
Abstract
The solubility of luminescent materials is a key parameter to improve the electroluminescent performances of solution-processed organic light-emitting diodes (OLEDs). The through-space charge transfer (TSCT) materials provide an alternative to introduce the solubilizing groups (SGs) to the linker. Herein, the tert-butyl and n-hexyl groups are introduced as SGs at C7 positions of spiro structure, named C6-DMB and tBu-DMB, away from the acceptor. This has no influence on the photophysical properties of the parent TSCT molecule. Highly efficient solution-processed OLEDs were demonstrated with the maximum external quantum efficiencies of 21.0% and 21.7%, respectively. To the best of our knowledge, these are champions in the state-of-the-art solution-processed OLEDs with TSCT emitters. This work confirmed our conjecture of constructing highly efficient soluble emitters by transforming an outstanding TSCT material from thermal evaporation to solution-processed OLEDs with SGs simply integrated on the ‘bridge’ linker.
Similar content being viewed by others
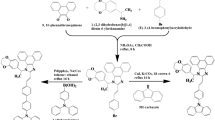

Introduction
Solution-processed organic light-emitting diodes (OLEDs) were first developed by Friend and co-workers over three decades ago1, which stimulated a series of thin-film technologies including spin-coating, blade coating, drop-casting, ink-jet printing, and roll-to-roll process, to reduce the material consumption, cut the cost and simplify the operation procedure2,3,4,5,6. The soluble emitters are evolved from conventional conjugated fluorescent materials to the emerging thermally activated delayed fluorescence (TADF)7 materials since the latter can harvest singlet and triplet excitons simultaneously by reverse intersystem crossing (rISC) process3. To achieve 100% exciton utilization during the TADF process, the materials should have a tiny energy gap (ΔEST) between their lowest singlet (S1) and triplet (T1) excited states to increase the population of the emissive states by strong coupling between S1/T1 states. In this regard, the separation of donor/acceptor is required in molecular construction to minimize the exchange integral which is crucial to reducing the ΔEST8,9,10,11,12,13.
TADF materials employed with solution-processed technology should possess good solubility to achieve high-efficiency devices, which can be achieved by the introduction of solubilizing groups (SGs) (Fig. 1)4,14,15,16. In general, the SGs will affect the highest occupied molecular orbital (HOMO) or lowest unoccupied molecular orbital (LUMO) if they are appended on either donor or acceptor, and thus vary the emissive colors of solution-processable TADF materials. The derivatized SGs on the π-linker will force the D-A structure further twisted which is accompanied by the bathochromic shift owing to the enhanced charge transfer (CT) feature17. Besides the traditional D-π-A type TADF molecules, now the noncovalent D/A TADF molecules have attracted researchers’ attention. Like the development of solution-processable TADF material, chemists first tried to modify the donor or acceptor to modulate solubility and, inevitably, tailor the electron-donating or -withdrawing effects18,19. Unlike the conjugated D/A interaction in the D-π-A structure, the linkers in noncovalent D/A TADF molecules were used to control their spatial distance and trigger through-space charge transfer (TSCT), which meant the charge transfer happened between two spatially isolated moieties without a chemical bond20,21,22,23,24. Theoretically, the linker is much more inert in noncovalent D/A interaction, i.e., an SG introduced to the linker can hardly influence the D/A intensities or vary the dihedral angles between them. However, this aspect hasn’t been verified in any other reports and thus needs further exploration.

Upper: design strategy; middle: examples for the introduction of SGs; bottom: the molecular structures in this work.
To this end, we designed two noncovalent D/A TADF emitters with good solubility, named C6-DMB and tBu-DMB. These materials are derived from our previous work, i.e., DM-B25, which was composed of 2,4,6-triphenyl-1,3,5-triazine (TRZ) and triphenylamine as D/A with a rigid ‘bridge’ linker based on the coplanar conformation due to the strong steric hindrance of C1-substituted spiro molecule26,27,28. The coplanar conformation made DM-B an obvious TSCT feature. In this work, we introduced SGs to the C7-site of the ‘bridge’ linker part and kept the other side of the fluorene away from the acceptors. The position where the n-hexyl chain (C6-DMB) or tert-butyl (tBu-DMB) is substituted has the least conjugation which can lessen the influence on the molecular skeleton to maintain the small ΔEST (0.17 eV) and high photoluminescence quantum yield (PLQYs) (89% and 98%, respectively). The corresponding solution-processed doped OLEDs with C6-DMB and tBu-DMB show the maximum EQEs of 21.0% and 21.7%, respectively. The electroluminescent performances are superior to the parent molecule DM-B, representing that the introduction of the SGs is an effective method to improve the device performances of solution-processed TADF TSCT emitters.
Results and discussion
Synthesis method
The solution-processed emitters, named C6-DMB and tBu-DMB, were synthesized following three steps, as shown in Fig. 2. The acceptors were connected with compounds 1 and 2, which had SGs already attached to them, by the Suzuki-Miyaura cross-coupling reaction to get the intermediates 3 and 4. Following nucleophilic addition and Friedel-Crafts cyclization, we got our target molecules, C6-DMB and tBu-DMB, with good solubility, enabling us to fabricate the solution-processed devices.

a Synthetic route and b theoretical calculation of C6-DMB and tBu-DMB. Conditions: (i) Pd(PPh3)4, K2CO3, toluene, ethanol, H2O, 105 °C; (ii) n-BuLi, THF, -78 °C; (iii) acetic acid, hydrochloric acid, 100 °C.
Theoretical calculations
To explain the relationship between the molecular structures and properties of the target molecules, theoretical calculations were performed with density functional theory (DFT) and time-dependent DFT (TD-DFT) under the ωb97xd29/def2-SVP30 level and ω-tuned29,30 TDA-ωb97xd29/def2-SVP30 level for the ground and excited states, respectively. The ω-tuned ranged separated function was suitable to analyze the charge transfer characteristics of TSCT emitters, based on our previous work25. The HOMO/LUMO distributions of C6-DMB and tBu-DMB are shown in Fig. 2b. The HOMOs of C6-DMB and tBu-DMB were mainly distributed on the triphenylamine part as the donor group, and the LUMOs were distributed on the TRZ parts as the acceptor group. The complete separation of HOMOs/LUMOs of C6-DMB and tBu-DMB led to the small ΔESTs (see Table 1), indicating the potential TADF feature. The independent gradient model (IGM)31 analysis was also performed based on the optimized ground state geometry, shown in Supplementary Fig. 1. The π-π interaction (the green color on the isosurface) between the D/A units occurred in both molecules.
Crystal structure
In addition to 1H-NMR, 13C-NMR, and mass spectrometry, we also obtained and characterized the single-crystal structures. The single crystals of C6-DMB and tBu-DMB were successfully obtained by slow volatilization from the dichloromethane solvent and X-ray structures were shown in Fig. 3a, b. Besides, the crystal structure of the parent molecule DM-B molecule was also shown in Fig. 3c for comparison. The distances between the TRZ plane and the triphenylamine plane connected to the ‘bridge’ fluorene for C6-DMB and tBu-DMB were 3.03 and 3.05 Å, respectively, smaller than that of DM-B (3.16 Å). These DM-B derivatives had smaller D/A distances in a face-to-face manner which led to better spatial interaction. The angles between the triphenylamine unit and the fluorene group in the C6-DMB and tBu-DMB were 133.9° and 127.2°, respectively, slightly larger than that of DM-B (123.4°). The dihedral angles between the TRZ plane and the fluorene group in C6-DMB and tBu-DMB are 68.4° and 66.6°, respectively, which were similar to that of the parent molecule DM-B (70°). Furthermore, the intermolecular distance was much closer in tBu-DMB (10.98 Å) than that of C6-DMB (21.09 Å). Therefore, the packing arrangements of tBu-DMB were tightly packed more closely than that of C6-DMB (Fig. 3d. e). This difference was caused by the more flexible and longer spatial structure of the n-hexyl group than the tert-butyl group.

Crystal structures of (a) C6-DMB, (b) tBu-DMB, (c) DM-B, and molecular packing arrangements of (d) C6-DMB and (e) tBu-DMB. Hydrogen atoms have been omitted for the sake of clarity. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centers supplementary publication no. CCDC-2153900 (C6-DMB), CCDC- 2153902 (tBu-DMB). These data can be obtained free of charge from The Cambridge Crystallographic Data Centre: https://www.ccdc.cam.ac.uk/structures-beta/.
Photophysical properties
As shown in Fig. 4a, b, the ultraviolet-visible (UV-vis) absorption spectra of C6-DMB and tBu-DMB at room temperature together with their fluorescence and phosphorescence spectra at 77 K in the toluene solution (10-5 M) were recorded. The photophysical properties of the two target products were approximately the same. Their absorption profiles both begin at about 330 nm, while their absorption peaks at about 300 nm represent the π-π* transition. In addition, the fluorescence peak wavelengths of C6-DMB and tBu-DMB at 77 K were 446.7 and 446.4 nm, respectively. The peak wavelengths of their phosphorescent spectra were 440.8 and 440.7 nm, respectively. Therefore, the ΔESTs of C6-DMB and tBu-DMB were both 0.17 eV, which meets the requirements of the TADF process. The ΔESTs were the same as that of the parent molecule DM-B (0.17 eV). To compare the utilization efficiency of excitons, their PLQYs of the emitters doped into a matrix of 1,3-di(9H-carbazol-9-yl)benzene (mCP) at a weight ratio of 30 wt% under nitrogen atmosphere were also measured, i.e., 89% and 98%, respectively, for C6-DMB and tBu-DMB, (see Supplementary Table 3). To more intuitively understand their TADF features, their transient photoluminescence properties of the doped emitting layers (30 wt%) at different temperatures ranging from 100 to 300 K were recorded, shown in Fig. 4c, d. C6-DMB and tBu-DMB both exhibited an obvious prompt decay component and a delayed fluorescence decay profile at room temperature. The prompt lifetimes (τps) of C6-DMB and tBu-DMB at room temperature are 102 and 99 ns, respectively. Moreover, their delayed lifetimes (τds) are 5.99 and 5.58 μs, respectively. When decreasing the temperature, their lifetimes of prompt and delayed fluorescence decline significantly. Their rISC rates (krISC) were 1.31 × 105 and 1.74 × 105 s-1, respectively, which were similar to the parent molecule DM-B (1.8 × 105 s-1). These results showed that both C6-DMB and tBu-DMB were highly emissive and featured obvious TADF properties. Regarding their similar lifetimes (τp and τd), it is confirmed that the introduction of SGs had a tiny effect on their TADF characteristics.

UV–vis absorption spectra, fluorescence, and phosphorescence spectra at 77 K of (a) C6-DMB and (b) tBu-DMB. Temperature-dependent transient PL decay characteristics of (c) C6-DMB and (d) tBu-DMB in doped (30 wt%) mCP films.
Electrochemistry properties
Cyclic voltammetry was used to test the electrochemical properties of C6-DMB and tBu-DMB under the condition of dehydrated dichloromethane (DCM) or N,N-Dimethylformamide (DMF) as the solvent, 0.1 M Bu4NPF6 as the supporting electrolyte, and the ferrocenium/ferrocene couple (Fc+/Fc) as the internal reference. The HOMO/LUMO energy levels of C6-DMB and tBu-DMB are summarized in Table 1, which are obtained by their first oxidation and reduction waves (Supplementary Fig. 2). According to the formula EHOMO = -−(Eox (vs. Fc+/Fc) + 4.8), their potential of HOMO energy levels (EHOMO) were -5.22 and -5.21 eV for C6-DMB and tBu-DMB, respectively. And their LUMO energy levels (ELUMOs) were calculated as -2.80 and -2.79 eV, respectively, according to the formula ELUMO = -(Ered (vs. Fc+/Fc) + 4.8). The above experimental values were consistent with the calculated values. These similar results indicated the slight influence of the SGs in both molecules on their electrochemical properties.
Thermal stability
To ensure their good stability for fabricating the devices, the thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were recorded, shown in Supplementary Fig. 3 and Table 1. The decomposition temperatures (Tds) of C6-DMB and tBu-DMB at weight loss of 5% were 416 and 405 °C, and the glass transition temperatures (Tgs) were located at 104 and 106 °C, respectively. These results revealed the good thermal stabilities of both molecules. These similar results also indicated the negligible influence of the C7-site SGs on the physical properties of both molecules.
Device performance
The high PLQYs as well as tiny ΔESTs of C6-DMB and tBu-DMB make them candidates for high-efficiency TADF OLEDs. In order to evaluate their electroluminescent (EL) characteristics, four solution-processed OLEDs with the doped emitting layers (EMLs) were fabricated with the structures: ITO/PEDOT:PSS (70 nm)/EML (40 nm)/DPEPO (10 nm)/TmPyPB (50 nm)/Liq (1 nm)/Al (100 nm) as shown in Supplementary Fig. 4. ITO was used as the anode material, poly(3,4-ethylenedioxythiophene)-poly(styrene sulfonate) (PEDOT: PSS) was used as the hole-injection layer, bis[2-(diphenylphosphino)phenyl] ether oxide (DPEPO) served as the hole blocking layer, 1,3,5-tri[(3-pyridyl)-phen-3-yl]benzene (TmPyPB) was utilized as the electron-transporting layer, and 8-hydroxyquinolinolato-lithium (Liq) was employed as the electron-injection layer. Finally, aluminum acted as the cathode. The EMLs were composed of C6-DMB or tBu-DMB respectively as dopants and 1,3-di(9H-carbazol-9-yl)benzene (mCP) as the host material. The doping ratios of 10 and 30 wt% were selected to optimize the performances. At a doping ratio of 30 wt%, the devices exhibited the same EL peak at 492 nm, shown in Fig. 5a. The maximum EQEs of the solution-processed devices with C6-DMB and tBu-DMB were 21.0% and 21.7%, respectively (Fig. 5f). The results indicated that C6-DMB and tBu-DMB had out-performed the state-of-the-art solution-processed TSCT TADF emitters. The maximum current efficiency (CE) and power efficiency (PE) of C6-DMB were 47.0 cd A-1 and 30.7 lm W-1, respectively, which were slightly higher than those of tBu-DMB (49.6 cd A-1 and 31.4 lm W-1, respectively), shown in Fig. 5d and e. When increasing the doping concentration of the emitters from 10 wt% to 30 wt%, the EQEs became higher. However, the corresponding Commission Internationale de L’Eclairage (CIE) coordinates were slightly red-shifted from (0.21, 0.34)/(0.21, 0.35) to (0.21, 0.38)25. The current density and luminance to voltage characteristics are shown in Fig. 5b, c, and Table 2.

a EL spectra; b current density to voltage curves; c luminance to voltage curves; d current efficiency to current density curves; e power efficiency to current density curves; f EQE to current density curves of doped devices.
Considering the bipolar nature of D/A structured TSCT emitters, the non-doped solution-processed devices utilizing C6-DMB or tBu-DMB as EMLs were also fabricated with structure: ITO/PEDOT:PSS (70 nm)/neat EML (40 nm)/DPEPO (10 nm)/TmPyPB (50 nm)/Liq (1 nm)/Al (100 nm). Compared with the doped devices, these non-doped devices exhibited the inferior EQEs of 14.3% and 8.7%, respectively, as shown in Fig. 6. tBu-DMB had a closer crystal packing mode than C6-DMB, owing to the flexible long chains. This accounts for its worse device performance because of the concentration quenching phenomenon of TADF materials. Their PEs and CEs were also significantly lower than those of the doped devices, attributed to the enhanced concentration quenching and imbalance of charge injection15.

a EL spectra; b current density to voltage curves; c luminance to voltage curves; d current efficiency to current density curves; e power efficiency to current density curves; f EQE to current density curves of non-doped devices.
In conclusion, we reported two TADF sky-blue solution-processable emitters, C6-DMB and tBu-DMB, by introducing different alkyl chains as SGs, which can maintain the efficient luminance properties. C6-DMB and tBu-DMB featuring TSCT, both showed similar PL spectra, identical ∆ESTs of 0.17 eV, and similar face-to-face distances between D/A of ~3.0 Å. The inherited properties from the parent molecule DM-B indicate that the introduction of alkyl chains at the C7-site of fluorene had negligible influence on their chemical, electrochemical, and TADF properties. Overall, the PLQY of tBu-DMB (98%) was higher than C6-DMB (89%). The solution-processed devices with C6-DMB and tBu-DMB possessed the superior EQEs of 21.0% and 21.7%, respectively, compared with the parent molecule DM-B (14%)18. It is noted that the utilization of SGs in fluorescence materials, like polyfluorene32,33, polycarbazole34, and perylene diimide35, also have little impact on their luminance performance. But this is not the case for conventional D-π-A TADF emitters as their functional units are susceptible to the SGs. We believe this approach of introducing SGs on the linker of the TSCT emitters would be applicable to many other TSCT emitters based on the simple engineering for solution-processed devices.
Methods
Synthetic method
C6-DMB (tBu-DMB): Under argon protection, n-BuLi was added dropwise to the dehydrated THF (25 mL) solution of 2-bromotriphenylamine in a 50 mL Shrek bottle at -78 °C. After stirring at −78 °C for 1 h, add compound 3 (compound 4). After stirring at room temperature for 2 h, the resulting mixture was quenched with methanol, and then concentrated by rotary evaporation and washed with methanol to obtain a crude product. The crude product was then dissolved in acetic acid (20 ml) and heated to 70 °C, and then 2 ml of concentrated hydrochloric acid (37 wt%) was added to the mixture, stirred overnight at 100 °C. The resulting mixture was cooled to room temperature and poured into ice water (100 mL). The precipitate was separated by filtration and purified with silica gel column chromatography (CH2Cl2/petroleum ether 10:1-3:1) to give C6-DMB (tBu-DMB) as yellow solid.
Device fabrication
The device facility is Suzhou Fangsheng FS-300. The ITO coated glass substrate was preliminarily cleaned in deionized water, acetone, and ethanol in an ultrasonic bath, respectively, and dried with N2. After treated with UV-ozone (UVO) for 20 min, the ITO substrate was coated with a layer of PEDOT: PSS (8000) at 4000 rpm for hole injection, and then the PEDOT: PSS layer was annealed at 120 °C for 10 min in air. Subsequently, the substrate was transferred into an N2-filled glovebox. The emitters and host dissolved and mixed with the desired weight ratios in chlorobenzene solution (10 mg ml-1) were then spin-coated onto PEDOT: PSS at 1000 rpm. After baking the emitting layer at 50 °C for 10 min, the sample was loaded into a vacuum chamber. The hole blocking layer (DPEPO), the electron transporting layer (TmPyPB), the electron injecting layer (Liq), and Al cathode were consecutively evaporated in the vacuum chamber, consecutively. Then, the device was sealed with curable UV resin in an N2-filled glove box.
Data availability
The datasets generated and analyzed during the current study are available from the corresponding authors on reasonable request.
References
Burroughes, J. H. et al. Light-emitting diodes based on conjugated polymers. Nature 347, 539–541 (1990).
Huang, T., Jiang, W. & Duan, L. Recent progress in solution processable TADF materials for organic light-emitting diodes. J. Mater. Chem. C 6, 5577–5596 (2018).
Liu, Y., Li, C., Ren, Z., Yan, S. & Bryce, M. R. All-organic thermally activated delayed fluorescence materials for organic light-emitting diodes. Nat. Rev. Mater. 3, 18020 (2018).
Choi, M.-C., Kim, Y. & Ha, C.-S. Polymers for flexible displays: from material selection to device applications. Prog. Polym. Sci. 33, 581–630 (2008).
Lian, C. et al. Flexible organic light-emitting diodes for antimicrobial photodynamic therapy. npj Flex. Electron. 3, 18 (2019).
Maasoumi, F. et al. An external quantum efficiency of >20% from solution-processed poly(dendrimer) organic light-emitting diodes. npj Flex. Electron. 2, 27 (2018).
Uoyama, H., Goushi, K., Shizu, K., Nomura, H. & Adachi, C. Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492, 234–238 (2012).
Zheng, Q. et al. Pure hydrocarbon host materials based on 9,9’-spirobifluorene/naphthalene hybrid. Chin. J. Org. Chem. 42, 572–579 (2022).
Qu, Y.-K., Zheng, Q., Fan, J., Liao, L.-S. & Jiang, Z.-Q. Spiro compounds for organic light-emitting diodes. Acc. Mater. Res. 2, 1261–1271 (2021).
Yang, S.-Y., Qu, Y.-K., Liao, L.-S., Jiang, Z.-Q. & Lee, S.-T. Research progress of intramolecular π-stacked small molecules for device applications. Adv. Mater. 34, 2104125 (2022).
Qu, Y.-K. et al. Steric modulation of spiro structure for highly efficient multiple resonance emitters. Angew. Chem. Int. Ed. 61, e202201886 (2022).
Wang, X.-Q. et al. Multi-layer π-stacked molecules as efficient thermally activated delayed fluorescence emitters. Angew. Chem. Int. Ed. 60, 5213–5219 (2021).
Li, H.-C. et al. Spatial donor/acceptor architecture for intramolecular charge-transfer emitter. Chin. Chem. Lett. 32, 1245–1248 (2021).
Bernius, M. T., Inbasekaran, M., O’Brien, J. & Wu, W. Progress with light-emitting polymers. Adv. Mater. 12, 1737–1750 (2000).
Zou, Y., Gong, S., Xie, G. & Yang, C. Design strategy for solution-processable thermally activated delayed fluorescence emitters and their applications in organic light-emitting diodes. Adv. Opt. Mater. 6, 1800568 (2018).
Shao, S. & Wang, L. Through-space charge transfer polymers for solution-processed organic light-emitting diodes. Aggregate 1, 45–56 (2020).
Cui, L.-S. et al. Controlling singlet–triplet energy splitting for deep-blue thermally activated delayed fluorescence emitters. Angew. Chem. Int. Ed. 56, 1571–1575 (2017).
Wang, T.-T. et al. π-Stacked thermally activated delayed fluorescence emitters with alkyl chain modulation. CCS Chem. 3, 1757–1763 (2021).
Peng, C.-C. et al. Highly efficient thermally activated delayed fluorescence via an unconjugated donor–acceptor system realizing EQE of over 30%. Adv. Mater. 32, 2003885 (2020).
Tsujimoto, H. et al. Thermally activated delayed fluorescence and aggregation induced emission with through-space charge transfer. J. Am. Chem. Soc. 139, 4894–4900 (2017).
Wada, Y., Kubo, S. & Kaji, H. Adamantyl substitution strategy for realizing solution-processable thermally stable deep-blue thermally activated delayed fluorescence materials. Adv. Mater. 30, 1705641 (2018).
Xue, Q. & Xie, G. Thermally activated delayed fluorescence beyond through-bond charge transfer for high-performance OLEDs. Adv. Opt. Mater. 9, 2002204 (2021).
Zheng, X. et al. Achieving 21% external quantum efficiency for nondoped solution-processed sky-blue thermally activated delayed fluorescence OLEDs by means of multi-(donor/acceptor) emitter with through-space/-bond charge transfer. Adv. Sci. 7, 1902087 (2020).
Hu, J. et al. Developing through-space charge transfer polymers as a general approach to realize full-color and white emission with thermally activated delayed fluorescence. Angew. Chem. Int. Ed. 58, 8405–8409 (2019).
Tang, X. et al. Highly efficient luminescence from space-confined charge-transfer emitters. Nat. Mater. 19, 1332–1338 (2020).
Poriel, C., Sicard, L. & Rault-Berthelot, J. New generations of spirobifluorene regioisomers for organic electronics: tuning electronic properties with the substitution pattern. Chem. Commu. 55, 14238–14254 (2019).
Sicard, L. et al. 1-Carbazolyl spirobifluorene: synthesis, structural, electrochemical, and photophysical properties. J. Phys. Chem. C 123, 19094–19104 (2019).
Sicard, L. et al. Spirobifluorene regioisomerism: a structure–property relationship study. Chem. Eur. J. 23, 7719–7727 (2017).
Chai, J.-D. & Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Lefebvre, C. et al. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 19, 17928–17936 (2017).
Chen, P. et al. Optimization of opto-electronic property and device efficiency of polyfluorenes by tuning structure and morphology. Polym. Int. 55, 473–490 (2006).
Wu, Z. et al. High-triplet-energy poly(9,9′-bis(2-ethylhexyl)-3,6-fluorene) as host for blue and green phosphorescent complexes. Adv. Mater. 20, 2359–2364 (2008).
van Dijken, A. et al. Carbazole compounds as host materials for triplet emitters in organic light-emitting diodes: polymer hosts for high-efficiency light-emitting diodes. J. Am. Chem. Soc. 126, 7718–7727 (2004).
Jiang, W., Li, Y. & Wang, Z. Tailor-made rylene arrays for high performance n-channel semiconductors. Acc. Chem. Res. 47, 3135–3147 (2014).
Acknowledgements
The authors acknowledge financial support from the National Natural Science Foundation of China (Nos. 51873139, 61961160731, 62175171, and 22175124) and the Natural Science Foundation of Jiangsu Province of China (BK20220057). This project is also funded by the Suzhou Science and Technology Plan Project (SYG202010), Suzhou Key Laboratory of Functional Nano & Soft Materials, Collaborative Innovation Centre of Suzhou Nano Science & Technology (Nano-CIC), the ‘111’ Project, and Joint International Research Laboratory of Carbon-Based Functional Materials and Devices.
Author information
Authors and Affiliations
Contributions
Q.Z., X.W., and Z.J. conceived the idea and designed the experiments. Q.Z. and X.W. synthesized the compounds under the supervision of Z.J. G.X. carried out the device characterizations. Q.Z. and X.W. cross-checked the LED measurement and photophysical experiments and analyzed the results Y.Q. and Q.Z. performed time-resolved photophysical properties and the computational experiments. Y.Q. conducted the crystal structure measurements and analyzed the results. G.X., L.L., and Z.J. supervised the work. Q.Z. and Z.J. analyzed all data and wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, Q., Wang, XQ., Qu, YK. et al. Solution-processable through-space charge-transfer emitters via solubilizing groups modification. npj Flex Electron 6, 83 (2022). https://doi.org/10.1038/s41528-022-00212-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41528-022-00212-5
