
Abstract
Challenges in the understanding of three-dimensional (3D) brain networks by simultaneously recording both surface and intracortical areas of brain signals remain due to the difficulties of constructing mechanical design and spatial limitations of the implanted sites. Here, we present a foldable and flexible 3D neural prosthetic that facilitates the 3D mapping of complex neural circuits with high spatiotemporal dynamics from the intracortical to cortical region. This device is the tool to map the 3D neural transmission through sophisticatedly designed four flexible penetrating shanks and surface electrode arrays in one integrated system. We demonstrate the potential possibilities of identifying correlations of neural activities from the intracortical area to cortical regions through continuous monitoring of electrophysiological signals. We also exploited the structural properties of the device to record synchronized signals of single spikes evoked by unidirectional total whisker stimulation. This platform offers opportunities to clarify unpredictable 3D neural pathways and provides a next-generation neural interface.
Similar content being viewed by others


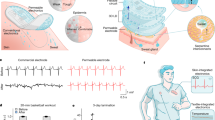
Introduction
The brain works with complex networks that contain thousands of neurons and plays a vital role in important life activities such as cognitive function, memory, or even communication1,2,3. Developments of neural interfaces capable of accurately mapping the complex activities of neural circuits are necessarily associated with the acquisition of clues to identifying diseases and unpredictable brain functions4,5,6. Among the methods of the neural interface, neuropathological analysis through an electrophysiological approach serves as a major strength as it provides an excellent temporal readout resolution compared to other approaches, such as magnetic resonance imaging, positron emission tomography, or confocal photon imaging of neuronal activity7,8,9,10,11. The electrophysiological approach to the diagnosis of neural diseases is broadly classified into three methods—(1) Electrocorticography (ECoG) arrays for surface recording12,13, (2) multi-shank Utah array to measure broadened intracortical area14, and (3) multi-channel Michigan probe15,16 to collect biological signals depending upon the layer of the deep target area of the brain.
Recently, while studies dealing with the correlation between signals from the surface and the intracortical area to find the pathological pathways of neural activities in the brain are attracting attention17,18, the demands for devices that measure these signals at the same time are also increasing19. The ultimate reason for the increased demands for this system is, that the clues of the electrophysiological signal provided by each region of the brain are totally different when diagnosing a neurological lesion. For example, ECoG arrays for surface recording provide high-resolution signals over a large area but cannot map the intracortical region of the brain. In contrast, penetrating probes such as the Utah Array or the Michigan probe for intracortical or deep brain recording do not provide clues about the propagation of expressed signals from inside the brain to the surface. This is because conventional ECoG surface array and penetrating probes sensing sites are designed and structurally aligned in a two-dimensional (2D) plane that collects information of only planar neural signaling and spikes at each insertion site.
Thus, several efforts to see brain signal transmission in three-dimensional (3D) have proceeded20,21,22,23,24,25. For example, Shin et al. implemented a multi-shank neural interface that provides neural signals in the deep brain in 3D by expanding the number of neural probes21. In addition, Liu et al. succeeded in the multi-site measurement of inner brain neural activity with an injectable, mesh-structured electrode array26. However, these studies failed to provide clues from the surface activity of the brain because they performed 3D recordings with a multi-channel electrode array in a limited space such as the ‘deep brain’. Other efforts have been made to prove the correlation between the surface and deep brain, by implanting ECoG array with inserting neural probe individually27,28. However, this recording system is severely restricted in temporal resolution because the time deviation of the electrophysiological signal from the respective devices occurs. Also, current implantable neural interface devices that rely primarily on rigid silicon-based probes or ECoG arrays formed from stiff materials have suffered from an inflammatory response, due to the mechanical mismatch between devices and soft brain tissue20,29,30. These rigid neural devices that interact with the soft brain exert great pressure on the tissue even with minute movements, leading to severe neuronal degeneration and tissue damage31,32,33,34,35. Therefore, the progress in the developments of flexible electronics concerning implantable neural interfaces establishes a platform with a negligible immune response and attenuated modulus between the interfaces. Flexible electronics provide conformal contact with living organs with minimal mechanical loading or a neural probe with a lower tethering force which has greatly raised the difficulty of analyzing the chronic function of neural activity36,37,38.
In summary, it is very important to develop a flexible device with extended recording coverage from the surface to the interior of the brain, because it gives an intuitive insight into neuro-dynamics. However, it was previously impossible to perform an accurate pathological study associated with 3D electrophysiological recording with current technology.
Here, we introduce the device achieved by extending the 3D structure of the neural interface in a way to accurately analyze complex neural signalings. The device establishes a single integrated platform with four flexible surface electrodes and penetrating probe by combining advantages of the aforementioned (1) ECoG arrays, (2) Utah array, and (3) Michigan probe for continuous and simultaneous recording of the surface and the intracortical activities of the brain. This system can simultaneously measure surface signals of the brain and neural activity occurring in the intracortical area, unlike previous studies that performed 3D ‘local’ electrophysiological recordings. In other words, a property of this system is that it performs neuronal transmission recording in different regions of the brain with a single device. A platform with a pop-up structure through the release of temporal elastomer substrate, forming flexible surface electrodes with penetrating probes simultaneously was elaborately designed. To implant the device into an intracortical area of the brain, penetrating shanks were temporarily hardened by coating the probes with biocompatible/bioresorbable stiffener, polyethylene glycol (PEG)39,40. It was confirmed that its mechanical/electrical properties were restored after the dissolution of PEG, confirming its biocompatibility in vivo and in vitro.
To determine whether the device can record electrophysiological signals in 3D structure, the local field potentials (LFPs) and the intracortical evoked single spikes of the somatosensory cortex through whisker stimulation in mice were simultaneously measured. Measurement of evoked single spikes with synchronized LFPs from unidirectional total whisker stimulation was also demonstrated, affirming the recording capability of our device in both the brain surface and the intracortical area.
Results
Design of the 3D structured and flexible neural electrode array
Figure 1a presents the schematic illustration of a foldable and flexible 3D neural mapping device. In electrophysiological heat mapping from this device, action potential signaling in neurons can be visually predicted as a blue- to the red-colored pathway as an example. This integrated 3D neural device includes the functions of ECoG Arrays, Utah array, and Michigan probe. These structural advantages that focus on the simultaneous recording of neural signals from both the surface and the intracortical area have shown the possibility of finding the signal correlation of the whole-brain neuronal pathway to generate a real 3D neural network mapping of the brain.

a Illustration of a 3D brain neural mapping device that is integrated from conventional 2D neural devices (ECoG, Utah array, and Michigan probe). b Flexible 2D fabrication process on a rigid substrate for the preparation of 3D pop-up structure (Steps 1–4) and the transformation from 2D planar-type into a 3D structure by pop-up process (Steps 5–8). Photographs of the front view (c) and the rotated view (d) of the 3D neural mapping device consisting of 9 electrodes of ECoG and 4 probes with 6 electrodes for each probe. Each electrode has a recording site of 20 × 20 µm2 with 500 µm intervals for the ECoG electrodes and 200 µm intervals for the probe electrodes. Each probe is ~1.5 mm in height with a thickness of <20 µm including both sides of the folded flexible substrates, scale bar, 250 μm. e Photograph of a foldable and flexible 3D neural mapping device wrapped around a glass rod with a radius of 5 mm, scale bar, 500 μm.
Figure 1b describes the fabrication process of the 3D neural mapping device. The entire procedure was divided into planar patterns (1–4) and structural pop-up processes (5–8). Benefiting from the lower mechanical mismatch between the device and the brain compared to the high Young’s modulus of conventional rigid neural devices, the polyimide (PI) substrate was prepared by the spin-coating process onto a temporal glass substrate. For the recording electrodes and the metal interconnect lines, gold (Au)/chromium (Cr) deposited by a thermal evaporator was patterned through a conventional photolithographic process. Subsequently, etching the device with a reactive ion etcher (RIE) formed the overall shape of the planar structure. UV curable epoxy (SU-8 2000.5, Kayaku Advanced Materials) was used for encapsulation except for electrophysiological signal sensing pads.
Steps 5–8 show the process of pop-up fabrication that mechanically assembles the 2D planar into the 3D structure (the detailed process is shown in Fig. S1). The device was first detached from the temporal glass substrate and then pre-transferred to the poly(vinyl alcohol) (PVA) tape using a polydimethylsiloxane (PDMS) stamp. And then, SiO2 was deposited only on the specific target bonding regions at the backside of the device using a shadow mask followed by the process of the oxygen plasma treatment on both the SiO2 layer and pre-stretched elastomer for siloxane covalent bonding (Si–O–Si)41,42,43,44 at the interface between them. By releasing the pre-stretched elastomer, a 3D pop-up structure was obtained.
As shown in Fig. 1c, d, the photographic images of the completed foldable and flexible 3D device show the integration of 2 × 2 types of a multi-shank array comprising four penetrating shanks with 6 electrodes with 200 µm spacing (three electrodes for each side with 400 µm spacing) per probe and 9 electrodes for ECoG with 500 µm spacing.
The recording site was 20 × 20 µm2 after encapsulation, designed not only to measure the LFP signal but also to enable a single spike recording. The thickness of the probe part where both sides were attached is <20 µm, including the 500 µm spacing between them. And the length of the folded probe part was 1.5 mm with 1.2 mm recording depth from the surface. To sufficiently reduce the electrochemical impedance of the sensing sites, ensuring a robust readout of single-unit activity, each site was also electroplated with platinum (Pt) black21,45. Also, the total thickness of the device with an elastomer layer for sample handling is 194.65 µm (Fig. S2). As shown in Fig. 1e, due to the thin geometry of the device, it could be wrapped around a 5 mm diameter rod without any mechanical failure.
Origami-based pop-up structure fabrication
The type of penetrating shanks directly inserted into the intracortical area must be carefully designed to minimize glial scarring caused by the penetrating probes during/after implantation into the brain. The focus was on minimizing the thickness and width of the multi-shank implanted in the brain and verified through the actual experimental process and simulation of finite element analysis (FEA). Figure 2a is a schematic illustration of the pop-up structure mechanism by partial bonding of the pre-stretched elastomer to surface area at both ends except for the folded parts. If the mechanical stress applied during bending is not concentrated at the tip when converting two needles into one penetrating shank, the blunt hinge is generated which frustrates the device implantation. To solve this problem, the part of the hinge which is the endpoint of the neural probe was etched to reduce arch-shaped diameter by extreme bending or folding at the endpoint to establish a structural similarity with the conventional neural probe46. The specific demonstration of the hinge etching area of the device is shown in Fig. S3. The 3D FEA simulation revealed the 3D electrode bending deformation as a function of the PI thickness in the central etched region. Since the PI thickness at the hinge was decreased by RIE using a metal mask, the folded 3D electrode exhibited a decreasing the tip head diameter and a higher height at the pop-up PI/SU8 bilayer, which was consistent with the experimental measurements. The effective bending stiffness \(\overline {EI}\) of a multilayered structure was associated with the plain-strain Young’s modulus \(\overline {E_i} = E_i/(1 - \nu _i^2)\) and the thickness hi of each layer as47,48
where \(b = \mathop {\sum }\limits_{i = 1}^N \bar E_ih_i\left( {\mathop {\sum }\limits_{j = 1}^i h_j - \frac{{h_i}}{2}} \right)/\mathop {\sum }\limits_{i = 1}^N \bar E_ih_i\) is the distance from the neutral mechanical plane to the bottom surface of the multilayered structure, Ei is Young’s modulus, and νi is Poisson’s ratio. With \(\nu _{{\rm {SU8}}} = 0.22\) and \(\nu _{{\rm {PI}}} = 0.34\), the effective bending stiffness \(\overline {EI}\) dramatically decreases as the PI thickness decreases in the PI/SU8 bilayer with the same thickness of 0.75 μm for the SU8 (Fig. 2b, Supplementary Table 1, and Fig. S4). Figure 2c shows the comparison between the photograph of the experiment and the FEA simulation as a function of the decrease in the etched ratio. The large local deformation at the tip of the thin bilayer structure (i.e., the thickness ratio of PI to bilayer < 0.42) led to crack formation as observed in the SEM image (Figs. 2c and S5). After considering the reduction in bending stiffness at the tip due to cracking, the FEA results provided a reasonably great concord with the experimental data. It is relevant to notice that although the crack formed at the tip of the probe did not correspond to the recording electrode area, the electrophysiological recording was not affected by deformation at the tip. In conclusion, the folded structure was fabricated with about 20 μm-thick probes that were completed in structural similarity to the conventional probe, which significantly reduced insertion damage to the brain with reduced volume inserted into the brain.

a Schematics of the pop-up fabrication process. The top view of the left schematic shows the bonding area (red), reactive ion etching (RIE) area (gray), and non-bonding area to be part of the pop-up probe (yellow). The side view schematic shows the device transferred in a pre-stretched elastomer with the RIE-etched area. The last schematic shows the folded probe after releasing. The red rectangle inset shows a hinge design including width of 90 µm and a cutting length of 100 μm, L2. The blue rectangle inset shows a reduction in thickness from 7.6 μm of total device thickness (t1) to 3.2 μm of etched hinge part thickness (t2). The green inset shows a magnified view of the fully folded area at the hinge. b Proportional relationships between bending stiffness with polyimide (PI) thickness at the hinge, t2 (black). And the red dashed line shows proportional relationships between the head diameter of the arc shape formed by the pop-up process with t2. c Photograph of the folded device with decreasing t2/t1 ratio from 1 to 0.42, scale bar, 250 μm (Top). The inset shows an scanning electron microscopy (SEM) image of the fully folded hinge with ~20 μm probe thickness, scale bar, 50 μm (Top). Finite element analysis (FEA) simulation of a gradual decrease in hinge thickness, scale bar, 250 μm (Bottom). The inset shows a magnified view of the fully folded hinge with Mises stress distributions, scale bar, 20 μm (Bottom). d Optical image of the device after PEG coating, scale bar, 500 μm. e Photograph of foldable and flexible 3D neural electrodes of 0% (left) and 100% (right) vertical insertion test into 0.7% aragose gel, scale bar, 500 μm. (f and g, ±s.d. n = 25 samples) Electrochemical impedance spectroscopy (EIS) of magnitude (left) and phase (right) of electrodes before (blue) and after (red) Pt black coating by electroplating, inset scale bar, 10 μm. h Electrochemical impedance magnitude and phase spectroscopy during the dissolution of coated polyethylene glycol (PEG) after insertion into the artificial cerebrospinal fluid (ACSF, pH 8.4 at 36.5 °C).
Although the above process provided optimal conditions for the multi-shank to penetrate brain tissue, the substrate composed of intrinsically flexible polymer poses the device implantation problem. To overcome this issue, the surface of penetrating shanks was coated with PEG 4000, which provides sufficient stiffness for the insertion of shanks in the intracortical area. Due to its well-known biocompatibility and dissolution characteristics in a biofluid environment, the device restored its electrical properties after the dissolution of the PEG coating on the surface of every multi-shanks39,40,49,50. Therefore, Fig. 2d presents a photograph of the PEG-coated part of penetrating shanks with ~40 μm thickness after coating (Fig. S6). As shown in Fig. 2e, the entire length of the probe was totally inserted until surface electrodes were contacted on the 0.7% agarose gel surface without device failure, demonstrating the potentially reliable brain insertion with its adequate rigidity (Fig. 2e, Supplementary Movies 1 and 2).
To investigate the electrical characteristics and the mechanical stability of the device, electrochemical impedance spectroscopy (EIS) was measured in vitro by immersing the electrode array in artificial cerebrospinal fluid (ACSF, pH 8.4 at 36.5 °C) and phosphate-buffered saline (PBS, pH 7.4 at 36.5 °C) to mimic in vivo environment (Figs. 2f, g, S7 and S8). To enhance the electrophysiological recording capability of the electrode, Pt black was electroplated to significantly reduce the impedance from ~1.3 Mohm to ~20 kohm (@1 kHz) at the Au layer. Also, an equivalent Randles circuit model is fitted to the EIS result of Au and Pt black including electrolyte resistance (Rs) series with double layer capacitance (Cdl) which is parallel with charge transfer resistance (RCT) and Warburg resistance (W). With 10 \(\mu {\rm {F}}/{\rm {cm}}^{2}\) and 34 \({\mu} {\rm{F}}/{\rm{cm}}^{2}\) values of Cdl for Au and Pt black, respectively, the EIS data is fitted well with the Randles circuit model (Fig. S9). Additionally, the electrochemical impedance spectra were analyzed depending on PEG dissolution to confirm that the electrical performance of the device was properly restored by automatically resolving PEG coating in a biological environment (Fig. 2h).
As expected, the device clearly showed the recovered electrochemical impedance (20 kohm at 1 kHz) which is a value before PEG coating on the multi-shank after the PEG was dissolved in biological fluid. The mechanical and electrical stability of the device was confirmed through the bending and stretching test for 1000 cycles (Fig. S12). The detailed experimental setup for the bending and stretching test is shown in Supplementary Figs. S10 and S11.
Biocompatibility test with the primary neuron cell and cell kit
Before the in vivo experiment to confirm the device’s functionality and safety after inserting the device, the biocompatibility of the electrode array was first tested. The biocompatibility test was conducted on PC 12 cells, which are traditionally used to interpret the function of nerve cells, and primary cortical neuron cells in the central nervous system51,52,53. As presented in Fig. 3a, the electrode array showed superior cell survival rates compared to negative and positive samples. Cytotoxicity of the device was then observed from changes in cell viability rate during cell growth when the compound extracted from the device was mixed with confluently cultured cells in a given ratio (50%, 75%, 100%) (Fig. S13). Surprisingly, even with a 100% extraction of device compound with cell culture media, the electrode array had a negligible effect on the growth of NIH 3T3 cells (Fig. 3b). Supplementary Table 2 shows the extraction of Pt black samples from device compounds showing cell spectrophotometry absorbance greater than 100%. However, this result is interpreted by statistical issues with cell culture, but it still intuitively guarantees its good biocompatibility.

a Viability of PC-12 cells cultured on the surface of Pt black electrode and glass substrates on days 1 and 7 via live/dead cells testing by laser scanning microscopy (Indicate: green (alive), red (dead)), scale bar, 50 μm. b NIH 3T3 cells viability assay for 75% extract at the top and 100% extract at the bottom using polyethylene (PE) film for negative control (blue), polyurethane (PU) for positive control (red), and Pt black electrodes (yellow) (n = 5, n is sample number; ns P = 0.0604 (75% extract, PE vs. Pt Black), ns P = 0.8324 (100% extract, PE vs. Pt Black)). Data are presented as mean ± SD. Statistical significance was tested with an unpaired two-tailed t-test. c and d Represent the viability of neurons cultured on the three substrates (glass substrate, Pt black pattern, and SU-8 substrate with Pt black pattern). c Representative confocal fluorescence images of live (green) and dead (red) cells stained with calcein-AM and PI on the three substrates on day 2, scale bar, 100 μm. d The viability of the cultured neurons cultured on the three substrates on day 2, using glass substrate for the control sample (blue), Pt black, and SU-8 Pt black electrodes (yellow) (n = 3, n is sample number; ns P = 0.0702 (Glass vs. PI), ns P = 0.0800 (Glass vs. SU-8, day 2)). Data are presented as mean ± SD. Statistical significance was tested with an unpaired two-tailed t-test.
Further biocompatibility test was carried out for primary cortical neurons because the main surrounding environment encountered by the device was the central nervous system (Figs. 3c and S14)54. The independent neuron cell culture was highlighted by dividing the electrode recording site (Pt black) and encapsulated UV curable epoxy (SU8 2000.5), which maintains a direct interface with brain cells. As expected, both the recording site and the epoxy environment showed high cell viability compared to the glass substrate (Fig. 3d). These results prove that the device reliably performs its functions without biological hindrance, suggesting the potential for chronic implants55,56,57,58.
Also, in vivo histology result to examine glial reaction after 2 days of device insertion into the brain was shown in Supplementary Fig. S15. It showed less immune response and glial scarring around the insertion site because of the flexibility of the device than other rigid neural implants20,59.
Recording of LFP signals and single spikes with whisker stimulation
To confirm the performance of the 3D neural electrode array, we conducted the in vivo electrophysiological recording. In this experiment, we targeted to show that the proposed system was able to not only measure neural activities from both intracortical brain regions and brain surface simultaneously but also reveal the functional neuronal connectivity from aformentioned area. It is well-known that the barrel cortex activity of a mouse is highly matched with whisker sensing function60,61. The insertion length of our multi-shank was appropriately designed to measure neural activity from five-barrel cortex layers, which has a depth of 1.5 mm from the somatosensory area. Thus, the four penetrating shanks (P1–P4) with each 6 electrodes were implanted into the intracortical brain region of the barrel cortex, and the surface electrode array was contacted to the surface of the barrel cortex. Then, the several right whiskers were stimulated with time intervals in one direction (Fig. 4a). Consequently, both (1) signals at different locations of intracortical brain and brain surface by one whisker stimulation and (2) patterns of signal changes when several whiskers with different locations were stimulated sequentially were successfully observed, and it could be called ‘unidirectional total whisker simulation’.

a Schematic illustration of the insertion area at the somatosensory region for recording neural signals by whisker stimulation using a 3D neural electrode array. The right side of the whiskers is uni-directionally stimulated while recording neural signals on the left hemisphere. As each colored cortex area matches particular whiskers, the device shows signal propagation between the surface of the cortex and intracortical brain area with unidirectional whisker stimulation. b Photograph of the PEG-coated 3D neural mapping device before (left) and partial (right) vertical insertion into the mouse brain, scale bar, 250 μm. c Photograph of top view image of the inserted 3D neural electrode array with four penetrating shanks with internal electrodes and a surface electrode array, scale bar, 250 μm. d Electrophysiological signals recorded from both internal electrodes which are inserted into the intracortical area and surface electrodes which are located on the surface of the barrel cortex. Neural signals are analyzed not only in a low-frequency LFP signal level but also in single spike levels. Unidirectional whisker stimulation lasted for 1 s and was marked with a blue box, LFP scale bar: 1 s vs. 500 μV, Spikes scale bar: 1 s vs. 200 μV.
The device was inserted into the barrel field in the primary somatosensory cortex (S1B) of the 3% isoflurane-anesthetized adult male wild-type mouse (C57BL/6; 8 weeks old). To ensure the insertion of the flexible shanks into the target region, the shanks were coated with PEG to prevent bending of the shank during the insertion process (Fig. 4b). The device was inserted with a speed of 0.2 mm/s, which was the optimized value considering cell damage and dissolution time of PEG coating. As a result, the four shanks were successfully inserted without unwanted bending (Fig. 4b), and the acryl handle attached to the surface substrate was removed to see whether the device is appropriately inserted on the target site through the transparent substrate of the device (Figs. 4c, and S16). After the insertion process, it was confirmed that the four shanks and conformal contact of 9 surface electrodes on the cortex surface were inserted well through the transparent substrate. Considering the sufficient PEG dissolution time after insertion and recovery period, a part of the 3D neural electrodes was fixed on the skull with dental cement and had a recovery period of 2 days. After this procedure, evoked neural signals from both the surface and intracortical regions of the somatosensory cortex were simultaneously recorded by whisker stimulation while maintaining the mouse in an anesthetized state. As a result, the change of LFP signals at all electrodes was recorded during ‘unidirectional total whisker stimulation’ lasted for 1 s and was repeated every 5 s (Figs. 4d, S18, and S19). As the synchronization of single spikes from neurons is known to induce the change of the local field potential62, further analysis of the recorded neural signals at a single neural spike level was conducted. Consequently, neural spikes were detected during whisker stimulation, which was highly synchronized with the amplitude change of the LFP signals. Recorded spikes were sorted based on the polarity and the amplitude through a customized spike-sorting algorithm20. More neural spikes from different neurons from a single electrode were recorded when the dynamic LFP signal changed. Especially at E5 (surface electrode), the amplitude of the LFP signal was the largest, thus the largest number of neural spikes was observed. Individually classified neural spikes with different colors easily indicated the number of neurons that generated neural spikes. The spikes of similar amplitude and shape could be regarded as spikes from the same neuron and marked with the same colors. A larger number of sorted neural spikes induced a higher change of neural spikes, thus showing that each neural spike contributed to the change of the LFP signals.
The waveforms and the raster plots of the sorted neural signals from 60 trials of ‘unidirectional total whisker stimulation’ reaffirmed that the evoked single spikes at various electrode locations were recorded (Fig. S21). In addition, the number of total spikes was recorded from several electrodes such as the surface electrode E5, the multi-shank electrode E23, E25, and E29 (Fig. S22). This detailed spike analysis shows that the device detected synchronized spikes caused by ‘unidirectional total whisker stimulation’ at each location of the device and the waveform and spike response patterns varied depending on the location. The recorded LFP signals were significantly changed, and the firing rate of single spikes was highly increased by whisker stimulations compared to spontaneous signals (Figs. S23 and S24). These results show that the foldable and flexible 3D electrode array could efficiently record both evoked and spontaneous neural signals.
As a result, the proposed device could investigate whether synchronized signals were evoked by ‘unidirectional total whisker stimulation’ by observing repeated changes in LFP signals and single spikes from both the intracortical area and surface regions of the brain simultaneously according to the presence of whisker stimulations.
3D Heat map analysis of recorded signals
Further analysis of the recorded electrophysiological signals with a 3D Heat map was introduced by showing the propagation of the brain signals from both the intracortical and surface region of the brain. As the whiskers were stimulated every 5 s with wooden sticks for 1 s in one direction, each whisker would be stimulated at time intervals. From the recorded LFP signals, two groups of peaks at the entire electrodes were identified (Fig. S25). Also, each time interval of 100 ms with the peak as a group of signals from a similar whisker region was categorized as Stim1 with the blue box and Stim2 with the red box, respectively (Fig. 5a). From the analysis of Stim1 and Stim2 in detail, several small peaks were found with five different frames on the peaks of each time section, marked with a dashed line (Fig. 5b, c). During the Stim1, each peak was estimated as a signal evoked by the stimulation of a particular whisker, and peaks appeared at an average time interval of about 10.8 ms, and the amplitude of the peaks varied from 250 to 800 μV. On the other hand, during the Stim2, peaks appeared at an average time interval of about 6.5 ms and the amplitude of the peaks ranged from 500 to 800 μV.

a Overlapped LFP signals from a total of 33 electrodes during the unidirectional whisker stimulation. The first peak of the overlapped signals is named Stim1 and marked with the blue box, and the second peak is named Stim2 and marked with the red box. b Overlapped LFP signals from whole electrodes during the time interval Stim1. 5 dashed line indicates the peak of the LFP signals evoked by each particular whisker stimulation. c Overlapped LFP signals from total electrodes during the time interval Stim2. 5 dashed line indicates the peak of the LFP signals. d 3D heatmap of the amplitude of the LFP signals at each time frame of the Stim1. The maximum amplitude of the LFP signals was 800 μV and represented as red color and the minimum value was −50 μV which was represented as a blue color. e 3D heatmap of firing rate and the number of the neuron during Stim1. The color of the firing rate 3D heatmap ranges from 0 (blue) to 70 Hz (red) and the color of the number of neurons 3D heatmap ranges from 0 (blue) to 4 (red). f 3D heatmap of amplitude changes by each frame of Stim2. As the maximum amplitude was 800 μV and the minimum value was −50 μV, the colors of the electrodes are matched from red to blue. g 3D heatmap of firing rate and the number of neurons during Stim2. The color of the firing rate 3D heatmap ranges from 0 (blue) to 70 Hz (red) and the color of the number of neurons 3D heatmap ranges from 0 (blue) to 4 (red). (The 3D heatmap data of the E3 electrode was averaged with adjacent electrodes and the reference point (RP), represents the location of the penetrating shank P3.
To observe the propagation pattern of the LFP signal in the intracortical region and surface of the brain according to the stimulation of each specific whisker, 3D electrophysiological mapping depending on the amplitude of the LFP with colors at each frame is shown in Fig. 5d, f. The amplitude change of the LFP signal was plotted visually through 3D heat mapping in the color range from −50 to 800 μV in three types of view (Front, Back, and Top). From the analysis of the LFP signals recorded from electrodes at penetrating shank P3 during the Stim1, the amplitude of LFP signals was gradually increased as the one specific whisker stimulation accumulated in succession. The increase of LFP signal amplitude was propagated to the electrodes at P3 on the front side and the electrodes at P4 and the center of the brain surface region. During the Stim2, similar propagation was observed while the amplitudes and time intervals were quite different from the Stim1. These results highlight that the proposed device successfully record the patterns of evoked signals by different specific whisker stimulation.
In addition, the recorded single spikes during the Stim1 and Stim2 were plotted depending on the firing rate and the number of sorted spikes (Fig. 5e, g). In detail, transient and raster plots of the sorted spikes at the single spike level were also presented in Supplementary Fig. S26. During the Stim1 and Stim2, the firing rate recorded from electrodes at P3 got the highest value as the LFP changes started at electrodes at P3. These results reaffirmed that the LFP were evoked by the synchronizations of neuronal firing. In conclusion, the different propagation patterns have appeared in terms of LFP changes, firing rate, and the number of neurons in 3D heat map analysis with our device when each whisker is stimulated.
Discussion
This study demonstrates the spatiotemporal interpretation capability of electrophysiological recordings by transforming the existing neural implants into a 3D structure. The flexibility of the device provides a robust signal readout with minimal immune response and glial scarring with a negligible mechanical mismatch when directly inserted into the brain. The device fabrication process through the pop-up structure presented in this study was carefully optimized so that the mechanical and electrical characteristics were not violated through various simulations and experiments. To investigate whether this work could be used to record the 3D neuronal propagation, electrophysiological brain mapping using whisker stimulation of mice was first presented. Both surface recording of the brain and intracortical signal propagation was demonstrated in the distribution of single spike firing rate and the number of neurons. Furthermore, acutely measured simultaneous ECoG recordings of intracortical activity from a single spike suggest that this study can be exploited as a tool to identify neurological disorders.
Although the design of this device exceeded the structure of the existing planar recording method, this work also had difficulties in overcoming the limitation of the rather small number of recording channels due to the spatial limitation from the passive electrode array. Also, the insertion stability should be substantiated, maintaining its flexibility. Therefore, as a future study, the possibility of developing an actively multiplexed 3D array (e.g., flexible transistor array) to reduce the wire bundles and extensively increase the number of recording sites, which guarantees large-area, high-density measurement of neural activity will be further investigated. The method of increasing the number of multi-shanks in intracortical areas could also be an alternative approach capable of maximizing the structural advantages of the neural interface. Consequently, this research will provide avenues for therapeutic mechanisms and pathological study in neuroscience and biomedical practices.
Methods
2D planar fabrication process (Fig. S1.1)
For the flat polyimide substrate, 2 layers of liquid PI (poly (pyromellitic dianhydride-co-4,4’-oxydianiline), amic acid solution from Merck Sigma-Aldrich solutions) were spin-coated on glass substrate by a spin coater (ACE-200, Dong AH Trade Corp.) with the following process. The first layer was coated by 1500 rpm rotation speed, 110 °C baking for 1 min, and 150 °C baking for 5 min. Then the second layer was coated the same as the first layer followed by a vacuum oven for 2 h at 220 °C. Subsequently, using a thermal evaporator (KVE-T2000, Korea Vacuum Tech.), a metal layer of 5 nm Cr and 50 nm Au was deposited on the PI film substrate.
For the device electrode, metal line and interconnect pattern, a positive photoresist (S1805, Dow Chemical) was spin-coated at 3000 rpm and baked at 100 °C for 1 min on the Au/Cr metal layer. Subsequently, the Mask aligner & Exposure system (MDA-400S(IM), MIDAS system) was used for exposure for 6 s with the photomask. The photoresist was patterned by immersing it in the developer (AZ 300 MIF Developer, Merck & Co. Inc.) for 1 min. And then, Au and Cr were etched in sequence by Au etchant (AT-409LB, Jeonyoung Co., Ltd.) and Cr etchant (CT-1200S, Jeonyoung Co., Ltd.). After the patterning process, acetone cleaning and gentle O2 plasma were used to remove the remaining photoresist.
To transform the device into a pop-up design shape by etching the PI through the copper (Cu) mask, a positive photoresist (AZ5214-E, AZ Electronic Materials) was patterned reversely by following step (2000 rpm of rotation speed, pre-baking at 110 °C for 110 s, UV exposure for 7 s, reversal baking at 120 °C for 120 s, Flood exposure for 14 s). And then for the photoresist pattern, it was immersed in the AZ 300 MIF developer for 1 min. For the Cu mask, 100 nm Cu was deposited using a thermal evaporator. Thereafter, immersed in acetone for the lift-off process until the remaining photoresist is removed. Next, a RIE (Q190620-M01, Young Hi-Tech) was used for the PI etching process (30 min at flow: O2 100 sccm, power: 200 W).
For the hinge part etching process, the Cu mask (remained in the last step) was spin-coated with a positive photoresist (AZ5214-E, AZ Electronic Materials) (rotation speed: 2000 rpm, pre-bake: 110 s at 110 °C, exposure: 7 s). And immersion was continued for 1 min in AZ 300 MIF developer for the photoresist pattern. The Cu of the patterned hinge portion was etched by cu etchant (CE-100, Transene Company, Inc.). To remove residual photoresist, the device was cleaned with acetone. An RIE was then used for PI hinge etching (ratio 0.42 for 13 min at flow: O2 100 sccm, power: 200 W). The Cu mask was removed by Cu etchant and cleaned with deionized (DI) water.
For encapsulation without recording electrodes area, negative photoresist SU-8 2000.5 from (Kayaku Advanced Materials) was spin-coated with 1000 rpm rotation speed on the device surface. Then soft baking at 100 °C for 2 min, UV exposure for 6 s, and post-baking at 100 °C for 2 min. For patterning SU8, the device was further immersed in SU8 developer (Y020100 4000L1PE, Kayaku) for 90 s. Finally, another 15 min hard baking at 110 °C finished the fabrication process of the planar device.
Pop-up fabrication
To detach the device from the glass substrate, it was immersed for 15 min in DI water on the 80 °C hot-plate. And then picked up the device using a mixture (10:1) of PDMS stamp (DOW Corning SYLGARD 184 Silicone Elastomer Kit). Then, the remaining water was removed through N2 blow and attached to the device on PDMS onto PVA water-soluble tape (3M 5414 water-soluble wave solder tape) (Fig. S1.2). To deposit SiO2 for the oxide bonding layer only in a specific ECoG area, a polyimide mask was attached to the PVA tape with the alignment process. (Fig. S1.3) SiO2 at 50 nm was deposited on the ECoG area of the device by sputter (LSP-06, LAT) (Fig. S1.4).
For the device pop-up structure, the elastomer (Dragon Skin 10 1:1, SMOOTH-ON) was previously stretched on the stretching machine. Before bonding SiO2 with the elastomer, both sides of the elastomer and the SiO2 layer were treated for 5 min with UV/O3 treatment (CR3015M, UVSMT) (Fig. S1.5). Then the PI mask on the PVA tape were removed and the elastomer was bonded with SiO2. Dissolve the PVA tape through the DI water rinse. Subsequently, the elastomer was released for pop-up fabrication (Fig. S1.6).
Pt black electroplating
To improve the performance of electrophysiological recording, the Pt black was electroplated on the Au layer. The used Pt black electroplating solution mixture includes 0.025 N of HCl, 3% [w/v] of hex-chloroplatinic acid hydrate (HPCA), and 0.025% [w/v] of lead acetate soluted in di water. (all materials purchased from Sigma-Aldrich). And the three electrode configurations in the solution were performed with the device connected as a working electrode, Ag/AgCl electrode for the reference electrode, and Pt wire for the counter electrode. Pt black was selectively deposited on the exposed area of the Au recording electrode by a potentiostat (Gamry Reference 600+) with a chronoamperometry process (30 s at 0.2 V amplitude with a sample period of 0.01 s).
In vitro test for electrochemical impedance test setup
EIS was measured with a potentiostat. PBS powder purchased from Sigma-Aldrich was mixed with DI water and artificial cerebrospinal fluid (ACSF) was prepared for another EIS before measurement. And the EIS was conducted with three-electrode configurations at 36.5 °C (Pt black electrode for the working electrode, Ag/AgCl electrode for the reference electrode, and Pt wire for the counter electrode) (Fig. S6). The sweep frequency of the measurement was between 0.1 Hz and 100 kHz with a 10 mV RMS AC voltage and 10 points/decade. For the PEG 4000 (purchased from Sigma Aldrich) dissolution test, the PEG-coated device was immersed in prepared solutions. In both PBS and ACSF, EIS had adequate ability to record electrophysiological signals. In the case of the PEG degradation test, EIS was used in the same experimental protocols but with a sweep frequency set to 1 kHz for 300 s.
In vitro bending and stretching test
For the bending test, wrapping the device on the 5 mm glass rod was performed. After every 10 bending cycles, the EIS of the electrodes was measured with a potentiostat following the above protocols. Additionally, using a stretching machine, the device was stretched from 0% to 40% for one stretch test cycle. After every 10 order of cycles, the EIS of the electrodes was measured as a bending and stretching test.
FEA simulation setup
The FEA simulation involves buckling a PI/SU8 bilayer. Obtaining the engineering stress–strain curve from the experimental data63 yields 4.02 GPa Young’s modulus and 0.22 Poisson’s ratio for SU8 in the linear elastic zone64,65. The elastic–plastic homogeneous isotropic model66 relates the uniaxial stress \(\sigma _{{\rm {PI}}}\) to the strain ε of PI as
where \(E_{{\rm {PI}}} = 2.34\;{\rm {GPa}}\) is Young’s Modulus, \(\sigma _{\rm {y}} = 24\;{\rm {MPa}}\) is the yield stress, and N = 0.5 is the hardening exponent of PI, respectively. The 3D quadratic brick element C3D20R is used for both materials in the static simulation with geometric nonlinearity. After applying a small out-of-plane perturbation at the center, a displacement load of 2.6 mm is applied to one side of the bilayer with an initial length of 3 mm (fixed on the other end). As for the simulation with cracks, a notch 4 µm wide and 3 µm deep (common in the fabrication processes67,68,69) is included in the central region of the bilayer.
Preparation of substrates
Cover glasses with a diameter of 18 mm (The Paul Marienfeld GmbH & Co. KG, Germany) were used to attach electroplated Pt black electrodes to the Au layer. Sterile cover glasses of the same size, coated and uncoated with laminin (1 mg/mL), were worked as positive and negative controls respectively. All samples, including Pt black electrodes, were sterilized with EtO gas for 18 h before use.
Preparation of cells
Pheochromocytoma cells (PC-12 cells) were used for biocompatibility tests. Cells from passage no. 8 were cultured in Roswell Park Memorial Institute 1640 medium (HyClone, USA) supplemented with 10% v/v horse serum (Sigma-Aldrich, Germany), penicillin–streptomycin (Gibco, USA), and 5% fetal bovine serum (Gibco, USA) for 5–7 days until reaching 70–80% confluency.
Pre-incubation of substrates in cell culture media
Pt black electrodes and cover glasses were pre-incubated in cell culture media. To do this, 500 µL of media was pipetted on each sample in 12-well plates, and placed in a 5% CO2 incubator for 24 h at 37 °C.
Cell seeding on the samples
PC-12 cells were detached, counted, and plated on the surface of the samples at a concentration of 10,000 cells/cm2. Media change was performed daily.
Live/dead cells assay
On culture days 1 and 7, a live and dead cells test was performed to assess cell viability on Pt black electrodes and control glasses. For this, Calcein-AM and Ethidium Homodimer I (EthD-I) fluorescent dyes were used to distinguish live and dead cells, respectively. Cell visualization was conducted by laser scanning microscopy. The green color shows the live cells and the red color shows the dead cells. To preserve sterile conditions, samples for each test day were cultured separately in two well plates.
Cell culture
NIH 3T3 cells from passage number nine were cultured in Dulbecco’s modified Eagle medium high glucose (DMEM, Gibco, USA) supplemented with 1% penicillin–streptomycin (Gibco, USA) and 10% fetal bovine serum (Gibco, USA) for 5 days until reaching confluency.
Testing Pt black material property to reduce WTS-8 reagent in Cell Counting Kit-8 (CCK-8, SX900, Dojindo Laboratories, Japan) assay solution
Three-electrode samples were inserted into Eppendorf tubes of 1.5 mL each. 100 µL of NIH 3T3 media was pipetted into each tube so that the electrode sample was completely immersed. Then 10 µL of CCK-8 solution was pipetted and thoroughly mixed with the medium. Additionally, another tube was used as a blank (no electrode sample, only media). The tubes were placed in the 37 °C, 5% CO2 incubator for 3 h. The procedure was conducted following the manufacturer’s instructions for use. The color change was not detected after the incubation time, which means that the tested material does not interfere with the CCK-8 assay reagent.
Preparation of sample and control materials extractions
To conduct a cytotoxicity test, a high-density polyethylene film (PE, Hatano Research Institute, FDSC) and 0.1% ZDEC polyurethane film (PU, Hatano Research Institute, FDSC)were used together with the Pt black test material as a negative and positive control, respectively.
First, the extraction solutions were prepared. For this, the required surface area was calculated following the ISO (International Standard for cytotoxicity test, ISO 10993-5). For materials thinner than 0.5 mm, the extraction solution must be prepared in a ratio of 6 cm2 per 1 mL of solvent. According to the calculations, films of Pt black, PU, and PE material were cut, placed in 15 mL falcon tubes, and sterilized in EOG for 18 h. After sterilization, 1.5 mL of pre-warmed NIH 3T3 media was pipetted into each tube, sealed with parafilm, and placed in a 40 °C water bath for 72 h with constant shaking at 40 rpm.
Incubation of cells in extract solutions
A day before the incubation of NIH 3T3 cells in materials extracts, cells were detached and immersed in a 96-well plate in concentration \(1\times 10^4\;{\rm {cells/well}}\) for adaptation.
After 72 h of incubation of the extracts, the cell culture medium was removed from the well plate with NIH 3T3 cells and changed to 50% (50 µL of sample extract + 50 µL of culture media per well), 75% (75 µL of sample extract + 25 µL of culture media per well), and 100% solutions (only 100 µL of sample extract) of Pt black, positive and negative control extracts in five repetitions for each concentration value, and incubated for 24 h at 37 °C, 5% CO2. Blank wells contained only cell culture media, without cells.
After incubation, a cytotoxicity test was performed using the CCK-8 assay. In short, each well was added with 10 µL of CCK-8 reagent and then incubated at 37 °C, 5% CO2 for 4 h. Absorbance was measured in a microplate reader using a 450 nm filter (VersaMax Microplate Reader, Molecular Devices, USA). The cell survival rate was calculated following the formula:
where Ab is the absorbance of blank sample and Ac is the absorbance of the negative control sample.
Primary neural culture on three substrates
A previously reported protocol for a 2D neural culture was followed54. Firstly, three substrates (i.e., a glass substrate, PI substrate with Pt black pattern, and SU-8 substrate with Pt black pattern) were cleaned with ethanol three times every 30 min and sterilized under ultraviolet light overnight. 100 μg/mL of poly-d-lysine (PDL; Sigma-Aldrich, USA) was used for coating the sterilized samples in a CO2 incubator overnight. Then, primary neural cultures were formed by the sacrifice of pregnant Sprague Dawley rats (embryo 18; DBL Co., South Korea). From the brain of the embryo, the cerebral cortex was dissected. Then, the cortical neurons were isolated by neural tissue dissociation kits using the gentleMACS dissociator (Militenyi Biotec, Germany). And, plating the neurons with a density of 400 cells/mm2 on the three PDL-coated substrates (i.e., glass, PI, and SU-8 substrate) was conducted after manually counting with a trypan blue (GIBCO, USA) and a hemocytometer (INCYTO, South Korea). And then, the neurons were cultured in the neurobasal Plus medium, supplemented with 1% (v/v) penicillin–streptomycin (GIBCO, USA), 2 mM Glutamax-I (GIBCO, USA), and 2% (v/v) B27 Plus supplement (Invitrogen, USA), and cultured in a CO2 incubator at 37 °C.
Live/Dead cell viability test of cultured neurons
The viability test of the cultured neurons for 2 days was conducted in vitro. The samples were treated in 0.1 M PBS, including AM (calcein-AM; Thermo Fisher Scientific), 15 μM propidium iodide (Sigma-Aldrich, USA), and 1 μM CellTrace calcein green for 30 min at 37 °C. After 1× PBS washing, fluorescence images that displayed green color for live cells and red color for dead cells through a ×5 objective lens in a confocal laser scanning microscopy (LSM 700, Carl Zeiss, Germany) were acquired. For non-transparent samples such as PI and SU-8 substrates, their images were acquired by flipping the sample over.
The viability of the cultured neurons was calculated as
Using ImageJ2 (by the National Institutes of Health), the number of cells was semi-automatically counted.
Animal surgery
The adult wild-type mice (C57BL/6; 8 weeks old) used in the experiment were anesthetized with 3% isoflurane and fixed to the stereotaxic instrument (Model 940, David Kopf Instruments, Tujunga, CA) for surgery. To insert the 3D neural mapping device into the barrel field in the primary somatosensory cortex (S1B) (AP: −1.5 mm, ML: −3 mm from bregma), we calibrated the bregma with an insertion shuttle and marked the target location based on the atlas of Paxinos and Franklin. The skull was removed with a size of 3 × 3 mm2 around the target point, and the dura mater was also removed (Supplementary movie S3 and Fig. S17). Then, the device is implanted into the brain by a robotic stereotactic manipulator (Neurostar Drill Robot, Neurostar, Tubingen, Germany) at an insertion rate of 0.2 mm/s. After insertion of the device, dental cement was applied to fix it, and wait 2 days for recovery of the cell at the insertion area. And then we continued in vivo neural signal recording.
In vivo electrophysiological signal recording
With an Intan RHD 2132 recording system, the neural signals were recorded to amplify and digitize the recorded electrical signal through a 3D Neural mapping device (Fig. S20).
For whisker stimulation in mice, the right side of the entire mice whisker was touched with a small thin wood stick every 5 s. Each stimulation was timed with an application named Metronome: Tempo Lite (Frozen Ape Pte.Ltd., Singapore).
The neural signals were analyzed through the device using a custom spike-sorting algorithm in MATLAB (Mathworks, USA)20. This algorithm detected and sorted the recorded signal by setting the neural spike polarity and amplitude threshold. For signal analysis, the classification by setting the threshold of amplitude manually until sufficient spikes were detected and sorted was performed. Among the classified spikes, spikes whose interspike interval (ISI) was <2 ms were excluded.
The Firing rate was obtained by separating the number of sorted spikes during the sampling period by the duration of the period in seconds.
Immunostaining of brain tissues
To examine if the glial reaction is induced around the inserted probe array in the brain, we implanted the foldable and flexible 3D neural electrode array into the brain of an adult mouse (C57BL6; 8 weeks old). After waiting 2 days for recovery, the mouse was sacrificed by transcardial perfusion using 4% paraformaldehyde (PFA) in 1× PBS. After waiting 30 min, we carefully removed the probe. The extracted brain was additionally fixed in 4% PFA for 24 h at 4 °C. Next, the brain was cryoprotected with 30% [w/v] sucrose in 1× PBS for more than 24 h at 4 °C. Then, the cryoprotected brain was sliced in the horizontal plane into 40 μm-thick slices using a cryostat (Thermofisher, HM525). The 40 μm-thick brain slices were washed in 1x PBS three times and blocked in a blocking solution containing 0.3% [v/v] Triton X-100, 2% [w/v] donkey serum, and 2% goat serum in 1× PBS for 1 h at room temperature. After 10-min washing in 1× PBS three times, the 40 μm-thick slices were incubated in the prepared blocking solution containing primary antibodies (chicken anti-GFAP, 1:500, AB5541, Millipore; rabbit anti-Iba1, 1:300, 019-19741, Wako) for 18 h at 4 °C. After 10-min washing in 1× PBS three times, the slices were then incubated in the prepared blocking solution containing secondary antibodies (Donkey anti-Chicken Alexa 488, 1:500, 703-545-155, Jackson Lab; Donkey-anti-Rabbit IgG 594, 1:500, 711-585-152, Jackson Lab) in the prepared blocking solution for 2 h at room temperature. After 10-min washing in 1× PBS three times, the slices were mounted on microscope slides. We then imaged the slices through a ×20 objective lens on a confocal microscope (A1, Nikon, Japan).
Heatmapping and movie
All electrophysiological signal data from the 3D neural mapping device were recorded with the Intan RHD 2132 recording system. The raw signal was filtered as 20 kS s−1 per channel, 0.1–6 kHz bandpass filter, and 60 Hz Notch filter with the Intan software. Each recorded LFP signal evoked by whisker stimulation was filtered with a 300 Hz low pass filter using Matlab. The overlapped LFP signals of the electrodes were colored by large to small amplitude values, from red to blue.
Before plotting the 3D heatmap, the entire device design including all electrode points was designed in 3D using Rhino software. Each part of the device was plotted in 3D using Matlab software by loading the figure from rhino software as in.stl form, and the amplitude of LFP signal data corresponded to the location of each electrode to plot a heatmap. With two diagonal front and back views, the top view of the electrode was captured in each frame of the 100 ms overlapped LFP changes. All frames were edited using Premiere Pro software to produce a 100 ms 3D heatmap movie. (Supplementary movies S3 and S4).
Statistical analysis
Statistical analysis of graph data was plotted using Origin Lab. software. Electrochemical impedance and phase plot with and without Pt black, electroplating was represented as mean values ± s.d., n = 25 electrodes. Statistical significance for the all-cell viability test was tested with unpaired two-tailed t-tests. The statistical significance of the experimental results was evaluated using one-way ANOVA and performed using a statistical analysis program (Prism, GraphPad Software Inc., USA).
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code availability
The code that supports the figures of this study is available from the corresponding author upon reasonable request.
References
Heneka, M. T., McManus, R. M. & Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 19, 610–621 (2018).
Rizzolatti, G. & Sinigaglia, C. The mirror mechanism: a basic principle of brain function. Nat. Rev. Neurosci. 17, 757–765 (2016).
Valkenborghs, S. R. et al. The impact of physical activity on brain structure and function in youth: a systematic review. Pediatrics 144, e20184032 (2019).
Cho, Y., Park, S., Lee, J. & Yu, K. J. Emerging materials and technologies with applications in flexible neural implants: a comprehensive review of current issues with neural devices. Adv. Mater. 33, 2005786 (2021).
McGlynn, E. et al. The future of neuroscience: flexible and wireless implantable neural electronics. Adv. Sci. 8, 2002693 (2021).
Ferguson, M., Sharma, D., Ross, D. & Zhao, F. A critical review of microelectrode arrays and strategies for improving neural interfaces. Adv. Healthc. Mater. 8, 1900558 (2019).
Passaro, A. P. & Stice, S. L. Electrophysiological analysis of brain organoids: current approaches and advancements. Front. Neurosci. 14, 1405 (2021).
Landré, E., Chipaux, M., Maillard, L., Szurhaj, W. & Trébuchon, A. Electrophysiological technical procedures. Neurophysiol. Clin. 48, 47–52 (2018).
O’Neill, G. C. et al. Dynamics of large-scale electrophysiological networks: a technical review. Neuroimage 180, 559–576 (2018).
Weisenburger, S. & Vaziri, A. A guide to emerging technologies for large-scale and whole-brain optical imaging of neuronal activity. Annu. Rev. Neurosci. 41, 431–452 (2018).
Wang, T. et al. Three-photon imaging of mouse brain structure and function through the intact skull. Nat. Methods 15, 789–792 (2018).
Khodagholy, D. et al. Organic electronics for high-resolution electrocorticography of the human brain. Sci. Adv. 2, e1601027 (2016).
Beauchamp, M. S., Sun, P., Baum, S. H., Tolias, A. S. & Yoshor, D. Electrocorticography links human temporoparietal junction to visual perception. Nat. Neurosci. 15, 957–959 (2012).
Choi, J.-r, Kim, S.-M., Ryu, R.-H., Kim, S.-P. & Sohn, J.-w Implantable neural probes for brain–machine interfaces—current developments and future prospects. Exp. Neurobiol. 27, 453 (2018).
Patil, A. C. & Thakor, N. V. Implantable neurotechnologies: a review of micro-and nanoelectrodes for neural recording. Med. Biol. Eng. Comput. 54, 23–44 (2016).
Steinmetz, N. A., Koch, C., Harris, K. D. & Carandini, M. Challenges and opportunities for large-scale electrophysiology with neuropixels probes. Curr. Opin. Neurobiol. 50, 92–100 (2018).
Li, Y.-t, Ibrahim, L. A., Liu, B.-h, Zhang, L. I. & Tao, H. W. Linear transformation of thalamocortical input by intracortical excitation. Nat. Neurosci. 16, 1324–1330 (2013).
Lee, J., Ozden, I., Song, Y.-K. & Nurmikko, A. V. Transparent intracortical microprobe array for simultaneous spatiotemporal optical stimulation and multichannel electrical recording. Nat. Methods 12, 1157–1162 (2015).
Wei, X. et al. Nanofabricated ultraflexible electrode arrays for high‐density intracortical recording. Adv. Sci. 5, 1700625 (2018).
Shin, H. et al. Multifunctional multi-shank neural probe for investigating and modulating long-range neural circuits in vivo. Nat. Commun. 10, 1–11 (2019).
Shin, H. et al. 3D high-density microelectrode array with optical stimulation and drug delivery for investigating neural circuit dynamics. Nat. Commun. 12, 1–18 (2021).
Gu, Y. et al. Three-dimensional transistor arrays for intra-and inter-cellular recording. Nat. Nanotechnol. 17, 292–300 (2022).
Park, Y. et al. Three-dimensional, multifunctional neural interfaces for cortical spheroids and engineered assembloids. Sci. Adv. 7, eabf9153 (2021).
Chung, J. E. et al. High-density, long-lasting, and multi-region electrophysiological recordings using polymer electrode arrays. Neuron 101, 21–31 (2019). e25.
Jiang, S. et al. Spatially expandable fiber-based probes as a multifunctional deep brain interface. Nat. Commun. 11, 1–14 (2020).
Liu, J. et al. Syringe-injectable electronics. Nat. Nanotechnol. 10, 629–636 (2015).
Renz, A. F. et al. Opto‐E‐Dura: a soft, stretchable ECoG array for multimodal, multiscale neuroscience. Adv. Healthc. Mater. 9, 2000814 (2020).
Konerding, W., Froriep, U., Kral, A. & Baumhoff, P. New thin-film surface electrode array enables brain mapping with high spatial acuity in rodents. Sci. Rep. 8, 1–14 (2018).
Fiáth, R. et al. A silicon-based neural probe with densely-packed low-impedance titanium nitride microelectrodes for ultrahigh-resolution in vivo recordings. Biosens. Bioelectron. 106, 86–92 (2018).
Wang, B. et al. An implantable multifunctional neural microprobe for simultaneous multi-analyte sensing and chemical delivery. Lab. Chip. 20, 1390–1397 (2020).
Shi, J. & Fang, Y. Flexible and implantable microelectrodes for chronically stable neural interfaces. Adv. Mater. 31, 1804895 (2019).
Lee, Y. et al. A lubricated nonimmunogenic neural probe for acute insertion trauma minimization and long‐term signal recording. Adv. Sci. 8, 2100231 (2021).
Obidin, N., Tasnim, F. & Dagdeviren, C. The future of neuroimplantable devices: a materials science and regulatory perspective. Adv. Mater. 32, 1901482 (2020).
Gao, D., Parida, K. & Lee, P. S. Emerging soft conductors for bioelectronic interfaces. Adv. Funct. Mater. 30, 1907184 (2020).
Salatino, J. W., Ludwig, K. A., Kozai, T. D. & Purcell, E. K. Glial responses to implanted electrodes in the brain. Nat. Biomed. Eng. 1, 862–877 (2017).
Vomero, M. et al. On the longevity of flexible neural interfaces: establishing biostability of polyimide-based intracortical implants. Biomaterials. 281, 121372 (2022).
Vázquez-Guardado, A., Yang, Y., Bandodkar, A. J. & Rogers, J. A. Recent advances in neurotechnologies with broad potential for neuroscience research. Nat. Neurosci. 23, 1522–1536 (2020).
Frank, J. A., Antonini, M.-J. & Anikeeva, P. Next-generation interfaces for studying neural function. Nat. Biotechnol. 37, 1013–1023 (2019).
Seo, K. J. et al. Transparent, flexible, penetrating microelectrode arrays with capabilities of single‐unit electrophysiology. Adv. Biosyst. 3, 1800276 (2019).
Guan, S. et al. Elastocapillary self-assembled neurotassels for stable neural activity recordings. Sci. Adv. 5, eaav2842 (2019).
Yan, Z. et al. Mechanical assembly of complex, 3D mesostructures from releasable multilayers of advanced materials. Sci. Adv. 2, e1601014 (2016).
Yan, Z. et al. Three-dimensional mesostructures as high-temperature growth templates, electronic cellular scaffolds, and self-propelled microrobots. Proc. Natl Acad. Sci. USA 114, E9455–E9464 (2017).
Nan, K. et al. Compliant and stretchable thermoelectric coils for energy harvesting in miniature flexible devices. Sci. Adv. 4, eaau5849 (2018).
Fu, H. et al. Morphable 3D mesostructures and microelectronic devices by multistable buckling mechanics. Nat. Mater. 17, 268–276 (2018).
Lee, Y. J., Kim, H.-J., Do, S. H., Kang, J. Y. & Lee, S. H. Characterization of nerve-cuff electrode interface for biocompatible and chronic stimulating application. Sens. Actuators B Chem. 237, 924–934 (2016).
Rogers, J., Lagally, M. & Nuzzo, R. Synthesis, assembly and applications of semiconductor nanomembranes. Nature 477, 45–53 (2011).
Wang, S. et al. Mechanics of epidermal electronics. J. Appl. Mech. 79, 031022 (2012).
Cheng, H. & Wang, S. J. J. o. A. M. Mechanics of interfacial delamination in epidermal electronics systems. J. Appl. Mech. 81, 044501 (2014).
Liu, X. Y., Nothias, J.-M., Scavone, A., Garfinkel, M. & Millis, J. M. Biocompatibility investigation of polyethylene glycol and alginate-poly-l-lysine for islet encapsulation. ASAIO J. 56, 241–245 (2010).
Patel, P. R. et al. Insertion of linear 8.4 μm diameter 16 channel carbon fiber electrode arrays for single unit recordings. J. Neural Eng. 12, 046009 (2015).
Nguyen-Vu, T. B. et al. Vertically aligned carbon nanofiber arrays: an advance toward electrical–neural interfaces. Small 2, 89–94 (2006).
Kim, D. et al. Substance P/heparin‐conjugated PLCL mitigate acute gliosis on neural implants and improve neuronal regeneration via recruitment of neural stem cells. Adv. Healthc. Mater. 10, 2100107 (2021).
Tyson, J., Tran, M. & Slaughter, G. Biocompatibility of a quad-shank neural probe. Solid-State Electron. 136, 113–119 (2017).
Kook, G. et al. Wafer-scale multilayer fabrication for silk fibroin-based microelectronics. ACS Appl. Mater. Interfaces 11, 115–124 (2018).
Nemani, K. V., Moodie, K. L., Brennick, J. B., Su, A. & Gimi, B. In vitro and in vivo evaluation of SU-8 biocompatibility. Mater. Sci. Eng. C 33, 4453–4459 (2013).
Márton, G. et al. A multimodal, SU-8-platinum-polyimide microelectrode array for chronic in vivo neurophysiology. PLoS ONE 10, e0145307 (2015).
Song, E., Li, J., Won, S. M., Bai, W. & Rogers, J. A. Materials for flexible bioelectronic systems as chronic neural interfaces. Nat. Mater. 19, 590–603 (2020).
Cho, Y. U. et al. Ultra‐low cost, facile fabrication of transparent neural electrode array for electrocorticography with photoelectric artifact‐free optogenetics. Adv. Funct. Mater. 32, 2105568 (2022).
Lee, H. C. et al. Histological evaluation of flexible neural implants; flexibility limit for reducing the tissue response? J. Neural Eng. 14, 036026 (2017).
Idogawa, S. et al. Coaxial microneedle-electrode for multichannel and local-differential recordings of neuronal activity. Sens. Actuators B Chem. 320, 128442 (2020).
Sawahata, H. et al. Single 5 μm diameter needle electrode block modules for unit recordings in vivo. Sci. Rep. 6, 1–12 (2016).
Denker, M. et al. The local field potential reflects surplus spike synchrony. Cereb. Cortex 21, 2681–2695 (2011).
Xu, T. et al. Characterization of the mechanical behavior of SU-8 at microscale by viscoelastic analysis. J. Micromech. Microeng. 26, 105001 (2016).
Lorenz, H. et al. SU-8: a low-cost negative resist for MEMS. J. Micromech. Microeng. 7, 121 (1997).
Chiou, D.-Y., Chen, M.-Y., Chang, M.-W. & Deng, H.-C. J. J. J. o. A. P. Characterization and optimization design of the polymer-based capacitive micro-arrayed ultrasonic transducer. Jpn. J. Appl. Phys. 46, 7496 (2007).
Lu, N. Mechanics of Hard Films on Soft Substrates (Harvard University, 2009).
Mimoun, B., Pham, H. T., Henneken, V. & Dekker, R. Residue-free plasma etching of polyimide coatings for small pitch vias with improved step coverage. J. Vac. Sci. Technol. B Nanotechnol. Microelectron.: Mater. Process. Meas. Phenom. 31, 021201 (2013).
Zulfiqar, A., Pfreundt, A., Svendsen, W. E. & Dimaki, M. Fabrication of polyimide based microfluidic channels for biosensor devices. J. Micromech. Microeng. 25, 035022 (2015).
Hong, G., Holmes, A. S. & Heaton, M. E. Su8 resist plasma etching and its optimisation. Symposium on Design, Test, Integration and Packaging of MEMS/MOEMS 2003. Vol 10, 268–271 (2003).
Acknowledgements
This work acknowledges the support received from the National Research Foundation of Korea (Grant Nos. NRF-2019R1A2C2086085, NRF-2021R1A4A1031437, and NRF-2018M3A7B4071109). H.C. also acknowledges the support provided by the National Institutes of Health (Award Nos. R21EB030140, U01DA056242, and R61HL154215).
Author information
Authors and Affiliations
Contributions
Designing research: J.Y.L., S.H.P., Y.J.K., Y.U.C., I.J.C., K.J.Y. Device fabrication: J.Y.L., S.H.P., Y.U.C. FEA simulation: J.Y.L., S.H.P., Y.G., L.B., H.Y.C. Data analysis: J.Y.L., S.H.P., Y.J.K., Y.U.C. In vitro experiments: J.Y.L., S.H.P. In vivo experiments: J.Y.L., S.H.P., Y.J.K. Cell viability, biocompatibility test: H.G.S., A.Z., Y.M.J. Histology: E.B.H., M.H.N. Writing—original draft: J.Y.L., S.H.P., Y.J.K., Y.U.C. Writing—review & editing: J.Y.L., S.H.P., Y.J.K., Y.U.C., I.J.C., H.Y.C., K.J.Y.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, J.Y., Park, S.H., Kim, Y. et al. Foldable three dimensional neural electrode arrays for simultaneous brain interfacing of cortical surface and intracortical multilayers. npj Flex Electron 6, 86 (2022). https://doi.org/10.1038/s41528-022-00219-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41528-022-00219-y
This article is cited by
-
Fully bioresorbable hybrid opto-electronic neural implant system for simultaneous electrophysiological recording and optogenetic stimulation
Nature Communications (2024)
-
Fully implantable and battery-free wireless optoelectronic system for modulable cancer therapy and real-time monitoring
npj Flexible Electronics (2023)
