
Abstract
Carbonate stability plays a crucial role in clarifying the evolution and protection of the naturally formed corrosion scales on the steel surface in the application of geothermal production. In this paper, the stability of the corrosion scales from both micro and atomic level are studied via a combination of electrochemistry, surface analysis and first-principle calculation. The chemical and physical characterisation of various iron–calcium mixed carbonates are experimentally analysed and results are compared with the first-principle calculation. In the presence of Ca2+, the preferential loss of Ca during the dissolution experiments was observed, the interactions within the crystal weaken where Ca2+ co-precipitation, confirmed by a faster degradation rate for Ca0.51Fe0.49CO3 than FeCO3. This work reveals the degradation and protection performance of the naturally formed carbonate layers and provides insights into understanding the corrosion product stability and chemical breakdown of the corrosion scales.
Similar content being viewed by others


Introduction
Ensuring engineering integrity while reducing the cost is the eternal topic for all industries such as oil and gas, geothermal, and nuclear. The research and development of corrosion-resistant alloys (CRAs) can achieve the material integrity aspect while the rise in cost restricted the applications1. Carbon steel was widely used in the industry such as oil and gas or geothermal productions based on its low cost and excellent mechanical performance2,3,4,5,6. However, carbon steel was subject to corrosion in the presence of CO2, resulting in the fast iron dissolution, and followed by the development of the corrosion products gradually covering the surface and effectively reduce the corrosion behaviour2,7,8.
Iron carbonate (FeCO3), for example, formed on the surface was considered as a natural protective scale to prolong the lifetime of carbon steel2,7,9,10. A large amount of previous research has adverted the FeCO3 protection to the surface through forming FeCO3 scales under amorphous or crystalline state6,11,12. The coverage and compactness for the development of FeCO3 scales were fully discussed in the literature in terms of crystal sizes13,14,15, orientation16, solution pH12,17, and other factors12,18.
Recent studies found that simplified corrosion products such as FeCO3 can misestimate the corrosion product protection to the steel19,20,21,22. Esmaeely et al.20 revealed that the presence of high concentration Ca2+ to be responsible for the growth of Ca1-x FexCO3 on the surface. Similar work done by Hua et al.21 indicated that the associated effects of Cl-, Ca2+, Mg2+, and CO2 within brine solution on the growth of the corrosion scales, they reported that the presence of Ca2+ prefers to co-precipitation within the crystalline FeCO3 and accentuates the total penetration rate of carbon steel. Shamsa et al.22 verified the addition of Ca2+ led to the growth of a less protective Ca1−xFexCO3 scale which not only accelerated the pit growth but also increased the general corrosion rate.
The structures for various carbonates FeCO3(siderite), MgCO3 (magnesite) and CaCO3 (calcite) have been studied for decades since they are extensively present in Earth’s crust23. Faruque et al.24 reported the electronic and bonding properties of rhombohedral CaCO3 and found the indirect energy gap to be 5.07 eV. Brik25 revealed that the calcite conduction band is much narrower than the one of magnesite. Elizabeth et al.26 investigated the dolomite (CaMg(CO3)2) (1 0 4) phase and showed the Mg and Ca atoms have different bond distances with oxygen atoms and the main Ca–O interatomic distances turned to be greater after optimization of the surface. The analysis from Golosova et al.27 demonstrated that the anisotropic lattice compression of siderite was mainly contributed by the Fe–O bonds. Han et al.28 calculated the interactions of iron in Mg1−xFexCO3 from the iron-poor (x = 0.125) to the iron-rich (x = 1, FeCO3 siderite) and found that the volume difference concerning MgCO3 changes with Fe doping and resulted in the difference in relative enthalpy. Numerous studies reported that the effects of pressure, temperature, and metal ion doping concerning structure optimization and electron distribution for carbonates. However, rare results regarded the mixture carbonates as corrosion products and focused on their corrosion protection to the surface as well as their electron distribution changes during the corrosion reactions.
To reveal the protectiveness of the corrosion scales, samples covered by FeCO3 layers, iron-calcium carbonate layers, and calcium-containing corrosion product layers were prepared by high temperature and high-pressure corrosion tests. The dissolution of these complex compounds was established through exposed them to a CO2-saturated solution at and pH of 3.75. Linear polarisation resistance (LPR) and electrochemical impedance spectroscopy (EIS) methods were used for tracking the degradation of the corrosion scales. In addition, first-principle thermodynamics based on the density functional theory (DFT) were used to provide a detailed theoretical investigation on the required calcium effects of bond energy, atomistic and electronic structure of the crystals, and was associated with the corrosion product protection to the surface. It is crucial to study these effects on the true protective performance of the corrosion products and reveal the stability of the naturally formed corrosion scales to be effective in corrosion protection for carbon steel under geothermal applications.
Results
Stability properties of corrosion products caused by Ca doping
Supercells of Ca1−xFexCO3 doping with various Ca mole fractions were calculated and compared with pure siderite (FeCO3, x = 1) and pure calcite (CaCO3, x = 0), with the preferred orientation along the plane (1 0 4), as shown in Fig. 1a–e. The results summarised in Fig. 2 represent the characteristics of various Ca1−xFexCO3, Ca mole fraction contains from Ca-poor (x = 0) to Ca-rich (x = 1) and found the average Fe–O distance for FeCO3 is 2.1 Å, while the average bond length between Ca and O turns to be 2.4 Å for CaCO3 at the top surface. The replaced sites by Ca results in the weakening of interaction between Ca cations and C–O3 for Ca1−x FexCO3 (x > 0), the length for Ca–O bonds in Ca0.25Fe0.75CO3 and Ca0.5Fe0.5CO3 increased by compressing the bond length of Fe–O, which suggests the non-uniform bond energy within Ca1−xFexCO3. As the co-precipitation of Ca2+ approached 75%, the increase in the bond length of Ca–O dominated the lattice parameter and resulting in the total volume expansion and leading to the increase in the bond length of Fe–O as well, which suggests that the stability for both Ca–O and Fe–O bonds decreased for Ca1−xFexCO3 compared with pure FeCO3.

a FeCO3 siderite, b Ca0.25Fe0.75CO3, c Ca0.5Fe0.5CO3, d Ca0.75Fe0.25CO3, and e calcite.

The length calculations of Fe–O (purple) bonds and Ca–O (green) bonds at various Ca mole fraction on the top surface.
Since Fe atoms were replaced by Ca atoms within the crystal, the unit-cell volume of Ca1−xFexCO3 expends to a looser arrangement than FeCO3, and the unit-cell approaches the volume of CaCO3 as the increase in the mole fraction of Ca within the unit-cell.
The electron density for FeCO3 and Ca1−xFexCO3 structures are provided to visualize the effect of Ca2+ co-precipitation. It can be seen that C and O atoms join via high electron density and form a molecular structural unit connected by strong covalent bonds for FeCO3. In the regions around Fe ions in Fig. 3a, there are electrons overlaps to the adjacent oxygen, indicating the covalent contribution is seen between Fe and O atoms with the predominant of the ionic bond27,29.

a FeCO3 siderite, b Ca0.25Fe0.75CO3, c Ca0.5Fe0.5CO3, and d Ca0.75Fe0.25CO3. The calculation of electron density of various Ca1−x FexCO3 units on the top surface.
After the co-precipitation of Ca2+ as shown in Fig. 3b, no electron interaction is observed between Ca and O considering the relatively longer distance than that of Fe–O. However, the constraint bond exists between Fe and O atoms and enhances with the increase of Ca2+ co-precipitation level from 25 to 50%, accompanied by the rotation of the plane of C–O3 molecular structural units due to the repulsion of the calcium atoms (Fig. 3c). When the Ca contents approach 75%, the expanded volume resulted in a decrease in the interactions between cations and oxygen, such as Ca–O and Fe–O bonds (Fig. 3d). The difference of electron density between Ca–O bonds and Fe–O bonds suggests the preferential for Ca–O bonds break during the dissolution of iron–calcium mixed carbonate into ions.
Many studies have proved the growth of the carbonated corrosion scales by the precipitation/dissolution processes10,30,31,32. The steel substrate beneath the carbonates suffered localised corrosion since the naturally formed corrosion scales are porous and local dissolution of the corrosion scales can occur at the material interface. Figure 4 provides examples of Ca0.5Fe0.5CO3 dissolution by losing Ca2+ and Fe2+, respectively, which yields the dissolution pathway as follows:
where \({\mathrm{Ca}}_{1 - x}\mathrm{Fe}_x\mathrm{CO}_{3\left( {\mathrm{Ca} - \mathrm{vac}} \right)}\) and \({\mathrm{Ca}}_{1 - x}\mathrm{Fe}_x\mathrm{CO}_{3\left( {\mathrm{Fe} - \mathrm{vac}} \right)}\) are shown as Fig. 4a, b, and the dissolution energy is expressed as follows:

a losing Ca and b losing Fe. The two typical ways of Ca0.5Fe0.5CO3 dissolution.
The change in energy of dissolution by losing Ca2+ ion (785.5 kcal/mol) is smaller compared with that by losing Fe2+ ion (832.0 kcal/mol), suggesting the degradation of corrosion products by preferential breaking Ca–O bonds.
Effect of time on the dissolution of corrosion product scales
Once the presence of Ca2+ ions was found in the fluids of geological formation, the precipitation of calcium-enriched carbonates was proposed to influence the morphology and structure, as well as affecting corrosion protection to the steel surface compared with the naturally formed FeCO3. To confirm the DFT calculation described above, revealing the degradation mechanisms of pure FeCO3 and iron-calcium mixed carbonates, carbon steel covered by various corrosion products were prepared in a simulated geothermal environment with/without the presence of Ca2+.
Figure 5 shows the SEM images and XRD patterns of the prepared corrosion products formed in 60 °C conditions with/without the addition of Ca salts, and Supplementary Figs 3b and 4 illustrate the thickness of the corrosion scales prior to dissolution experiments. The presence of Ca2+ in the solution resulted in the peaks for naturally formed corrosion products shift to the left from FeCO3 (purple dash lines) and toward a mixture of Fe–Ca carbonates (green dash lines), which consistent with our previous studies21,22. The shifted peaks from XRD measurement correspond with the increase of d-spacing for the plane (1 0 4) for iron-calcium mixed carbonate according to Bragg’s law as shown in Eq. (4):
where n is a positive integer and λ is the wavelength of the incident X-ray wave. The mole fraction of calcium in the iron-calcium mixed carbonate was 0.51 according to the relationship between d-spacing and calcium proportion, the calculations are shown in Supplementary Eq. (7).

The formation of the corrosion products in 60 °C conditions with/without the addition of Ca. a SEM-FeCO3, b SEM-Ca0.51Fe0.49CO3, and c XRD patterns.
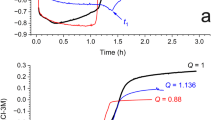
Polarisation resistance (\(R_{\mathrm{p}}\)) can reflect the dissolution speed of the corrosion scales with time. The obtained in situ \(R_{\mathrm{p}}\) measurements by the LPR technique are plotted in Fig. 6. The recorded \(R_{\mathrm{p}}\) during the corrosion scale dissolution were determined from the slope of the line:

Dissolution measurements of carbon steel covered with (purple) FeCO3 or (green) Ca0.51Fe0.49CO3 scale over time in a CO2-saturated 3 wt.% NaCl solution at 15 °C.
Purple and green symbols represent \(R_{\mathrm{p}}\) for carbon steels covered by pure FeCO3 and Ca0.51Fe0.49CO3 scales exposed to the CO2-saturated 3 wt.% NaCl solution at 15 °C respectively. For both FeCO3 and Ca0.51Fe0.49CO3 scales covered samples, high Rp values were measured at the beginning. The initial Rp for naturally formed FeCO3 was higher than that of Ca0.51Fe0.49CO3, suggesting that the sample covered with FeCO3 has a lower initial corrosion rate. For stage 2, a sharp Rp reduction was recorded for the sample covered with Ca0.51Fe0.49CO3 due to the scale dissolved fast with immersion time in comparison with the sample covered with FeCO3. A relatively stable Rp was recorded at stage 3 after 1000 min for the sample covered by Ca0.51Fe0.49CO3 and similar values were measured after 6000 mins for sample surface covering FeCO3, suggesting the dissolution of the corrosion scales consisted of Ca0.51Fe0.49CO3 was easier compared with pure FeCO3 under the same experimental conditions.
Figure 7 presents the EIS measurements of samples exposed to the solution at each dissolution stage. The impedance measured in stage 1 suggests that the coverage of pure FeCO3 on the entire surface acting as a diffusion barrier and protecting to the surface, while the Ca0.51Fe0.49CO3 scales present a porous feature (fitting as Warburg impedance, ZW). The aggressive ions penetrate easily through this porous layer, resulting in the fast converting to activation dissolution in stage 1.

(a), (c) and (e) for carbon steel covered with FeCO3, (b), (d) and (f) for carbon steel covered with Ca0.51Fe0.49CO3 scales at various stages along with Fig. 6.
Stage 2 corresponded to a rapid decline in Rp over the exposure time, indicating the corrosion product dissolution occurred on the surface33. In Fig. 7b, the Nyquist plots for FeCO3-covering samples shown in stage 2 present the diffusion-controlled feature at low frequencies, revealing the generation of gaps between FeCO3 crystals through the dissolution processes, which as expected that the protection for naturally formed scales is dependent on the localised corrosion scale dissolution. However, the diffusion control disappeared on Nyquist plots for Ca0.51Fe0.49CO3-covering samples, replacing with the measured inductance arcs and suggesting the large decrease in Rp through dissolving the corrosion scales34, accelerating the penetration where the adsorption and desorption of dissolved cations occur constantly.
For stage 3, the measured semi-circles for both FeCO3 and Ca0.51Fe0.49CO3-covering samples became smaller, suggesting that the corrosion rates increased due to the corrosion scales dissolved from the surface. A similar observation was seen from the XRD in Supplementary Fig. 2, the most intense Bragg reflection (1 0 4) Miller planes for Ca0.51Fe0.49CO3 showed a decrease in intensity. It is interesting to note that there is no significant difference in the intensity for FeCO3-covering sample, and the measured semi-circle of the FeCO3-covering sample was larger than that of Ca0.51Fe0.49CO3-covering samples, suggesting that the crystalline FeCO3 dissolved slower than Ca0.51Fe0.49CO3. The co-precipitation of Ca2+ ions into the crystalline carbonate has weakened the bonds between Ca2+ and CO32- and favoured the Ca–O bonds break compared to FeCO3 during the dissolution, which is in agreement with DFT calculations as shown in Fig. 2.
Characterisation of the corrosion product morphology during the dissolution experiment
Figure 8 shows the morphology of corrosion scales before and after the dissolution experiments. Figure 8a represents the dense and uniform cubic FeCO3 crystals and compared with the relative complex structures of Ca0.51Fe0.49CO3 (Fig. 8c) crystals formed on the surface with the presence of 1000 ppm Ca2+ ions in the solution. The co-precipitation of Ca2+ ions into crystalline transforms the cubic morphology into globular structure crystals. Some pores were observed between these globular crystals, consistent with the impedance measured in stage 1 during the dissolution process, smaller semicircle and higher corrosion rate were obtained compared to that of sample covered with FeCO3 as shown in Fig. 7.

a Crystalline FeCO3 before dissolution experiment, b crystalline FeCO3 after the dissolution experiment at 15 °C; c Ca-containing corrosion scales before dissolution experiment, d Ca-containing corrosion scales after the dissolution experiment at 15 °C.
Figures 8b and d represent the dissolved corrosion products after 120 hrs which correspond to stage 3 in Figs 6 and 7. The morphology in Fig. 8b indicates that the gaps appeared and are revealed as a result of the preferential dissolution occurring at the boundaries between FeCO3 crystals. For the dissolution experiment of Ca0.51Fe0.49CO3, the surface as shown in Fig. 8d illustrates porous corrosion products. The high magnification SEM images show the crystals were partially dissolved on the surface and resulted in porous characteristics and poor protection which agree with the EIS measurements as shown in Fig. 7d and f.
Supplementary Fig. 3 indicates the detected elemental compounds of Ca0.51Fe0.49CO3 scales before and after dissolution by using EDS. The EDS results indicate that the proportion of Ca within the corrosion products was about 48.7% before the dissolution, this value decreased to 3.5% after dissolution experiments. The results are supported by DFT calculation which suggests the weak bonds between Ca2+ and CO32- and the degradation of Ca0.51Fe0.49CO3 by preferential breaking Ca-O bonds. It is interesting to note that the presence of Ca-rich areas was detected locally and mainly found in the pores after 120 h of dissolution (Supplementary Fig. 3e), suggesting that the growth of FexCa1−xCO3 scales contains ununiform Ca mole fraction and results in the preferential dissolution occurred locally on the surface.
Discussion
The present study helps fill the current knowledge gaps that the degradation processes of naturally formed corrosion scales occur from the micro-level to the molecular level on the surface. Combining the first-principle DFT calculations and experimental validation is an effective way that can be used to elucidate the dissolution of corrosion product scales and protection performance to the steel surface. Ca0.51Fe0.49CO3 with a complex structure that considers the preferential orientation, composition, and interactions that yield insights into the molecular mechanisms of sensitive sites for degradation. The results of our first-principle calculations reveal that the degradation of Ca0.51Fe0.49CO3 by preferential breaking Ca-O bonds and the required breaking energy to lose Ca is smaller than Fe in FeCO3 structure (Fig. 4). The propensity of Ca dissolution within the Fe–Ca mixed carbonates accelerates with the increase of Ca2+ co-precipitation (Supplementary Fig. 5) and the obtained results are validated with experimental measurements. In the absence of Ca2+, the results suggest that the preferential dissolution occurs at the grain boundary, indicating the weak sites for a protective FeCO3 layer. During the dissolution process of the sample was covered by dense FeCO3 crystals, the aggressive ions such as CO32−, Cl− can penetrate through the weak sites, reaching the steel substrate as shown in Fig. 8. In the presence of H+, the fast dissolution occurs at Ca2+ co-precipitation sites and acts as pathways for the penetration of aggressive ions, which promotes the subsequent local corrosion processes and develops into pores/gaps on the surface. In both cases, the preferential dissolution occurs at the grain boundary or locally, indicating a high risk of potential to induce localised corrosion on the surface, resulting in a major threat to the pipeline integrity. The current study yields atomistic insights into the interactions and weak spots involved in complex corrosion products and can serve as the input data to guide the corrosion prediction of carbon steel used in geothermal production applications.
In summary, we compare the characteristics of the naturally formed corrosion product layer in CO2-saturated formation water in the presence or absence of Ca2+ ions, and focus on the degradation mechanism for iron-calcium mixed carbonates layers and compared with pure iron carbonate scale in acidification conditions. The following main conclusions can be made:
-
1.
The co-precipitation of Ca2+ into iron carbonate increases the bond length between cation and oxygen and expands the lattice volume.
-
2.
The Ca2+ co-precipitated position acts as weak sites and is favourable to breakdown, leading to local dissolution and accelerating the penetration rate of carbon steel.
-
3.
Although both Fe–O and C–O bonds within Ca1−xFexCO3 can be broken in a condition at a lower pH, FeCO3 presents better stability than Ca0.51Fe0.49CO3. The weak bonds between Ca2+ and CO32− promote the preferential dissolution in Ca-containing region, leaving a porous corrosion product layer consist of a high proportion of Fe.
-
4.
The experimental results indicated that the presence of Ca-rich areas was detected locally and mainly found in the pores after the dissolution experiments, suggesting that the growth of Ca1−xFexCO3 scales contains ununiform Ca mole fraction and results in the preferential dissolution occurred locally on the surface.
Methods
Preparation of naturally formed corrosion scales
Carbon steel specimens (API 5L X65) were cut from a bar (15-mm diameter, BAO steel) and the thickness is 6 mm. The elemental composition of carbon steel specimens consisted of 0.260 wt.% C, 0.32 wt.% Si, 0.43 wt.% Mn, 0.008 wt.% P, 0.005 wt.% S, 0.490 wt.% Cr, 0.8 wt.% Mo, 0.037 wt.% Ni, 0.080 wt.% V, \(\le\)0.010 wt.% Ti, and balance with Fe. The specimens used for the electrochemical testing were sealed in resin to leave one surface with an exposure area of 1.77 cm2 to the solution. All these specimens were ground the entire surface from 120 to 800 grit SiC papers, rinsed with distilled water, acetone, and ethanol, then dried with compressed nitrogen gas. Prepared specimens were stored in a vacuum desiccator and used within 10 min. The in situ autoclave setup is provided in Supplementary Fig. 1. Three immersion specimens and three electrochemical specimens were placed within the autoclave separately for each test, generating a volume to surface ratio of 33 ml/cm2. The autoclave sealed at ambient temperature and the whole system was flushed with high-pressure CO2 to remove the remaining O2, then pumping in the prepared solutions (solution 1: CO2-saturated 3 wt.% NaCl, 0.08 wt.% NaHCO3 solution; solution 2: CO2-saturated 1.54 wt.% NaCl, 1.83 wt.% CaCl2·2H2O, 0.08 wt.% NaHCO3 solution, maintained Cl- concentration constant and O2 < 10 ppb), followed by pressurising the autoclave to 20 bar with CO2, then heated to 60 °C and the test duration is 72 h of exposure test to generate pure FeCO3 layer or a mixture of Ca1−xFexCO3 layer on the sample surface.
The naturally formed corrosion products were observed by scanning electron microscope (SEM, Hitachi, Regulus 8100). All images were collected at an accelerating voltage of 15 kV and a working distance of ~15.3 mm. The total composition of naturally formed corrosion scales was collected using X-Ray Diffraction (XRD, Bruker D8 ADVANCE) over a range of 2θ = 20–70° at a step size of 0.033° per second. The compositions of corrosion scales provided with a HORIBA EX-350 AZtecEnergy energy disperse spectroscopy (EDS) system using spot scanning and mapping.
Dissolution of the naturally formed corrosion scales
The prepared specimens covered by corrosion scales were exposed to a CO2-saturated 3 wt.% NaCl solution at 15 °C and pH 3.75. During the dissolution process, LPR was employed for tracking the dissolution of the corrosion scales and performed from −10 to 10 mV vs OCP at a scanning rate of 0.167 mV/s by Gamry workstation in a conventional three-electrode cell. The prepared specimens were used as working electrode, platinum foil as the counter, and saturated calomel electrode (SCE) as a reference electrode, respectively. The EIS measurements at various dissolution stages were performed, which were monitored in the CO2-saturated solution at OCP over a frequency range of 100 kHz to 10 mHz using 10 mV sinusoidal potential modulation. The electrochemistry data was analysed with surface morphology and corrosion product compositions at various dissolution stages via SEM, XRD, and EDS.
First-principle calculation
First-principles thermodynamics based on DFT were used to calculate the optimised atomistic and electronic structure of the naturally formed corrosion products. To simplify the model, typical corrosion products were selected, which are FeCO3 (siderite), Ca0.25Fe0.75CO3, Ca0.5Fe0.5CO3, Ca0.75Fe0.25CO3, and CaCO3 (calcite) based on the experimental results via XRD, more details calculation of Ca and Fe mole fraction within the crystals can be found in our previous publications21,22.
For siderite and calcite (inorganic crystal structure database, ICSD), supercells were constructed in (1 0 4) cleave surface. The Ca0.25Fe0.75CO3, Ca0.5Fe0.5CO3, and Ca0.75Fe0.25CO3 supercells were constructed by replacing the Fe atoms in the interlayers with Ca atoms. Based on the geometry optimization model proposed by CASTEP, the generalized gradient approximation (GGA) exchange-correlation energy functional was used with the Perdew-Burke-Ernzerhof (PBE) method and the Kurdjumov-Sachs (KS) relationship. For the Fe-containing systems, a Hubbard U of 4.5 eV was employed for the correction of Fe 3d-electrons. The ultrasoft pseudopotential (USPP) was performed to describe the effective parameter of interaction between electrons and ion core, the cutoff energy is 380 eV for a plane-wave basis with a k-point set of 33334. The ionic relaxation was considered as convergence when the total energies below 10−5 eV and the force on every atom is less than 0.03 eV/Å.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
References
Ikeda, A., Mukai, S. & Ueda, M. Corrosion behavior of 9 to 25% Cr steels in wet CO2 environments. Corrosion 41, 185–192 (1985).
Nesic, S., Postlethwaite, J. & Olsen, S. An electrochemical model for prediction of corrosion of mild steel in aqueous carbon dioxide solutions. Corrosion 52, 280–294 (1996).
Springer, R. D., Wang, Z., Anderko, A., Wang, P. & Felmy, A. R. A thermodynamic model for predicting mineral reactivity in supercritical carbon dioxide: I. Phase behavior of carbon dioxide–water–chloride salt systems across the H2O-rich to the CO2-rich regions. Chem. Geol. 322, 151–171 (2012).
Sun, W. & Nešic, S. Kinetics of corrosion layer formation: part 1—iron carbonate layers in carbon dioxide corrosion. Corrosion 64, 334–346 (2008).
Zheng, L., Landon, J., Matin, N. S. & Liu, K. FeCO3 coating process toward the corrosion protection of carbon steel in a postcombustion CO2 capture system. Ind. Eng. Chem. Res. 55, 3939–3948 (2016).
Hua, Y., Barker, R., Charpentier, T., Ward, M. & Neville, A. Relating iron carbonate morphology to corrosion characteristics for water-saturated supercritical CO2 systems. J. Supercrit. Fluids 98, 183–193 (2015).
Dugstad, A., Hemmer, H. & Seiersten, M. Effect of steel microstructure on corrosion rate and protective iron carbonate film formation. Corrosion 57, 369–378 (2001).
Dugstad, A. Mechanism of Protective Film Formation During CO2 Corrosion of Carbon Steel. in NACE-98031 11 (NACE International, 1998).
Crolet, J., Thevenot, N. & Nesic, S. Role of conductive corrosion products in the protectiveness of corrosion layers. Corrosion 54, 194–203 (1998).
Hua, Y. et al. The formation of FeCO3 and Fe3O4 on carbon steel and their protective capabilities against CO2 corrosion at elevated temperature and pressure. Corros. Sci. 157, 392–405 (2019).
Hua, Y., Barker, R. & Neville, A. Comparison of corrosion behaviour for X-65 carbon steel in supercritical CO2-saturated water and water-saturated/unsaturated supercritical CO2. J. Supercrit. Fluids 97, 224–CO237 (2015).
Sk, M. H. et al. Local supersaturation and the growth of protective scales during CO2 corrosion of steel: effect of pH and solution flow. Corros. Sci. 126, 26–36 (2017).
Guo, S., Xu, L., Zhang, L., Chang, W. & Lu, M. Corrosion of alloy steels containing 2% chromium in CO2 environments. Corros. Sci. 63, 246–258 (2012).
Guo, S., Xu, L., Zhang, L., Chang, W. & Lu, M. Characterization of corrosion scale formed on 3Cr steel in CO2-saturated formation water. Corros. Sci. 110, 123–133 (2016).
Gao, M., Pang, X. & Gao, K. The growth mechanism of CO2 corrosion product films. Corros. Sci. 53, 557–568 (2011).
Ahmad, E. A. et al. Corrosion protection through naturally occurring films: new insights from iron carbonate. ACS Appl. Mater. Interfaces 11, 33435–33441 (2019).
Tanupabrungsun, T., Brown, B., & Nesic, S. Effect of pH on CO2 corrosion of mild steel at elevated temperatures in NACE-2348 1 (NACE International, 2013).
Han, J., Yang, Y., Brown, B. & Nesic, S. Electrochemical investigation of localized CO2 corrosion on mild steel. in NACE-07323 (NACE International, 2007).
Tavares, L. M., da Costa, E. M., de Oliveira Andrade, J. J., Hubler, R. & Huet, B. Effect of calcium carbonate on low carbon steel corrosion behavior in saline CO2 high pressure environments. Appl. Surf. Sci. 359, 143–152 (2015).
Esmaeely, S. N., Young, D., Brown, B. & Nešić, S. Effect of incorporation of calcium into iron carbonate protective layers in CO2 corrosion of mild steel. Corrosion 73, 238–246 (2017).
Hua, Y., Shamsa, A., Barker, R. & Neville, A. Protectiveness, morphology and composition of corrosion products formed on carbon steel in the presence of Cl-, Ca2+ and Mg2+ in high pressure CO2 environments. Appl. Surf. Sci. 455, 667–682 (2018).
Shamsa, A. et al. The role of Ca2+ ions on Ca/Fe carbonate products on X65 carbon steel in CO2 corrosion environments at 80 and 150 °C. Corros. Sci. 156, 58–70 (2019).
Powell, R., Condliffe, D. M. & Condliffe, E. Calcite–dolomite geothermometry in the system CaCO3–MgCO3–FeCO3: an experimental study. J. Metamorph. Geol. 2, 33–41 (1984).
Hossain, F. M., Murch, G. E., Belova, I. V. & Turner, B. D. Electronic, optical and bonding properties of CaCO3 calcite. Solid State Commun. 149, 1201–1203 (2009).
Brik, M. First-principles calculations of structural, electronic, optical and elastic properties of magnesite MgCO3 and calcite CaCO3. Phys. B Condens. Matter 406, 1004–1012 (2011).
Escamilla-Roa, E., Sainz-Díaz, C. I., Huertas, F. J. & Hernandez-Laguna, A. Adsorption of molecules onto (1014) dolomite surface: an application of computational studies for microcalorimetry. J. Phys. Chem. C 117, 17583–17590 (2013).
Golosova, N. et al. Magnetic and structural properties of FeCO3 at high pressures. Phys. Rev. B 96, 134405 (2017).
Hsu, H. & Huang, S.-C. Spin crossover and hyperfine interactions of iron in (Mg, Fe) CO3 ferromagnesite. Phys. Rev. B 94, 060404 (2016).
Shi, H., Luo, W., Johansson, B. & Ahuja, R. First-principles calculations of the electronic structure and pressure-induced magnetic transition in siderite FeCO3. Phys. Rev. B 78, 155119 (2008).
Yue, X. et al. Evolution and characterization of the film formed on super 13Cr stainless steel in CO2-saturated formation water at high temperature. Corros. Sci. 163, 108277 (2020).
Yue, X., Zhang, L., Sun, C., Xu, S., Wang, C., Lu, M., ... & Hua, Y. A thermodynamic and kinetic study of the formation and evolution of corrosion product scales on 13Cr stainless steel in a geothermal environment. Corros. Sci. 169, 108640 (2020).
Yue, X. et al. Influence of a small velocity variation on the evolution of the corrosion products and corrosion behaviour of super 13Cr SS in a geothermal CO2 containing environment. Corros. Sci. 178, 108983 (2020).
Zhu, J., Xu, L. & Lu, M. Electrochemical impedance spectroscopy study of the corrosion of 3Cr pipeline steel in simulated CO2-saturated oilfield formation waters. Corrosion 71, 854–864 (2015).
Cao, C. N. & Zhang, J. Q. An Introduction of Electrochemical Impedance Spectroscopy Science. vol. 9.5 (Science Press, 2002).
Acknowledgements
This work is supported by the Postdoctor Research Foundation of Shunde Graduate School of University of Science and Technology Beijing (No. 2020BH013).
Author information
Authors and Affiliations
Contributions
X.Y. performed the density functional theory calculations and analyzed the results with the help of Y.H. and L.Z. X.Y. and Y.H. wrote the manuscript and performed the experiments. Data interpretation was discussed among all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yue, X., Zhang, L. & Hua, Y. Fundamental insights into the stabilisation and chemical degradation of the corrosion product scales. npj Mater Degrad 5, 8 (2021). https://doi.org/10.1038/s41529-021-00152-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-021-00152-x
