
Abstract
In this work, the use of atomic emission spectroelectrochemistry (AESEC) coupled to electrochemical impedance spectroscopy (EIS) is presented as a method of revealing dissolution mechanisms. To illustrate the method, the dissolution kinetics of Al cations from an Al-Zn pure phase (Zn-68 wt.% Al) was investigated in an alkaline solution. In the cathodic potential domain, a nearly direct formation of dissolved Al3+ was observed, while in the anodic potential domain the Al dissolution occurred by migration across a ZnO/Zn(OH)2 film. It was demonstrated that this methodology can be applied to a nonstationary system during a potentiostatic experiment for a lower Al content phase (Zn-22 wt.% Al). The nature of the charge transfer mechanisms depended on the applied potential and could be identified by comparing the direct current and alternating current faradaic yield using AESEC-EIS.
Similar content being viewed by others

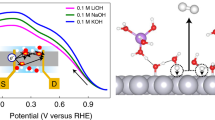

Introduction
Since its introduction to the corrosion field by Epelboin et al.1, electrochemical impedance spectroscopy (EIS) has proven itself an essential and ubiquitous technique in corrosion research. For steady-state corroding systems, EIS may provide a direct measurement of the corrosion rate1, and the form of the EIS spectrum may reveal the underlying mechanisms resolved over a wide frequency range of time constants2. The advantage of the technique is that it involves only a small amplitude electrochemical perturbation of the system avoiding many of the problems associated with linear scan techniques. The difficulties of EIS alone are the inability to identify the specific faradaic reactions that occur during charge transfer and the requirement of steady-state system. To resolve these difficulties a variety of couplings, such as EIS–Raman spectroscopy3 have been developed. The nature of the faradaic processes may be revealed by correlating the Raman emission with the modulated current and frequency. The theoretical development of such techniques has been addressed in the development of a generalized EIS transfer function4.
Atomic emission spectroelectrochemistry (AESEC) provides a direct measurement of elemental dissolution rates5. One of the difficulties of this technique is that dissolution may be directly related to a faradaic process, weakly related as in the case of anodic dissolution by way of an oxide intermediate, or unrelated as when dissolution is due to a non-faradaic process. The correlation of electrochemical current transients with dissolution transients is one means of distinguishing between these possibilities as demonstrated by Jiang et al.6,7 for the dissolution of Zn in the presence of conversion coatings. The coupling of EIS and AESEC allows for a more sophisticated analysis of dissolution kinetics; the oscillating elemental dissolution rates may be related to the oscillating current and low-frequency EIS data may be decomposed into elemental faradaic processes. Further, nonsteady-state systems may be investigated by considering the kinetic data in the time domain. A detailed model for AESEC-EIS was developed by Shkirskiy and Ogle8, directly correlating the elemental dissolution rate and electrical current for pure Zn at open circuit potential. Zn dissolution in 0.1 M NH4Cl occurred without any significant intermediate film formation as evidenced by the Zn dissolution rate being in-phase with the alternating current (AC) component of the electrical current. In 0.5 M NaCl solution, no correlation between the AC and the Zn dissolution rate was observed, indicating that oxide film formation and dissolution were decoupled faradaic and non-faradaic processes, respectively. The goal of this article is to extend the previous work to a multielement system and to use the EIS spectra to distinguish different mechanisms of dissolution by taking advantage of the time-resolved measurement of each element of a multielement system that AESEC technique provides.
The Al-Zn alloy system in alkaline solution was considered an ideal system for an early demonstration of the method. The system is industrially significant: Al-Zn alloys are commonly used as galvanic coatings on steel and occur as separate phases in Zn containing Al alloys, such as the 6000 series, and may be exposed to alkaline solution in their lifetime either during surface treatment by alkaline etching, or during corrosion when the cathodic reaction leads to local pH changes. The mechanisms of anodic and cathodic dissolution of multiphase Zn-5 wt.% Al alloy coatings on steel were previously investigated9,10. A complex interaction between the Zn and Al was observed; however, interpretation of the mechanisms of these interactions was hindered due to the multiphase nature of the commercial coating material. Therefore, to simplify, we isolated the chemistry of a single phase; the α-phase of Al-Zn (Zn-68 wt.% Al). The anodic dissolution of Al and Zn were investigated as a function of potential in slightly alkaline (pH = 10.1)11 and alkaline (pH = 12.8)12 electrolytes. In the latter, the temperature dependence of anodic dissolution was also investigated13.
In summary of these results, three potential domains were identified where the dissolution of the material occurred by very different mechanisms. Based on the individual Zn and Al dissolution rate results and the electrochemical current, it was found that Al underwent a direct, potential independent dissolution mechanism in the cathodic potential domain by reacting with H2O, and without the formation of a significant intermediate oxide film. Metallic Zn accumulated on the surface in a mechanism of cathodic dealloying. In the anodic domain, Zn was transformed into an oxide film and Al dissolution occurred by ionic transfer across the film. The previous work used the AESEC technique to distinguish the different dissolution mechanisms, such as noise signal and potential dependency of each dissolution rate. As the two mechanisms are very different, it was felt that EIS coupled to AESEC might further confirm these mechanisms and allow a higher degree of precision on the mechanistic details. In this work, a direct comparison between electrical current and elemental dissolution rate of each element during the AESEC-EIS measurement is given. In this way, the dissolution mechanisms of individual alloying elements may be resolved.
Results
Overview of Al5.2Zn mechanisms as a function of potential
The sensitivity of the Al-Zn dissolution mechanism to potential is evident from the AESEC-linear sweep voltammetry (AESEC-LSV) curve of Al5.2Zn (Zn-68 wt.% Al) phase in 0.1 M NaOH solution, shown in Fig. 1. Elemental dissolution rates (jM) and the convoluted electrical current density (je*) as a function of potential are given with 0.5 mV s−1 scan rate. As previously described, the polarization curve shows three distinctive potential domains, to which can be ascribed three unique mechanisms of dissolution. The cathodic dealloying domain occurs between −1.72 V to −1.35 V vs. Hg/HgO, where the dealloyed metallic Zn-enriched layer, Zn(0), is formed by the preferential Al dissolution12. It is characterized by an intense Al dissolution rate, jAl, and a Zn dissolution rate, jZn, close to the detection limit. A direct reaction between Al and H2O was inferred with significant hydrogen evolution reaction (HER)14 and accumulation of metallic Zn.

Elemental current densities (jM, M = Zn or Al), and convoluted electrical current density (je*) as a function of potential in 0.1 M NaOH, pH = 12.80, Ar deaerated electrolyte at T = 25 °C. Vertical dashed lines are selected potential values in the (a): cathodic dealloying, (b): anodic, and (c): intermediate domain.
Al dissolution rate was potential independent and in fact, did not reflect or correlate in anyway with the electrochemical current the magnitude of which was much lower and changed sign during this potential domain.
In the anodic domain (from −0.9 V to −0.4 V vs. Hg/HgO) where a potential independent Zn dissolution is observed, a totally different mechanism is apparent evidenced by the nearly congruent dissolution of Zn and Al, and the significantly decreased amplitude of perturbations in the jAl signal. The direct current (DC) faradaic yield of elemental dissolution may be determined as ηDC = jΣ/je* when cathodic current contribution is negligible, where jΣ = jZn + jAl. In this case, the dissolution rate follows the electrical current with nearly a 100% DC faradaic yield (ηDC = 0.98) of dissolution. The potential independence of Zn and Al dissolution rates suggests an oxide film formation type of mechanism and indeed, Raman spectroscopy and scanning electron microscopy (SEM) analysis also demonstrated the formation of ZnO layer in this potential domain11,12.
The elemental dissolution rates in the intermediate domain (from the onset of Zn dissolution −1.35 V vs. Hg/HO to the starting potential of anodic domain −0.90 V vs. Hg/HgO), is more complex. It involves the accumulated metallic Zn-enriched layer that was formed in the cathodic domain by the preferential Al dissolution. At the onset of Zn dissolution, there is a notable decrease in the jAl, that we previously termed a negative correlation effect (NCE) in ref. 12. The jZn reached its maximum value in approximately the same potential domain as jAl obtained its minimum value.
AESEC-EIS
To corroborate and gain further insight into the proposed dissolution mechanisms, potentiostatic EIS was performed simultaneously with AESEC in the three potential domains, at potentials indicated as (a), (b), and (c) in Fig. 1. The Nyquist plots are presented in Fig. 2, and the corresponding AESEC-EIS dissolution profiles are given in Fig. 3a–c. The effective oxide capacitance (Cδ) values were determined from the Cole–Cole representation of the complex capacitance curve at high frequency. In this way, the capacitance may be evaluated regardless of any time constant distribution within the oxide film15. The capacitance values, the constant phase element (CPE) parameters (α and Q), and the corresponding effective oxide layer thicknesses (δ) for each potential domain are summarized in Table 1.

Potentiostatic EIS of the Al5.2Zn phase at a −1.70 V vs. Hg/HgO, b −0.80 V vs. Hg/HgO, and c −1.20 V vs. Hg/HgO. Arrows are indicating frequency values (Hz). SEM images after the EIS experiment at each potential domain are given.

jM, je*, and j∆ during AESEC-EIS measurement in 0.1 M NaOH, Ar deaerated electrolyte including potentiostatic hold (Eap), EIS, and open circuit measurement (Eoc). a Eap = −1.70 V vs. Hg/HgO, b Eap = −0.80 V vs. Hg/HgO, and c Eap = −1.20 V vs. Hg/HgO. jM values are normalized based on the molar composition for b and c.
The cathodic dealloying domain (−1.72 V to −1.35 V vs. Hg/HgO)
EIS in the cathodic dealloying domain (Fig. 2a) revealed a single time constant, suggesting that H2O reduction reaction was the dominate charge transfer reaction. The dissolution profile obtained by AESEC (Fig. 3a) again demonstrates the potential independence of Al dissolution, as the potential cycles at low frequency do not appear as oscillations in the elemental dissolution rates. This is an important conclusion because it indicates that the EIS spectra are revealing information on the interfacial electrochemical processes that may not directly affect the dissolution rate.
The analysis of the high-frequency loop shows a CPE behavior, where α = 0.73 and Q = (1.07 ± 0.20) × 10–3 Ω−1 cm−2 sα−1, estimated from a graphical analysis16. Assuming that this time constant is attributed to a 2D frequency distribution17, Brug’s relationships18 allow an estimation of the equivalent capacitance from the CPE parameters, in this case the double layer capacitance, Cdl = 191 μF cm−2. This value is about an order of magnitude larger than the expected value for a double layer capacitance and could represent the response of a very thin, perhaps a nonuniform oxide film. It is reasonable to suppose that Al(OH)3 forms as a short-lived intermediate in Reaction 1 as previously discussed12,13. Reaction 1 may be broken down into more elementary steps as follows:
AESEC directly measures Reaction 4, which involves no electron transfer and in principle should be potential independent. The EIS results indicate that water reduction was potential dependent (Fig. 2a). This confirms the conclusions of ref. 13; the rate-determining step (RDS) is the dissolution of the Al(OH)3 layer (Reaction 4) as previously reported19,20,21,22. At steady state, Reactions 3 and 4 must occur at identical rates. The reaction rate is probably determined by the film properties that control the access of H2O to metallic Al, thereby controlling the cathodic reaction rate (Eq. 2). The presence of the film may not be directly detected by AESEC due to the intense dissolution rate and cathodic current; however, the film was detected indirectly by EIS. The cathodic current resulting from the HER may be determined simply as j∆ = je* − jΣ = jc, shown in Fig. 3a.
The anodic domain (−0.9 V to −0.4 V vs. Hg/HgO)
In the anodic domain, the polarization curve (Fig. 1) demonstrates that Zn and Al dissolution rates, and the electrochemical current are stable and nearly independent of potential. In the EIS spectrum (Fig. 2b), three time constants are observed. The high-frequency capacitive loop may be attributed to the charge transfer resistance of Zn and/or Al oxidation in parallel with the interfacial capacitance. In this system, independent anodic dissolutions from Zn and Al simultaneously occur at different rate, and thus the admittance is the sum of these contributions. This may explain why the two entangled time constants are observed in the high-frequency domain. The low-frequency loop shows a nearly 45° phase shift that suggests a diffusion process23,24. The time constant observed at intermediate frequency (f = ~1 Hz) is poorly resolved due to overlap with the high- and low-frequency contributions, and will not be discussed.
The high-frequency capacitive loop is flattened and this can be described by a CPE in parallel with the charge transfer resistance. We assume that this CPE is due to the distribution of properties in a thin oxide film16, for instance, the distribution of resistivity according to a power-law model25,26. From the analysis of the complex capacitance calculated from the impedance data15, the high-frequency limit of the of the capacitance was Cδ = 0.65 ± 0.05 μF cm−2. Such a low value is characteristic of the dielectric response of a thin oxide film. Assuming a dielectric constant ε = 8.8 ± 0.8 for ZnO (ref. 27) the thickness of the thin oxide film, δ, formed at the electrode interface is given by:
where ε0 is the vacuum permittivity (8.85×10−14 F cm−1). This equation yields δ = 12 ± 1 nm. Such a value confirms the hypothesis that the interfacial capacitance is governed by the thin oxide film contribution (i.e., the double layer capacitance that is in series with the capacitive contribution of the oxide film, has in this case a negligible contribution).
The presence and characteristics of the oxide film may be further refined by consideration of the low-frequency capacitive loop. For the AESEC-EIS (Fig. 3b), very slight oscillations are observed in the Zn and Al dissolution rates at low frequency (magnified curve is given in the inset). The faradaic yield of dissolution for the total current, ηDC = 0.96 ± 0.03 close to 1, in agreement with ref. 12. The faradaic yield of the AC component, ηAC, is defined as:
From the data in Fig. 3b, ηAC = 1.01 ± 0.05, reasonably identical to ηDC within experimental error. Note that error bar of ηAC was obtained from different frequency domains. It appears to be distributed essentially in the Al dissolution although slight peaks above background are observed for Zn dissolution as well.
The second capacitive loop in Fig. 2b shows a typical diffusion-controlled charge transfer mechanism through an oxide layer24. The characteristic frequency, fc = 0.032 Hz, at the apex of the time constant of the diffusion process in the Nernst layer may be expressed as:
where δ is the thickness of the corrosion product, that can be in a first approximation evaluated to be at least the thickness previously obtained from the CPE parameter (δ = 12 nm). The diffusion coefficient may be calculated from Eq. 7, D = 1.15 × 10−13 cm2 s−1. Such a low value may be due to an underestimated δ value and surface area because the calculation did not consider the porosity of the oxide, as well as the accumulation of corrosion products at the electrode surface. It may be attributed to a diffusion of species inside of a micrometric diffusion layer of corrosion product. In this case, the diffusion and migration caused by the large electric filed inside the film should be taken into account.
Figure 4 shows the oscillation trends of the je, je*, jM, and applied potential (E) at a given frequency of AESEC-EIS result from Fig. 3. To facilitate the comparison, each profile is presented in arbitrary units. The phase shift of jM and je* vs. E (\(\phi _{{\mathrm{j}}_{\mathrm{M}}}\)) results from the residence time distribution of the flow cell5, which means that it is not frequency dependent but time dependent. The actual phase shift between E and je (ϕ) was nearly zero in all cases as shown in Fig. 4.

jM, je, je*, and potential (E) as a function of time at a given frequency for a Eap = −0.80 V vs. Hg/HgO and b Eap = −1.20 V vs. Hg/HgO from Fig. 3b, c, respectively.
The impedance contribution from each elemental dissolution in real part, Zr (jM), is given in Table 2, calculated at each frequency domain as:
For Eap = −0.80 V vs. Hg/HgO (Fig. 4a), jZn oscillation is less evident than at Eap = −1.20 V vs. Hg/HgO (Fig. 4b), probably indicating a weak potential dependent Zn dissolution in the anodic potential domain due to Zn-based corrosion product formation. The high Zr (jZn) values at Eap = −0.80 V vs. Hg/HgO could also be explained by the weak potential dependency of Zn dissolution at this potential. For Eap = −1.20 V vs. Hg/HgO, Zr (jZn) + Zr (jAl) ≈ Zr–Re, which may indicate that jZn and jAl contributed to the total impedance with a parallel relationship (Re is the electrolyte resistance). It is consistent with the previously proposed dissolution model of the Al5.2Zn phase, in which Al dissolution occurs through a porous Zn(0) layer in parallel with Zn dissolution12. It should be mentioned that the dissolution is not a simple mechanism as it usually involves the adsorbed intermediate species that may result in a delay between the je* and jM.
The correlation between the AC current and the dissolution rates is highlighted by the Lissajous analysis in Fig. 5a (Eap = −0.80 V vs. Hg/HgO) and Fig. 5b (Eap = −1.20 V vs. Hg/HgO) at f = 0.004 Hz. The je* curve is shown on the top, both for the forward and reverse potential scans. For the jM vs. E curves in the middle, je* assuming congruent dissolution of Al and Zn are also shown. For example, in Fig. 5a, Al dissolution was congruent as jAl ≈ (je*/1.13), whereas Zn dissolution was not as jZn < (je*/8.8). The Zn dissolution rate correlated directly with potential although it was less obvious than the Al dissolution rate–potential correlation (also see Fig. 4a). In principle, at this potential, there is no significant cathodic reaction, so we conclude that the AC current is primarily going to the formation and reduction of ZnO or Zn(OH)2. It was previously shown that the ηAC ≈ ηDC ≈ 1 at Eap = −0.80 V vs. Hg/HgO, supporting this assumption. It is worth noting the weak correlation of Zn dissolution rate and potential since the overall faradaic yield of dissolution based on the DC current density and elemental dissolution rates was nearly 100% (ηDC = 0.96 ± 0.03) at this potential. In this case, it may be concluded that the DC component was due to the dissolution of the corrosion products6,7,28.

je*, jAl, jZn, and j∆ as a function of IR drop compensated applied potential (E–jeRe) at a Eap = −0.80 V vs. Hg/HgO and b Eap = −1.20 V vs. HgO. One cycle including upper and lower potential scans, indicated by arrows, is given.
The kinetic parameters for ZnO formation and reduction may be estimated from the jΔ vs. E Lissajous plot, shown in the bottom of Fig. 5. The Zn dissolution in the anodic potential domain (Fig. 5a) may be a series of elementary reactions as:
The intermediate species, such as ZnOH or ZnOHads (refs 29,30,31) are not considered as the time scale of the AESEC experiment is too slow to measure these species. The formation rate of Zn(OH)2 or ZnO (jΔ) is potential dependent (Reactions 9 and 11) given that the Lissajous plot of jΔ vs. E showed linearity, neglecting the contribution of cathodic current at this potential. The anodic Tafel slope of jΜ (ba, jM) may be calculated as:
where jM+ and jM− represent jM measured at the highest and lowest potential, respectively, obtained from Fig. 5. ΔE is the difference between the highest and lowest potential. \({{b}}_{{\mathrm{a}},\,{{j}}_\Delta }\) = 94 mV decade−1 and \({{b}}_{{\mathrm{a,}}\,j_{{\mathrm{Zn}}}}\) = 2350 mV decade−1 are calculated by Eq. 13. It is demonstrated that the formation of ZnO (Reaction 11) is faster than the dissolution (Reaction 12), resulting in the growth of the ZnO layer12,13.
The quantity of oxide formed during the AESEC-EIS experiment may be estimated by the dissolution profile obtained after releasing the potential to open circuit at the end of the experiment. The Eap = −0.80 V vs. Hg/HgO AESEC-EIS profile of Fig. 3b illustrates this idea. At the end of the experiment, the presence of oxide dissolution is indicated by a plateau in the Eoc vs. time profile at approximately Eoc = −1.5 V vs. Hg/HgO. The removal of the oxide and the exposure of the underlying metallic substrate is indicated by the sudden drop in potential to <−1.6 V vs. Hg/HgO. The removal of the oxide film gives rise to a transient dissolution of Zn, which decays steadily but at a rate significantly slower than the residence time distribution associated with the flow cell. The latter is indicated by the drop of the convoluted electrochemical current, je*. Integration of the jZn yields a thickness of ZnO layer δ = 11 nm, assuming a uniform ZnO layer formation over the geometrical surface area and a ZnO density of 5.6 g cm−3. This is in the same range as the oxide thickness estimated from EIS of 12 ± 1 nm (Table 1).
The intermediate domain (−1.35 V to −0.9 V vs. Hg/HgO)
In the intermediate domain, both jZn and jAl shows obvious in-phase oscillations, with the low-frequency AC current (Fig. 3c). The EIS response in Fig. 2c shows three capacitive time constants and one inductive loop. An inductive loop has been reported for both Zn and Al dissolution. In the case of Zn, this loop is usually observed in the intermediate frequency range24,32 whereas for Al, it can be only be seen in the low-frequency domain in an alkaline solution33,34. We thus conclude that the inductive loop observed for f < 10−2 Hz may be attributed to the relaxation of the surface intermediates involved in the dissolution mechanism of Al and Zn (ref. 35). The third capacitive loop is similar to the one observed at Eap = −0.80 V vs. Hg/HgO and may be ascribed to diffusion processes inside a thin layer. The characteristic frequency is also similar to the previous case in Fig. 2b, confirming that convection–diffusion-controlled mechanism by the flux of the electrolyte in the AESEC experiment.
The second capacitive loop (10−1 Hz < f < 100 Hz) was also distributed, and its analysis is complicated by the fact that it is convoluted with other processes at both higher and lower frequency. However, it may be ascribed to Zn dissolution24. The high-frequency time constant was analyzed as previously described. From the CPE behavior, the oxide film thickness was estimated at δ = 2.4 nm. The analysis of the high-frequency loop for each potential showed that the thin film formed on the alloy surface has a thickness that strongly depends on the applied potential.
In Fig. 3c, a peak of Zn dissolution is observed after the potential release, albeit a much smaller peak than at Eap = −0.80 V vs. Hg/HgO (Fig. 3b). Nevertheless, this peak cannot be unequivocally attributed to oxide dissolution as there is no clearly defined potential plateau following the potential release. Assuming that this peak was due to oxide dissolution, an estimated thickness of 2.5 nm is obtained with the same assumptions as before, in good agreement with that obtained from the EIS analysis of 2.4 ± 0.3 nm (Table 1).
The low-frequency Lissajous plots at Eap = −1.20 V vs. Hg/HgO (Fig. 5b) show linear behavior within experimental error, indicating that the elemental dissolution current (jM) for each species was in-phase with the potential. In this case, the linearity of the Lissajous plots of jZn and jΔ is clearly demonstrated, indicating that both reactions are charge transfer limited. The cathodic current would make a negligible contribution to je*, as Eap was ~+400 mV than the zero current potential (Ej = 0). The Zn oxidation reaction may be written by the combination of Reactions 9 and 10 as:
and the formation of the ZnO (jΔ) is via Reaction 11. \({\mathrm{b}}_{{\mathrm{a,}\,j}_\Delta } = 68\,{\mathrm{mV}}\) decade−1 and \({\mathrm{b}}_{{\mathrm{a,}}\,j_{{\mathrm{Zn}}}} = {\mathrm{864}}\,{\mathrm{mV}}\) decade−1 were obtained by Eq. 13 from Fig. 5b. Given that the Tafel slope of jZn at Eap = −1.20 V vs. Hg/HgO (864 mV decade−1) is lower than that at Eap = −0.80 V vs. Hg/HgO (2350 mV decade−1), it could be concluded that the dissolution of Zn is faster in the former case, resulting in a relatively thinner ZnO layer formation.
In this case, both Zn and Al dissolution were not faradaic in that je*/1.13 ≠ jAl and je*/8.8 ≠ jZn (Fig. 5b). It showed a significant difference between ηDC = 0.92 ± 0.02 and ηAC = 0.65 ± 0.03. It was proposed that the AC component is due to non-faradaic reactions at the metal/oxide interface and the DC component to the faradaic dissolution of the film at the oxide/electrolyte interface6,7,28. For the AC case in the intermediate domain, it may be concluded that the formation of the corrosion product would be the RDS, given that ηDC >> ηAC, consistent with the Tafel slope analysis. A simplified elemental dissolution at each interface in the anodic potential domain is illustrated in Fig. 6. Note that the potential dependent and potential independent mechanisms are observed for two different elements of a material/electrolyte combination. In the previous work with AESEC-EIS, they were observed for pure Zn in two different electrolytes8.

Simplified model of elemental dissolution in anodic potential domain at each interface of AlxZn in 0.1 M NaOH solution.
It is worth noting that jZn was higher than its congruent dissolution level (je*/8.8), whereas jAl was lower than je*/1.13. The excess Zn dissolution could lead to a restrained Al dissolution, referenced as the NCE12.
Al0.7Zn (Zn-22 wt.% Al)
In this section, we will demonstrate the AESEC-EIS methodology with a nonstationary Zn-Al system during potentiostatic measurement36. An AESEC-EIS curve (Fig. 7a), magnified oscillation trends at f = 0.010 Hz (Fig. 7b) and a corresponding Nyquist plot (Fig. 7c) for the Al0.7Zn (Zn-22 wt.% Al) nominally pure phase at Eap = −1.36 V vs. Hg/HgO (i.e., +150 mV from Ej = 0) in the same electrolyte are given. It is demonstrated from Fig. 7a that the elemental dissolution mechanism varied throughout the time period of the experiment at a constant potential. Initially, Zn dissolution was intense at approximately the same level as Al. However, for t > 2700 s (f < 0.005 Hz), jZn decreased to under the detection limit and Al dissolution changed from a charge transfer to a chemical dissolution mechanism. The latter is indicated by the disappearing oscillation of jAl, and the significantly increased jAl noise signal resulted from bubble formation by the HER12,14. The non-charge transfer Al dissolution mechanism is also revealed in the Nyquist plot (Fig. 7c), where an unambiguous diffusive impedance trend was observed (ϕ = 45°) in this frequency domain. ZnO/Zn(OH)2 formation and dissolution would occur for f ≥ 0.007 Hz as Reactions 9–12.

a AESEC-EIS curve at Eap = −1.36 V vs. Hg/HgO in 0.1 M NaOH, Ar deaerated electrolyte, b oscillation trends at f = 0.010 Hz from a, and c corresponding Nyquist plot.
Oscillations were observed in jZn beginning at a relatively higher frequency domain (f ≈ 0.030 Hz), where no jAl oscillation was observed. Oscillations in jAl disappeared with the dissolution of ZnO/Zn(OH)2 for t > 2700 s, indicating that Al dissolution occurred through this Zn-based corrosion product layer (see also Fig. 6). The phase shift of E vs. je (ϕ) was not zero as summarized in Table 3, whereas ϕ ≈ 0 for Al5.2Zn (Fig. 4). The measured total impedance was close to the sum of each elemental contribution calculated from Eq. 8, Zr– Re ≈ Zr (jZn) + Zr (jAl), similar to the Al5.2Zn in the same potential domain, which can be an indicative of the parallel relationship between Zn and Al dissolution. The phase shift between jZn and jAl was 180° (Fig. 7b), whereas in-phase relation was monitored for Al5.2Zn (Fig. 4). The out-of-phase of jZn and jAl should have been explained by the fact that Al dissolution is related to the cathodic reaction at open circuit potential28. However, this is unlikely in the present work because this phase shift was observed at a relatively positive potential, where the cathodic current contribution is negligible. One possible explanation is that in this potential domain, the slope of the Al dissolution rate–potential curve is negative36 resulting in a negative low-frequency impedance37,38.
For f ≤ 0.007 Hz, jZn decreased to under the detection limit as all the previously formed ZnO/Zn(OH)2 dissolves. Previously for Al5.2Zn phase, an obvious Zn dissolution peak was observed during the spontaneous dissolution after the AESEC-EIS experiments in Fig. 3b, c. It was attributed to the residual ZnO/Zn(OH)2 dissolution formed during the potentiostatic experiment. In Fig. 7, no jZn peak was monitored in Eoc indicating that the oxide was completely dissolved near t = 2700 s.
Discussion
In this work, we have demonstrated the application of combined AESEC-EIS to identify and quantify different anodic dissolution processes for a multielement system, Al-Zn pure phase in 0.1 M NaOH. The elemental dissolution mechanism at each potential domain was elucidated by the AESEC-EIS technique.
The AESEC method provided immediate information on the elemental dissolution rates and transient behavior, while EIS gave information on the physical properties of intermediate oxide layers and kinetic information for water reduction in the cathodic domain. In the cathodic potential domain (Eap = −1.70 V vs. Hg/HgO), Al dissolution was found to be potential independent and occurred with the formation of a negligible oxide layer.
By comparing the DC and AC faradaic yield, information on the localization of charge transfer reactions could be obtained. In the anodic domain (Eap = −0.80 V vs. Hg/HgO), ηDC ≈ ηAC indicating that the formation and dissolution of the corrosion products occur at the nearly same rate, consistent with previous observations12,13. In the intermediate domain (Eap = −1.20 V vs. Hg/HgO), formation would be the RDS as ηDC >> ηAC. In the former case, the oxidation of metal occurred through a corrosion product layer. In the latter case, the oxidation occurred directly from the metal/oxide complex. It was further demonstrated by the Tafel slope of jZn from the elemental Lissajous analysis (Fig. 5). In this way, AESEC-EIS can be utilized to distinguish the different elemental dissolution kinetics. Diffusion processes were identified although the precise origin was not ascertained, i.e., across corrosion product films or within intergranular crevices and pits.
Elemental impedance contribution, Zr (jM), demonstrated the potential dependent elemental dissolution model of Al-Zn pure phase proposed in ref. 12. In the intermediate potential domain, Al dissolution occur through a porous Zn(0)-enriched layer in parallel with Zn dissolution as Zr (jZn) + Zr (jAl) ≈ total measured impedance (Zr– Re).
For Al5.2Zn, Al and Zn dissolution rates were in-phase regardless of the applied potential. For Al0.7Zn and other low Al content alloys and phases36, Al and Zn dissolution rates showed a 180° phase shift. However, the origin of the phase shift is not ascertained to the satisfactory level.
Methods
Materials
Nominally pure phases of Al-Zn (Zn-68 wt.% Al and Zn-22 wt.% Al), provided and characterized by the Department of Metals and Corrosion Engineering, University of Chemistry and Technology, Prague, were investigated in this work. Zn-68 wt.% Al phase is denoted as Al5.2Zn and Zn-22 wt.% Al as Al0.7Zn, based on their molar compositions to facilitate the congruent dissolution analysis. The chemical composition of these materials were 67.6 wt.% Al and 32.4 wt.% Zn (Al5.2Zn), and 21.4 wt.% Al and 78.6 wt.% Zn (Al0.7Zn), respectively, measured by atomic absorption spectroscopy. The Al5.2Zn contains mainly α-phase of Al with only a trace of η-phase of Zn characterized by X-ray diffraction spectroscopy (XRD). A single β-phase of Al was characterized for Al0.7Zn after solution annealing and water quenching. However, this phase decomposes at room temperature into α-phase of Al and η-phase of Zn, also characterized by XRD. All samples were ground with Si-C paper up to P4000 under ethanol, dried with flowing N2 gas then stored in a humidity chamber of 50% relative humidity with saturated Mg(NO3)2·6H2O during 24 h, providing a reproducible native oxide formation on the surface after the sample polishing process36,39. All the experiments were performed in a 0.1 M NaOH (pH = 12.80) at T = 25 °C, prepared from analytical grade materials using deionized water obtained by a MilliporeTM system (18.2 MΩ cm). The electrolytes were deaerated by Ar gas for 30 min prior to the tests and maintained during the experiments.
Atomic emission spectroelectrochemistry
The AESEC technique has been described in detail elsewhere5,40. The working electrode was in contact with the flowing electrolyte in a specially designed flow cell5,41 with conventional three-electrode system; a Hg/HgO in 0.1 M NaOH (−165 mV vs. SHE) as a reference electrode and a Pt foil as a counter electrode. The elements released from the working electrode were transported to an Ultima 2 C Horiba Jobin-Yvon inductively coupled plasma atomic emission spectrometer (ICP-AES). The concentrations of Zn and Al were determined from the emission intensity at 213.86 nm (Zn with a Paschen-Runge polychromator) and at 167.08 nm (Al with a monochromator) wavelength, respectively, using standard ICP-AES calibration techniques.
A Gamry Reference 600TM potentiostat was used to perform EIS and LSV. The electrochemical response (namely the electrical current density, je, and the electrode potential, E) were recorded in real-time with the elemental dissolution rates, using the analog data acquisition and the software interface (QuantumTM, Horiba Jobin-Yvon) of the AESEC technique. The EIS was performed at different potentials from 100 kHz to 0.004 Hz with eight points per decade of frequency and applying a 10 mVrms sinewave perturbation. All the AESEC-EIS experiments were repeated at least three times.
Data analysis of the AESEC technique
The atomic emission intensity at a characteristic wavelength (λ) of the element M, IM,λ, was recorded by the ICP-AES as a function of time downstream from an electrochemical flow cell. The elemental concentration (CM) is calculated as:
where IM,λ° is the background signal, and is κλ the sensitivity factor of M, obtained from a standard ICP calibration method. The elemental dissolution rate (vM) can be calculated from Eq. 15 with the flow rate of the electrolyte (f = 2.8 mL min−1) and the exposed surface area A (1.0 cm2) as:
The vM was converted to an equivalent elemental current density (jM) to facilitate comparison with the electrical current density (je) measured by the potentiostat, in a relationship with
where F is the Faraday constant and z is the valance of the dissolving species (Al3+, Zn2+). It is often useful to present je* that represents the measured je after a numerical convolution with the residence time distribution in the flow cell (a lognormal distribution), thereby allowing a direct comparison between the instantaneous values of je* and jM (ref. 5). Cathodic reactions and the formation of insoluble or slightly soluble species are not directly detected by ICP-AES. If surface charging is neglected, the faradaic component of these hidden processes may be determined by a mass–charge balance as:
Surface characterization
The sample surface after each potentiostatic AESEC-EIS experiment was characterized by SEM using a Zeiss LEO 1530TM microscope with field emission gun source at 15 keV, and with 15 mm working distance. An equal mix of secondary electron and back-scattered electron detector was used.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper. The Supplementary Data will be available upon reasonable request.
References
Epelboin, I., Keddam, M. & Takenouti, H. Use of impedance measurements for the determination of the instant rate of metal corrosion. J. Appl. Electrochem. 2, 71–79 (1972).
Keddam, M., Mattos, O. R. & Takenouti, H. Mechanism of anodic dissolution of iron-chromium alloys investigated by electrode impedances - II. Elaboration of the reaction model. Electrochim. Acta 31, 1159–1165 (1986).
Wang, X. et al. A new transfer function in electrochemistry: dynamic coupling between Raman spectroscopy and electrochemical impedance spectroscopy. Electrochim. Acta 55, 6299–6307 (2010).
Gabrielli, C. & Tribollet, B. A transfer function approach for a generalized electrochemical impedance spectroscopy. J. Electrochem. Soc. 141, 1147–1157 (1994).
Ogle, K. Atomic emission spectroelectrochemistry: real time rate measurements of dissolution, corrosion and passivation. Corrosion 75, 1398–1419 (2019).
Jiang, L., Wolpers, M., Volovitch, P. & Ogle, K. The degradation of phosphate conversion coatings by electrochemically generated hydroxide. Corros. Sci. 55, 76–89 (2012).
Jiang, L., Wolpers, M., Volovitch, P. & Ogle, K. An atomic emission spectroelectrochemical study of passive film formation and dissolution on galvanized steel treated with silicate conversion coating. Surf. Coat. Tech. 206, 3151–3157 (2012).
Shkirskiy, V. & Ogle, K. A novel coupling of electrochemical impedance spectroscopy with atomic emission spectroelectrochemistry: application to the open circuit dissolution of zinc. Electrochim. Acta 168, 167–172 (2015).
Vu, T. N., Volovitch, P. & Ogle, K. The effect of pH on the selective dissolution of Zn and Al from Zn–Al coatings on steel. Corros. Sci. 67, 42–49 (2013).
Vu, T. N., Mokaddem, M., Volovitch, P. & Ogle, K. The anodic dissolution of zinc and zinc alloys in alkaline solution. II. Al and Zn partial dissolution from 5% Al–Zn coatings. Electrochim. Acta 74, 130–138 (2012).
Han, J. & Ogle, K. Cathodic dealloying of α-phase Al-Zn in slightly alkaline chloride electrolyte and its consequence for corrosion resistance. J. Electrochem. Soc. 165, C334–C342 (2018).
Han, J. & Ogle, K. The anodic and cathodic dissolution of α-phase Zn-68Al in alkaline media. Corros. Sci. 148, 1–11 (2019).
Han, J., Thierry, D. & Ogle, K. Temperature dependence of the passivation and dissolution of Al, Zn and α-phase Zn-68Al. Corrosion 75, 68–78 (2019).
Han, J. & Ogle, K. Hydrogen evolution and elemental dissolution by combined gravimetric method and atomic emission spectroelectrochemistry. J. Electrochem. Soc. 166, C3068–C3070 (2019).
Benoit, M. et al. Comparison of different methods for measuring the passive film thickness on metals. Electrochem. Acta 201, 340–347 (2016).
Orazem, M. E., Pébère, N. & Tribollet, B. Enhanced graphical representation of electrochemical impedance data. J. Electrochem. Soc. 153, B129–B139 (2006).
Hirschorn, B. et al. Determination of effective capacitance and film thickness from constant-phase-element parameters. Electrochim. Acta 55, 6218–6227 (2010).
Brug, G. J., Van Den Eeden, A. L. G., Sluyters-Rehbach, M. & Sluyters, J. H. The analysis of electrode impedance complicated by the presence of a constant phase element. J. Electroanal. Chem. 176, 275–295 (1984).
Zhang, J., Klasky, M. & Letellier, B. C. The aluminum chemistry and corrosion in alkaline solutions. J. Nucl. Mater. 384, 175–189 (2009).
Chu, D. & Savinell, R. F. Experimental data on aluminum dissolution in KOH electrolytes. Electrochim. Acta 36, 1631–1638 (1991).
Moon, S. M. & Pyun, S. I. The corrosion of pure aluminium during cathodic polarization in aqueous solutions. Corros. Sci. 39, 399–408 (1997).
Pyun, S. & Moon, S. Corrosion mechanism of pure aluminium in aqueous solution”. J. Solid State Electrochem. 4, 267–272 (2000).
Cachet, C., Ströder, U. & Wiart, R. The kinetics of zinc electrode in alkaline zincate electrolytes. Electrochim. Acta 27, 903–908 (1982).
Cachet, C., De Pauli, C. P. & Wiart, R. The passivation of zinc in slightly alkaline solutions. Corros. Sci. 25, 493–502 (1985).
Hirschorn, B. et al. Constant-phase-element behavior caused by resistivity distributions in films: II. Applications. J. Electrochem. Soc. 157, C458–C462 (2010).
Hirschorn, B. et al. Constant-phase-element behavior caused by resistivity distributions in films: I. Theory. J. Electrochem. Soc. 157, C452–C457 (2010).
Madelung, O., Rössler, U. & Schulz M. Zinc oxide (ZnO) dielectric constants: Landolt-Börnstein – Group III Condensed Matter, Vol. 41B (Springer, Berlin Heidelberg, 1999).
Shkirskiy, V., Maltseva, A., Ogle, K. & Volovitch, P. Environmental effects on selective dissolution from ZnAlMg alloy under low frequency alternating current perturbations. Electrochim. Acta 238, 397–409 (2017).
Bockris, J. O. ’M., Nagy, Z. & Damjanovic, A. On the deposition and dissolution of zinc in alkaline solutions. J. Electrochem. Soc. 119, 285–295 (1975).
Cachet, C., Saïdani, B. & Wiart, R. The behavior of zinc electrode in alkaline electrolytes I. A kinetic analysis of cathodic deposition. J. Electrochem. Soc. 138, 678–687 (1991).
Thomas, S., Birbilis, N., Venkatraman, M. S. & Cole, I. S. Corrosion of zinc as a function of pH. Corrosion 68, 015009-1-015009-9 (2012).
Armstrong, R. D. & Bell, M. F. The active dissolution of zinc in alkaline solution. Electroanal. Chem. Interf. Electrochem. 55, 201–211 (1974).
de Wit, J. H. W. & Lenderink, H. J. W. Electrochemical impedance spectroscopy as a tool to obtain mechanistic information on the passive behaviour of aluminium. Electrochim. Acta 41, 1111–1119 (1996).
Lnderink, H. J. W., Linden, M. V. D. & de Wit, J. H. W. Corrosion of aluminium in acidic and neutral solutions. Electrochim. Acta 38, 1989–1992 (1993).
Gudić, S., Radošević, J. & Kliškić, M. Study of passivation of Al and Al–Sn alloys in borate buffer solutions using electrochemical impedance spectroscopy. Electrochim. Acta 47, 3009–3016 (2002).
Han, J. The electrochemistry and dealloying of Zn-Mg/Zn-Al pure phases and their significance for corrosion of Zn-Al-Mg alloys. PSL Research University, tel-02860750 (2018).
Epelboin, I., Keddam, M. & Lestrade, J. C. Faradaic impedances and intermediates in electrochemical reactions. Faraday Discuss. Chem. Soc. 56, 264–275 (1973).
Annergren, I., Keddam, M., Takenouti, H. & Thierry, D. Modeling of the passivation mechanism of Fe-Cr binary alloys from ac impedance and frequency resolved rrde-I. Behavior of Fe-Cr alloys in 0.5M H2SO4. Electrochim. Acta 41, 1121–1135 (1996).
Stoulil, J. et al. Electrochemical properties of corrosion products formed on Zn-Mg, Zn-Al and Zn-Al-Mg coatings in model atmospheric conditions. Mat. Corros. 66, 777–782 (2015).
Ogle, K. & Weber, S. Anodic dissolution of 304 stainless steel using atomic emission spectroelectrochemistry. J. Electrochem. Soc. 147, 1770–1780 (2000).
Ogle, K., Lodi, P. & Storhaye, A. Méthode d’analyse d’un échantillon métallique par dissolution de sa surface, et dispositif pour sa mise en œuvre. Patent, FR2689244A1 (1992).
Acknowledgements
The authors would like to appreciate to Dr. Jan Stoulil, University of Chemistry and Technology in Prague, Czech Republic for supplying Zn-22 wt.% Al and Zn-68 wt.% Al samples.
Author information
Authors and Affiliations
Contributions
J.H. carried out the AESEC-EIS experiments and coordinated analysis between two techniques. V.V. was responsible for the EIS data interpretation. K.O. conceptualized the AESEC-EIS analysis. All authors contributed to write the paper and revision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, J., Vivier, V. & Ogle, K. Refining anodic and cathodic dissolution mechanisms: combined AESEC-EIS applied to Al-Zn pure phase in alkaline solution. npj Mater Degrad 4, 19 (2020). https://doi.org/10.1038/s41529-020-0123-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-020-0123-0
