–16Cr–3W–2Cu–1C (mass%), and demonstrated its high hardness and corrosion resistance in a sulfuric acid solution23. The formation of a relatively large fraction of M23C6 carbide particles and the martensitic matrix in the developed steel made its hardness (>700 HV) comparable to that of conventional HSS. More importantly, the addition of trace Cu significantly increased the corrosion resistance of this steel, whereas its Cu-free counterpart (Fe–16Cr–3W–1C) exhibited accelerated anodic dissolution of its martensitic matrix despite the high Cr content. Consequently, the corrosion rate of the developed steel was approximately two orders of magnitude lower than that of the Cu-free steel. These characteristics are favourable for manufacturing metal components used in harsh tribocorrosive environments, such as injection moulding of super engineering plastics (a corrosive medium with hard glass reinforcements24) and other applications in the chemical and energy fields. A previous study23 also suggested that the corrosion of these steels occurred via the formation of microcorrosion cells consisting of noble carbide and less noble martensite coupling. However, the corrosion mechanism of Cu-doped steels with multiple carbide precipitates has not been fully elucidated, although the effect of Cu addition on the corrosion behaviour of austenitic, ferritic, and martensitic stainless steels was examined in detail25,26,27,28,29,30,31.
In this study, Fe–(13, 16)Cr–3W–2Cu–1C (mass%) steels with different bulk Cr concentrations were prepared and the surface evolution during immersion in a H2SO4 solution was examined by scanning electron microscopy (SEM), electron probe microanalysis (EPMA), scanning transmission electron microscopy (STEM), X-ray photoelectron spectroscopy (XPS), and SKPFM to achieve a better understanding of their corrosion properties. We focused on the effect of quenching temperature (800–1100 °C) at which the steel samples were austenised prior to quenching. The corrosion mechanism of the studied steels was discussed by considering their surface characteristics and bulk microstructures. Hereafter, the manufactured steels are denoted as 13Cr and 16Cr.
Results
As-quenched microstructures
Figure 1 depicts the SEM-backscattered electron (BSE) images of the as-quenched microstructures of the studied steels. It shows that precipitates (displayed as bright particles) were formed in all samples. Although two types of particles with different sizes were present in the as-quenched samples, the fraction of the finer particles significantly decreased after the heat treatment at 1100 °C. The black regions in Fig. 1 represent the inclusions or holes that were formed by the detachment of particles during sample preparation for microstructural observations. No significant differences in the precipitation behaviours of different steels were observed at each quenching temperature.

SEM-BSE images for the a–d 13Cr and g–h 16Cr steels heat treated at a, g 800 °C, b, f 900 °C, c, g 1000 °C, and d, h 1100 °C.
Figure 2a, b displays the SEM-BSE images and the corresponding EPMA elemental maps of the 16Cr steel samples heat treated at 800 and 1100 °C, respectively. The observed precipitates are enriched with Cr, W, and C elements, indicating the presence of carbide. The chemical compositions of the carbide particles do not vary significantly with their size and quenching temperature. Conversely, Fe and Cu are distributed throughout the steel matrix. Precipitation of fine Cu particles occurred in the sample heat treated at 800 °C.

SEM-BSE images and the corresponding EPMA elemental maps of the 16Cr steels heat treated at a 800 and b 1100 °C.
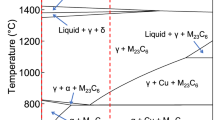
Figure 3a, b depicts the neutron diffractograms of the 13Cr and 16Cr steels, respectively. Overall, both steels demonstrated similar dependences on the quenching temperature, and all the analysed samples exhibited the diffraction peaks of a body-centred cubic structure. The broadening of these peaks indicates the formation of the α′-martensite phase with a high dislocation density. Electron backscatter diffraction (EBSD) measurements (Supplementary Fig. 1) captured the lath martensitic matrix in all the specimens, whereas the ferrite grains in the as-received bars remained intact during heat treatment at 800 °C. The small peaks were assigned to the M23C6 carbide phase with the Fm\(\bar 3\)m space group, and no other carbide phases were detected. Notably, higher carbide peak intensities were observed for the samples heat treated at 800 °C, whereas the retained γ-phase is detected exclusively in the samples heat treated at 1100 °C. Overall, the observed constituent phases were consistent with the results of thermodynamic calculations (Supplementary Fig. 2).

Neutron diffractograms of the a 13Cr and b 16Cr steels heat treated at different temperatures. Intensity is displayed on the logarithmic scale.
Corrosion behaviour
Figure 4a, b depicts the weight losses of the 13Cr and 16Cr steels as functions of the immersion time. Overall, the corrosion weight losses of the studied steels increased with the immersion time, and lower weight losses are observed for the samples quenched from higher temperatures. Notably, the 16Cr steel demonstrated a higher corrosion resistance than that of its 13Cr counterpart at each quenching temperature (this trend was more pronounced at higher temperatures). The corrosion rates of the 13Cr and 16Cr steel samples are plotted as functions of the immersion time in Fig. 4c and d, respectively. These plots show that the samples heat treated at lower temperatures exhibit higher corrosion rates. The 16Cr samples quenched from 1000 and 1100 °C exhibited extremely low corrosion rates at the initial immersion stage, which then became mostly invariant with further increases in the immersion time.

a, b Weight loss and c, d corrosion rate plotted as functions of the immersion time for the a, c 13Cr and b, d 16Cr steels in the 0.5 M H2SO4 solution. e SEM images of the 13Cr and 16Cr steels heat treated at different temperatures after the 7-h immersion.
Figure 4d, e shows the surface characterisation data obtained for the 13Cr and 16Cr steels after 7 h of immersion, respectively. All samples underwent localised corrosion to form porous surfaces. Notably, the corrosion products spalled off from the corroded surfaces of the 13Cr steel samples. However, this phenomenon became insignificant at higher quenching temperatures. In contrast, the 16Cr steel, which exhibited less pronounced spallation, produced similarly corroded surfaces regardless of the quenching temperature. Although smoother (less corroded) surfaces were observed for the 16Cr steel samples, they possessed granular morphologies on the micrometre scale.
Surface evolution
The sample surfaces were also examined by SEM at the initial immersion stage. Figure 5a, b shows the SEM images of the surfaces of the 16Cr steels quenched from 800 and 1100 °C, respectively, after 1 h of immersion in the 0.5 M H2SO4 solution. The sample heat treated at 800 °C exhibited a visibly corroded surface, and its corrosion products were partially present in the matrix, whereas no apparent changes were detected for the carbide phase with flat ground surfaces. In contrast, the corrosion of the martensitic matrix in the 1100 °C sample was remarkably inhibited. However, the carbide/martensite interfaces were preferentially attacked to form gaps, which was not obvious in the 800 °C sample. These phenomena can be clearly observed from the high-magnification images of the 800 and 1100 °C samples depicted in Fig. 5c, d, respectively. Notably, for the 1100 °C sample, fine particles were observed on not only the martensitic matrix but also the carbide surface (Fig. 5d). In contrast, the formation of such particles in the sample quenched from 800 °C exclusively occurred on the matrix surface, as shown in Fig. 5c. According to the EPMA elemental maps in Fig. 5e, the corrosion products that are observed on the matrix surface of the 800 °C sample were enriched with S and O. In contrast, more uniform elemental distributions were observed in each constituent phase of the sample quenched from 1100 °C (Fig. 5f). Notably, the formation of negligible amounts of such corrosion products is confirmed from the S and O maps because the 1100 °C sample exhibits higher corrosion performance than the 800 °C sample. The Cu particles formed on the steel surface are not captured clearly in the EPMA maps. Nonetheless, a higher intensity of Cu visible uniformly at the martensite surface of the 1100 °C sample implies enhanced surface Cu accumulation although the substrate chemical composition may affect the EPMA results.

a–d SEM images and e, f EPMA elemental maps of the surface regions of the 16Cr steel samples quenched from a, c, e 800 °C and b, d, f 1100 °C after immersion in the 0.5 M H2SO4 solution for 1 h. High-magnification images of panels a and b are shown in panels c and d, respectively. In the EPMA maps, the intensity range in e and f was the same for each element.
After the 3-h immersion, the sample quenched from 800 °C exhibited a remarkable dissolution of the steel matrix, resulting in the formation of a porous granular surface with visible asperities (Fig. 6a). In contrast, the surface of the 1100 °C sample was smoother and less corroded than that of the sample quenched from 800 °C (Fig. 6b). Figure 6c shows that the sample quenched from 1100 °C had a characteristic surface consisting of fine feather-like corrosion products. Notably, submicron-sized particles, together with the abovementioned feather-like substances, were also observed on the polished surface of a carbide particle (Fig. 6d).

a, g Fe 2p, b, h Cr 2p, c, i W 4f, d, j Cu 2p, e, k C 1s, and f, l O 1s peaks obtained for the outermost surfaces of the 16Cr steel samples heat treated at a–f 800 °C and g–l 1100 °C before and after the 7-h immersion in the 0.5 M H2SO4 solution.
Figure 6e, f shows the cross-sections of both samples obtained by the focused ion beam (FIB) technique after the 3-h immersion, which contain the porous surfaces of the martensitic matrices. The porous surface of the 800 °C sample is much thicker than that of its 1100 °C counterpart (the latter also has gaps, which occurred toward the inner part, around carbide particles). The fine particles on the carbide surface have been successfully incorporated into the FIB sample, as shown in Fig. 6f. Figure 6g depicts the corresponding cross-sectional annular bright-field and high-angle annular dark-field (HAADF) STEM images obtained for the outermost surface region of the 1100 °C sample. The brighter contrast of the particles in the HAADF-STEM image originated from the elements with higher atomic numbers. The obtained STEM-energy-dispersive X-ray spectroscopy (EDS) elemental maps revealed that these particles mainly consisted of Cu atoms, whereas the other alloying elements were uniformly distributed across the carbide phase beneath the Cu particles.
XPS
Supplementary Figure 3a, b depicts the XPS survey scan profiles recorded at various immersion times for the outermost surfaces of the 16Cr steel samples quenched from 800 and 1100 °C, respectively. The peaks corresponding to the metallic elements in the studied steel (Fe 2p, Cr 2p, W 4f, and Cu 2p) were detected. The evolution of the Cu 2p peak after the immersion was more evident for the 1100 °C sample, whereas O 1s peaks were detected for all samples regardless of the immersion time. Conversely, the intensities of the C 1s and N 1s peaks decreased with increasing immersion time, although the C 1s peak was retained after the 7-h immersion.
Figure 7 displays the high-resolution scan profiles of the Fe 2p, Cr 2p, W 4f, Cu 2p, C 1 s, and O 1s peaks and the peaks obtained for the outermost surfaces of the 16Cr steel samples before and after 7 h of immersion in the 0.5 M H2SO4 solution. The Cr 2p1/2 peak was used to analyse the Cr components because the Cr 2p3/2 peak overlapped with the Cu LMM Auger peak. Overall, the differences caused by the bulk Cr concentration (i.e., 13 and 16 mass%) were negligible. The envelope peaks obtained for metallic elements were deconvoluted into the peaks of the metallic and oxidation states. Thus, Fe atoms existed predominantly in the form of Fe2+ and Fe3+ ions before and after the immersion, respectively. Meanwhile, the Cr3+ component, which was observed on the sample surfaces before the immersion, decreased during the immersion process. The metallic (W0) and oxidised (W6+) states were primarily detected on the as-polished and immersed surfaces, respectively. In general, it was difficult to distinguish between the Cu0 and Cu+ components because their binding energies (BEs) were very close. Nonetheless, the immersion in sulfuric acid produced both Cu0/Cu+ and Cu2+ species on the outermost specimen surfaces, whereas negligible Cu amounts were detected on the surfaces prior to the immersion. In contrast, the C 1s peaks deconvoluted into the C–C/C–H, C–O, and C=O components at each condition. Moreover, a peak assignable to carbide species was detected as well, which was consistent with the microstructural observations (Figs. 1–3). Finally, immersion in the H2SO4 solution increased the amounts of both the OH− and O2− components, whereas the highest fraction of OH− ions was observed on the outermost surfaces. The BEs, full-width at half-maximum (FWHM), and concentrations of each state in the samples, which are summarised in Supplementary Table 1, did not show significant differences. Notably, Cu was dominantly distributed at the outmost surface in both cases. A small amount of sulfate (<1 at%) was detected as a corrosion product.

SEM images of the surface regions of the 16Cr steel samples quenched from a 800 and b 1100 °C after immersion in the 0.5 M H2SO4 solution for 3 h. Magnified SEM images of the corroded surfaces of the c martensitic matrix and d carbide particles in the 1100 °C sample. Cross-sectional SEM images of the e 800 °C and f 1100 °C samples obtained during FIB sampling. g STEM observations and the corresponding EDS mapping data obtained for the sample quenched from 1100 °C.
The high-resolution XPS profiles of the 16Cr steel samples were obtained at various depths from the corroded surface after 7 h of immersion (Fig. 8). The sputtering time where each spectrum was obtained is indicated for the samples. The sample quenched from 1100 °C contained a considerable amount of the Fe3+ component (as compared with that of the 800 °C sample) even in the inner surface region, although the metallic component became dominant in the inner regions of both samples. The Cr0 fractions of these samples increased in the bulk regions; however, neither Cr0 nor Cr3+ species were detected on their outermost surfaces. A transition from the oxidised state (W6+) to the metallic state was observed deeper from the surface for both samples. However, the sample quenched from 1100 °C contained both the W0 and W6+ components in the inner region. In contrast, the BE of the Cu peak did not change significantly after moving from the outermost surface to the metal substrate. Thus, Cu most likely existed in the metallic state (Cu0) rather than in the oxidised state (Cu+). The Cu0 component was predominantly observed for both samples, although small amounts of Cu2+ species were also detected on the outmost surfaces. While the high carbon content on the outermost surface can be considered a contamination, the obtained XPS profiles exhibited carbide peaks in the inner regions of the samples, especially those quenched from 800 °C. The OH− and O2− components were detected exclusively on the outermost surfaces, whereas the amount of O2− species increased in the inner region. Interestingly, the O2− component in the sample quenched at 1100 °C retained a relatively high intensity within the analysed depth as compared with that of the other sample.

a, g Fe 2p, b, h Cr 2p, c, i W 4f, d, j Cu 2p, e, k C 1s, and f, l O 1s peaks for the 16Cr steel samples heat treated at a–f 800 °C and g–l 1100 °C after the 7-h immersion in the 0.5 M H2SO4 solution. The sputtering time where each spectrum was acquired is indicated for each.
The depth-wise elemental distributions obtained for the 16Cr steel samples exhibited significant variations in surface chemical states with the quenching temperature (800 and 1100 °C), as shown in Fig. 9. The Fe concentration, which was equal to 10–20 at% on the outermost surfaces of both samples, increased to ~30 and ~45 at% in the inner regions of the samples quenched from 800 and 1100 °C, respectively. The higher Fe content in the corroded surface regions of the sample quenched from 1100 °C as compared with that of the sample quenched from 800 °C reflects the distribution of the Fe3+ component. Although the Cr concentrations in both samples increased from the outermost surface to the inner part, the maximum value obtained for the sample quenched from 800 °C was approximately 30 at% Cr, which was more than three times higher than that obtained for the sample quenched from 1100 °C. Both samples demonstrated a gradual increase in the W concentration with increasing depth from the outermost surface. Although high carbon concentrations due to contamination were detected on the outermost surfaces of both samples, their magnitudes in the inner regions of the heat treated at 800 and 1100 °C were ~15 and 5 at%, respectively. The higher Cr and W contents of the 800 °C sample mostly originated from the higher fractions of M23C6 carbide particles that remained intact during immersion. These samples also produced different oxygen concentration profiles: the higher quenching temperatures resulted in higher oxygen concentrations in the surface and subsurface regions after immersion. Although Cu enrichment was observed on the surfaces of both samples, higher Cu concentrations in the surface and subsurface regions were detected for the 1100 °C sample. The surface state variations became insignificant within the first 1–2 h of immersion, as shown in Supplementary Figs. 4 and 5.

Atomic fractions of Fe, Cr, W, Cu, C, and O elements in the 16Cr steel samples quenched from a 800 °C and b 1100 °C plotted as functions of the depth from the outermost surface after immersion in the 0.5 M H2SO4 solution for 7 h.
Volta potential mapping
Figure 10a–d shows the topography maps obtained for the surfaces of the 16Cr steel samples before and after the immersion in the 0.5 M H2SO4 solution for 1 h. Polishing prior to the immersion procedure produced nanoscale topographical heterogeneities between the constituent phases (Fig. 10a and b). Figure 10e–h, i–l presents the corresponding local Volta potential maps and the line profiles along the white lines. The Volta potentials of the as-polished states strongly depended on the constituent phase beneath the corresponding surfaces. Namely, the Volta potential of the martensitic matrix was higher than that of the carbide phase, which indicated its lower electrochemical nobility, and the difference (approximately 0.04 V) in the Volta potentials of the matrix and carbide phases suggested the existence of microgalvanic coupling between the constituent phases. Potential variations within the martensitic matrix likely originated from the inter-lath boundaries21. Overall, the quenching temperature did not affect the Volta potential values of each phase.

a–d Topography and e–h the corresponding Volta potential difference maps obtained before and after immersion of the 16Cr steel samples quenched from 800 and 1100 °C in the 0.5 M H2SO4 solution for 1 h. i–l Line profiles of the Volta potential constructed along the white lines in panels e–h.
The surface roughness after the immersion was much higher than those of the as-polished states (Fig. 10c, d). Thus, to reduce the noise level, the probe–surface distance was increased during the Volta potential measurements. Although it is difficult to compare the results obtained for the as-polished and immersed samples because different measuring conditions were utilised, a measurable Volta potential difference (~0.07 V) between the matrix and the carbide phase was detected for the 800 °C sample after the immersion. Notably, the immersed 1100 °C sample exhibited a negligible Volta potential difference between its microstructural constituents, indicating that the microgalvanic coupling disappeared during immersion. Thus, the Volta potential distribution obtained after the immersion demonstrated a significant quenching temperature dependence.
Discussion
Herein, we investigated the corrosion behaviour of the Cu-doped martensitic steel with multiple carbide precipitates by varying its bulk Cr content (13 and 16 mass%) and quenching temperature (800–1100 °C). The as-quenched microstructures were primarily mixtures of the martensitic matrix and M23C6 carbide phase, whereas the ferrite and retained austenite phases were also detected after quenching from 800 and 1100 °C, respectively. The experimental results revealed that the corrosion performance of the studied steels strongly depended on the quenching temperature: lower weight losses after the immersion in the 0.5 M H2SO4 solution were obtained for the samples heat treated at higher temperatures (Fig. 4). The anodic dissolution of the martensitic matrix in these steels originated from the electrochemical potential difference between the martensite and carbide phases23. Thus, higher quenching temperatures, which lower the carbide fraction and consequently the number density of galvanic coupling, can increase the corrosion resistance (Fig. 1).
In contrast, as described in a previous study23, the higher corrosion resistances of the studied steels compared to that of their Cu-free counterpart can be explained by the gradual enrichment of the corroded surface with Cu atoms in the corrosive medium. The results of the present work demonstrate that samples quenched from higher temperatures have more surface Cu enrichment (Fig. 9). Moreover, according to the SEM observations (Figs. 5 and 6), Cu enrichment involved the formation of nanoscale Cu particles on the corroded surfaces although the EPMA elemental maps revealed uniform distributions of Cu at the microscale. Interestingly, the quenching temperature significantly affected the surface Cu enrichment. Thus, the 1100 °C sample contained Cu particles both on the martensitic matrix and carbide surfaces, whereas in the 800 °C counterpart, Cu particles existed exclusively on the matrix surface.
The surface evolution ultimately affected the electrochemical potential difference (Fig. 10). The Volta potential distributions of the 800 and 1100 °C samples obtained prior to the immersion were very similar, exhibiting higher and lower values for the matrix and carbide phases, respectively. A change in the matrix constituent from the ferrite/martensite phase to the martensite with retained austenite that occurred at higher quenching temperatures did not considerably alter the Volta potential value. The potential difference remained significant on the surface of the 800 °C sample after immersion. In contrast, the potential differences between the constituent phases were negligible after the immersion of the studied steels quenched from 1100 °C. This phenomenon could be explained by the Cu deposition on the surfaces of both the martensitic matrix and carbide phase in the 1100 °C sample, which was inconsistent with the preferential Cu enrichment of the steel matrix of the 800 °C sample. Thus, the microgalvanic corrosion between the constituent phases was inhibited, which increased the corrosion resistance of the 1100 °C sample as compared with that of the 800 °C sample.
Meanwhile, preferential dissolution was observed at the matrix/carbide interfaces in the 1100 °C sample (Figs. 5d and 6f), although the anodic dissolution of the martensitic matrix was significantly suppressed. Cr depletion is not obvious in the corroded surface (Fig. 5f). In contrast, Anantha et al.21 reported that the large potential gradients along the carbide/matrix interfaces in a martensitic stainless steel made it more susceptible to corrosion during exposure to corrosive electrolytes. Such potential gradients existed in the 1100 °C sample only along the martensite/carbide interfaces after the immersion (Fig. 10h). Thus, the different degrees of Cu enrichment of the studied steels strongly affected their corrosion characteristics, causing a shift from microgalvanic corrosion to preferential corrosion at the carbide/matrix interfaces. The selective corrosion attacks can be detected as the higher fractions of the O2− component as well as the oxidised states of Fe and W elements in the inner sample part (Figs. 8 and 9).
Several researchers have investigated the mechanism of corrosion resistance enhancement in Cu-bearing steels. Notably, anodic dissolution was suppressed by Cu deposition on the specimen surface immersed in a corrosive medium25,28,32,33. The observed precipitation of Cu particles on the carbide surfaces of the steels fabricated in the present study indicated that Cu was deposited from the corrosive medium. The feather-like substances, which were associated with the Cu particles on the corroded surfaces, could be related to Cu deposition, although it was difficult to characterise this process experimentally. In contrast, Pardo et al.25 suggested that the deposition of the Cu layer considerably reduced the hydrogen overpotential, thereby favouring the partial regeneration of the passive layer comprising Cr2O3 species. Oguzie et al.28 also reported that Cu addition facilitated the passivation of austenitic, ferritic, and martensitic stainless steels in a 0.1 M H2SO4 solution because Cu addition increased the donor and acceptor densities that enhanced the stability of a passive oxide film. However, XPS revealed that the corroded surface of the carbide-containing steels in the present work was not covered with a Cr2O3 film after immersion in 0.5 M H2SO4 solution (Fig. 8b, h). Thus, the surface characteristics of the studied steels were significantly different from those of the conventional Cr-containing alloys, in which passivation by the Cr2O3 film strongly influenced the corrosion behaviour. These results indicated that the presence of a large number of carbide precipitates resulted in a significantly different corrosion behaviour of the prepared steels compared to that of conventional stainless steels.
The other factors influencing the corrosion behaviour of the studied steels include the bulk Cr concentration because a higher corrosion resistance was obtained at higher Cr contents. Arguably, the dealloying process, aimed to create the Cu-enriched surface at the initial immersion stage, should be controlled by the Cr-containing oxide film. The low corrosion rates obtained at the early immersion stage of the 16Cr steel samples quenched from 1000 and 1100 °C (Fig. 4d) also support this hypothesis. Furthermore, the formation of Cu particles on the outmost specimen surface may be influenced by its surface chemical composition, as the Cu particles on the martensite surface were finer than those on the carbide surface (Fig. 5d). The Cu-phase precipitation in the steel matrix at 800 °C could also affect the Cu deposition on the sample surface. Position-sensitive surface and microelectrochemical characterisation techniques34 can be potentially used to establish a direct correlation between the surface evolution and substrate microstructure.
Methods
Sample preparation
Fe–(13, 16)Cr–3W–2Cu–1C (mass%) steels with different bulk Cr concentrations were produced using an industrial apparatus (Eiwa Co., Ltd, Japan). Ingots (~30 kg) were prepared with various steel compositions (see Supplementary Table 2) by high-frequency vacuum induction melting.
The obtained ingots were first subjected to a homogenising heat treatment (1200 °C, 4 h) and then hot-forged into bars with cross-sections of approximately 80 mm × 80 mm after heating to 1200 °C. Thereafter, the steels were solution-treated (1150 °C, 2 h) followed by hot forging (cross-section: approximately 50 × 50 mm2). Hot calibre rolling was subsequently performed to produce bars with diameters (φ) of 30 mm. To prevent hot cracking during hot working, the bars were wrapped with ceramic insulating wool (IBIWOOL-E, IBIDEN, Japan) and cooled slowly in air. Finally, the bars were annealed at 850 °C for 2 h followed by furnace cooling. Thus, the as-received steels mainly contained ferrite grains and carbide precipitates.
The obtained bars were cut into cylindrical samples (φ = ~10 mm) via electrical discharge machining (EDM, HS-300, Brother Industries, Japan). Each sample was encapsulated in a quartz tube containing Ar gas and heat treated in a muffle furnace (800–1100 °C, 2 h, FR302, Yamato Scientific, Japan), followed by water quenching.
Thermodynamic calculations
Thermo-Calc software (ver. 2017a) with a database for steels (TCFE9) (Thermo-Calc Software, Sweden) was used to examine the equilibrium phase distributions and chemical compositions of the constituent phases in the studied steels.
Microstructural characterisation
SEM observations were performed using an S-3400N system (Hitachi High-Technologies, Japan) operated at 15 kV. Elemental maps were acquired through field-emission electron probe microanalysis (FE-EPMA; JXA-8430F, JEOL, Japan) performed at 15 kV. EBSD measurements were conducted on a field-emission SEM instrument (FE-SEM, XL30S-FEG, FEI, USA) operated at 20 kV. EBSD data were collected and analysed using a TSL-OIM system (ver. 7.3, EDAX, USA). Samples for microstructural characterisation were cut from the as-quenched samples by EDM. They were subsequently ground with emery paper (up to #3000, Struers, Japan), polished with a 1-μm alumina suspension (AP-A, Struers, Japan), and finished with a 0.04-μm colloidal silica suspension (OP-S, Struers, Japan).
To investigate the constituent phases of the prepared samples, time-of-flight neutron diffraction measurements were conducted using the BL20 (iMATERIA) neutron diffractometer at the Materials and Life Science Experimental Facility (MLF) of the Japan Proton Accelerator Research Complex (J-PARC), Japan35. Bulky samples with relatively large volumes (diameter: 10 mm and length: 30 mm) were prepared from the hot-rolled bars and then heat treated as described above.
Static immersion tests
The corrosion behaviour of the studied steels in 0.5 M H2SO4 solution was evaluated by conducting immersion tests in accordance with the ASTM G31 standard36 under the conditions specified in a previous study23. The 0.5 M H2SO4 solution was prepared from H2SO4 (FUJIFILM Wako Pure Chemical, Japan) and deionised water. The samples were cut to sizes of approximately φ10 mm × 2 mm, and their surfaces were ground using emery paper (up to #1000, Struers, Japan). Next, they were immersed in the H2SO4 corrosion medium (30 mL) at an approximate temperature of 25 °C using Teflon crucibles. Subsequently, the samples were removed from the crucibles, cleaned with distilled water, dried, and weighed using an analytical balance (AUW320, Shimadzu, Japan). This process was repeated every hour for a total of 7 h. The weight loss of each sample was determined via the following equation:
where i is the index of the immersion period; mi and m0 are the sample masses before and after the ith immersion period, respectively; and A is the surface area of the specimen prior to the immersion test. The corrosion rate (in mg cm−2 h−1) at a given immersion time was calculated from the weight loss data.
Surface characterisation
The corroded surfaces after the immersion tests were examined by FE-SEM (15 kV, JSM-7100F, JEOL, Japan) and FE-EPMA (JXA-8430F, JEOL, Japan). Their cross-sections were prepared by the FIB technique (Versa 3D Dual Beam, FEI, USA) and characterised by STEM and EDS elemental mapping conducted on a dual spherical aberration-corrected S/TEM instrument (TITAN3 G2 60-300, FEI, USA) operated at 300 kV. Pt was deposited to protect the corroded surface during sample preparation by FIB. SEM images were also acquired with the FIB instrument at an acceleration voltage of 5 kV.
To analyse the surface states of the steels before and after the immersion tests, XPS measurements were performed on an Axis Ultra DLD spectrometer (Shimadzu-Kratos, UK) using a monochromatised Al Kα X-ray source (1486.7 eV). General survey and high-resolution spectra were acquired over a 300 μm × 700 μm area at step sizes of 1 and 0.1 eV, respectively. The chemical states of the analysed elements were determined using the CasaXPS software (ver. 2.3.15; details of the XPS procedure are provided in our previous study37). The depth profiles of the alloying elements were recorded by Ar gas ion sputtering. After the immersion tests, the samples were first cleaned with distilled water and then air-dried prior to XPS measurements.
SKPFM measurements were conducted using an atomic force microscope (Multi-Mode 8, Bruker AXS, Germany) in the PeakForce tapping mode to obtain distributions of the Volta potential (the contact potential difference between the Pt reference tip and the sample surface) before and after immersion in the 0.5 M H2SO4 solution. The measured area was set to 20 μm × 20 μm for each specimen. To minimise surface roughness, the samples were mirror-polished with a colloidal silica suspension (OP-S, Struers, Japan) prior to immersion.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author upon reasonable request.
Change history
17 September 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41529-021-00197-y
References
ASTM A600-92a, Standard Specification for Tool High Speed (ASTM International, 2016).
Grzesik, W. Advanced Machining Processes of Metallic Materials (Elsevier, 2017).
Kattamis, T. Z. & Suganuma, T. Solidification processing and tribological behavior of particulate TiC-ferrous matrix composites. Mater. Sci. Eng. A 128, 241–252 (1990).
Pagounis, E. & Lindroos, V. K. Processing and properties of particulate reinforced steel matrix composites. Mater. Sci. Eng. A 246, 221–234 (1998).
Godec, M. et al. Characterization of the carbides and the martensite phase in powder-metallurgy high-speed steel. Mater. Charact. 61, 452–458 (2010).
Hwang, K. C., Lee, S. & Lee, H. C. Effects of alloying elements on microstructure and fracture properties of cast high speed steel rolls part I: microstructural analysis. Mater. Sci. Eng. A 254, 282–295 (1998).
Ding, P., Shi, G. & Zhou, S. As-cast carbides in high-speed steels. Metall. Trans. A 24, 1265–1272 (1993).
Stiller, K. et al. High resolution microanalytical study of precipitation in a powder metallurgical high speed steel. Acta Metall. 32, 1457–1467 (1984).
Simchi, A. & Asgharzadeh, H. Densification and microstructural evaluation during laser sintering of M2 high speed steel powder. Mater. Sci. Technol. 20, 1462–1468 (2004).
Brett, C. M. A. & Melo, P. I. C. Influence of anions on the corrosion of high speed steel. J. Appl. Electrochem. 27, 959–964 (1997).
Alves, V. A., Brett, C. M. A. & Cavaleiro, A. Influence of heat treatment on the corrosion of high speed steel. J. Appl. Electrochem. 31, 65–72 (2001).
Navas, C., Conde, A., Fernández, B. J., Zubiri, F. & de Damborenea, J. Laser coatings to improve wear resistance of mould steel. Surf. Coat. Technol. 194, 136–142 (2005).
Kwok, C. T., Cheng, F. T. & Man, H. C. Microstructure and corrosion behavior of laser surface-melted high-speed steels. Surf. Coat. Technol. 202, 336–348 (2007).
Ehiasarian, A. P., Münz, W. D., Hultman, L., Helmersson, U. & Petrov, I. High power pulsed magnetron sputtered CrNx films. Surf. Coat. Technol. 163–164, 267–272 (2003).
Zhang, S. & Zhu, W. TiN coating of tool steels: a review. J. Mater. Process. Technol. 39, 165–177 (1993).
Fedrizzi, L., Rossi, S., Cristel, R. & Bonora, P. L. Corrosion and wear behaviour of HVOF cermet coatings used to replace hard chromium. Electrochim. Acta 49, 2803–2814 (2004).
Michalski, J., Lunarska, E., Wierzchon, T. & AlGhanem, S. Wear and corrosion properties of TiN layers deposited on nitrided high speed steel. Surf. Coat. Technol. 72, 189–195 (1995).
ASTM A1010/A1010M, Standard Specification for Higher-Strength Martensitic Stainless Steel Plate, Sheet, and Strip (ASTM International, 2018).
Bonagani, S. K., Bathula, V. & Kain, V. Influence of tempering treatment on microstructure and pitting corrosion of 13 wt.% Cr martensitic stainless steel. Corros. Sci. 131, 340–354 (2018).
Lu, S. Y. et al. The effect of tempering temperature on the microstructure and electrochemical properties of a 13 wt.% Cr-type martensitic stainless steel. Electrochim. Acta 165, 45–55 (2015).
Anantha, K. H. et al. Correlative Microstructure Analysis and In Situ Corrosion Study of AISI 420 Martensitic Stainless Steel for Plastic Molding Applications. J. Electrochem. Soc. 164, C85–C93 (2017).
Lo, K. H., Shek, C. H. & Lai, J. K. L. Recent developments in stainless steels. Mater. Sci. Eng. R 65, 39–104 (2009).
Zhang, C., Yamanaka, K., Bian, H. & Chiba, A. Corrosion-resistant carbide-reinforced martensitic steel by Cu modification. npj Mater. Degrad. 3, 30 (2019).
Mallick, P. K. Fiber-Reinforced Composites: Materials, Manufacturing, and Design (CRC Press, 2007).
Pardo, A. et al. Influence of Cu and Sn content in the corrosion of AISI 304 and 316 stainless steels in H2SO4. Corros. Sci. 48, 1075–1092 (2006).
Hong, I. T. & Koo, C. H. Antibacterial properties, corrosion resistance and mechanical properties of Cu-modified SUS 304 stainless steel. Mater. Sci. Eng. A 393, 213–222 (2005).
Ujiro, T., Satoh, S., Staehle, R. W. & Smyrl, W. H. Effect of alloying Cu on the corrosion resistance of stainless steels in chloride media. Corros. Sci. 43, 2185–2200 (2001).
Oguzie, E. E. et al. The effect of Cu addition on the electrochemical corrosion and passivation behavior of stainless steels. Electrochim. Acta 55, 5028–5035 (2010).
Banas, J. & Mazurkiewicz, A. The effect of copper on passivity and corrosion behaviour of ferritic and ferritic-austenitic stainless steels. Mater. Sci. Eng. A 277, 183–191 (2000).
Ren, L., Nan, L. & Yang, K. Study of copper precipitation behavior in a Cu-bearing austenitic antibacterial stainless steel. Mater. Des. 32, 2374–2379 (2011).
Pardo, A., Merino, M. C., Carboneras, M., Coy, A. E. & Arrabal, R. Pitting corrosion behaviour of austenitic stainless steels with Cu and Sn additions. Corros. Sci. 49, 510–525 (2007).
Hong, J. H., Lee, S. H., Kim, J. G. & Yoon, J. B. Corrosion behaviour of copper containing low alloy steels in sulphuric acid. Corros. Sci. 54, 174–182 (2012).
Hermas, A. A., Ogura, K. & Adachi, T. Accumulation of copper layer on a surface in the anodic polarization of stainless steel containing Cu at different temperatures. Electrochim. Acta 40, 837–844 (1995).
Klemm, S. O., Schauer, J. C., Schuhmacher, B. & Hassel, A. W. A microelectrochemical scanning flow cell with downstream analytics. Electrochim. Acta 56, 4315–4321 (2011).
Ishigaki, T. et al. IBARAKI materials design diffractometer (iMATERIA)-Versatile neutron diffractometer at J-PARC. Nucl. Instrum. Methods Phys. Res. Sect. A 600, 189–191 (2009).
ASTM G31-21, Standard Guide for Laboratory Immersion Corrosion Testing of Metals (ASTM International, 2021).
Yamanaka, K. et al. Corrosion mechanism of an equimolar AlCoCrFeNi high-entropy alloy additively manufactured by electron beam melting. npj Mater. Degrad. 4, 24 (2020).
Acknowledgements
The authors acknowledge Masahiro Yamazaki (Eiwa Co., Ltd) for providing the steel specimens and Issei Narita, Kazuyo Omura, Yumiko Kodama, and Yuichiro Hayasaka (Institute for Materials Research, Tohoku University) for providing technical assistance with the microstructural/surface characterisations. This work was supported by the Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (JSPS) [Grant No. 20H02472]; Research Promotion Grant from the Iron and Steel Institute of Japan (ISIJ); Research Grant from the Die and Mould Technology Promotion Foundation; and Research Society for Quantum-beam Analysis of Microstructures and Properties of Steels (ISIJ). EPMA and XPS were conducted at the Cooperative Research and Development Centre for Advanced Materials, Institute for Materials Research, Tohoku University [Proposal Nos. 19G0414 and 20G0416]. The neutron experiment at the MLF of J-PARC was performed as per proposal 2019PM2006.
Author information
Authors and Affiliations
Contributions
K.Y. and A.C. conceived the concept of the research and designed the experiments. K.Y., M.M., Y.O., and S.S. performed the experimental analysis. K.Y. contributed to the preparation of the alloy specimens. K.Y. wrote the manuscript. M.M. and S.S. commented and made revisions on the manuscript. All the authors contributed to the interpretation of the experimental data and discussed the results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yamanaka, K., Mori, M., Yoshida, K. et al. Surface evolution and corrosion behaviour of Cu-doped carbide-reinforced martensitic steels in a sulfuric acid solution. npj Mater Degrad 5, 43 (2021). https://doi.org/10.1038/s41529-021-00187-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-021-00187-0
This article is cited by
-
Long-term corrosion monitoring of carbon steels and environmental correlation analysis via the random forest method
npj Materials Degradation (2022)
-
Unveiling nano-scaled chemical inhomogeneity impacts on corrosion of Ce-modified 2507 super-duplex stainless steels
npj Materials Degradation (2022)
