
Abstract
Astronauts have been previously shown to exhibit decreased salivary lysozyme and increased dental calculus and gingival inflammation in response to space flight, host factors that could contribute to oral diseases such as caries and periodontitis. However, the specific physiological response of caries-causing bacteria such as Streptococcus mutans to space flight and/or ground-based simulated microgravity has not been extensively investigated. In this study, high aspect ratio vessel S. mutans simulated microgravity and normal gravity cultures were assessed for changes in metabolite and transcriptome profiles, H2O2 resistance, and competence in sucrose-containing biofilm media. Stationary phase S. mutans simulated microgravity cultures displayed increased killing by H2O2 compared to normal gravity control cultures, but competence was not affected. RNA-seq analysis revealed that expression of 153 genes was up-regulated ≥2-fold and 94 genes down-regulated ≥2-fold during simulated microgravity high aspect ratio vessel growth. These included a number of genes located on extrachromosomal elements, as well as genes involved in carbohydrate metabolism, translation, and stress responses. Collectively, these results suggest that growth under microgravity analog conditions promotes changes in S. mutans gene expression and physiology that may translate to an altered cariogenic potential of this organism during space flight missions.
Similar content being viewed by others

Introduction
Streptococcus mutans is a primary causative agent of dental caries, demonstrated by its isolation from carious tooth lesions in humans,1 its ability to initiate caries in germ-free rodent models of infection,2 and the established link between high levels of this bacterium in the oral cavity and active caries.3 Although preventable by fluoridation of water and various dental procedures,4, caries remains the most common chronic disease among children, and remains untreated in up to 30% of adults over 35 years-old.5 S. mutans transient bacteremias can also initiate endocarditis in at-risk patient groups.6 Successful colonization of the oral cavity and persistence in multi-species dental plaque by S. mutans is inherently dependent on both its ability to form biofilm and its rapid metabolism of carbohydrates by the glycolytic pathway. The low pH that results from accumulation of acidic metabolic end-products favors the growth of S. mutans and other acid-tolerant species relative to healthy plaque microflora, as well as the eventual demineralization of tooth enamel and the development of caries. Adherence of S. mutans to the tooth surface and subsequent biofilm development are mediated by several bacterial factors, including production of sucrose-dependent extracellular glucan polymers by glucosyltransferase enzymes,7 specific cell surface adhesins,8, 9 the major autolysin AtlA,10 the quorum-sensing competence (com) system,11 and extracellular DNA.12
To protect the health of astronauts during space flight missions, it is critical to understand the effects of this environment on both host biology and the host microbiota. Simulated microgravity exposure has been shown to cause increased mandibular and alveolar bone loss13 and decreased saliva flow,14 significant predisposing host factors that could contribute to development of caries and/or periodontal disease. Traditionally, the approach to maintaining astronaut oral health during space flight missions has been routed in preventative dentistry, whereby astronauts are subjected to rigorous preflight dental screening and treatment.15 However, in the face of future long-term space flight missions, research and discovery of new methods for maintaining oral hygiene and dental health under a microgravity environment are required.15 Previous studies on the effects of spaceflight on the oral microbiota have primarily focused on culture-dependent methods of quantifying the numbers of oral bacteria in human subjects before and after spaceflight.16,17,18 Furthermore, astronauts have been shown to exhibit decreased salivary lysozyme and increased dental calculus and gingival inflammation in response to space flight.16 However, assessment of the physiology and virulence potential of oral pathogens such as S. mutans under controlled microgravity analog conditions has been relatively lacking. In this respect, rotating wall vessel bioreactors are a common technology used to grow bacteria under microgravity analog conditions in ground-based studies. When completely filled with media (lacking air bubbles or “head space”) and rotated on the axis perpendicular to the Earth’s gravitational vector, these reactors maintain a low-shear environment (<1 dyn/cm2) whereby gravitational vectors are randomized over the surface of the cells or particles contained within the vessel.19 As such, this simulated environment models “weightlessness” by counteracting gravitational forces that would otherwise promote cellular sedimentation.19,20 The low-shear force experienced in this microgravity analog system boasts the additional advantage of mimicking the low-shear force of saliva flow (<0.8 dyn/cm2) experienced by plaque bacteria in the oral cavity.21 In this study, high-aspect rotating vessels (HARVs) were therefore used to ascertain the effects of simulated microgravity on S. mutans gene expression, physiology, oxidative stress resistance, and competence, using both culture-dependent and “-omic” (metabolomics and RNA-seq) approaches.
Results
S. mutans HARV growth curves and cell aggregation
To probe the effect of simulated microgravity growth on S. mutans, continuous growth curves (whereby cell samples were removed from the same HARV at different time points) were performed (n = 3 independent experiments each) in biofilm media (BM) containing 11 mM glucose and 10 mM sucrose over a 24 h period. In this analysis, no statistically significant differences in CFU/ml at each time point were observed (Supplemental Fig. S1). An initial decrease in CFU/ml was observed in both the simulated microgravity and normal gravity HARV cultures between t = 0 (time of inoculation) and t = 2 h growth, which was likely a function of sucrose-dependent cell aggregation. Very small differences in cell viability patterns in early stationary phase (6–8 h growth) suggest that the simulated microgravity HARV culture may have entered stationary phase slightly sooner than the normal gravity HARV culture in this experiment. As well, the simulated microgravity culture persisted in stationary phase until approximately 12 h growth, followed by a somewhat rapid death phase, with a 1-log loss of viability between 12 and 24 h growth. In contrast, the viability of normal gravity culture underwent a more gradual decrease in cell viability between 12 and 24 h of growth.
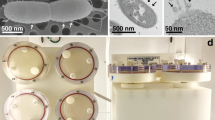
Monitoring of cell growth by CFU/ml in the continuous growth curve experiments described above required constant disruption of the HARV cultures, making it difficult to assess potential macroscopic differences in cell aggregation in the simulated microgravity and normal gravity conditions. Therefore, these growth curves were also performed using an “end-point” approach, whereby simulated microgravity and normal gravity HARV cultures (n = 2 independent experiments per time point per growth condition) were grown undisturbed in replicate HARV vessels for t = 4, 6, 8, 12, 24, and 48 h (Fig. 1). Using this end-point approach, no significant differences in CFU/ml were observed between the simulated microgravity and normal gravity HARV cultures at the assessed time points (Fig. 1), and the initial drop in CFU/ml observed early in the growth curves of the continuous cultures (Supplemental Fig. S1) was not observed in these end-point cultures. As well, both the simulated microgravity and normal gravity cultures underwent the same pattern of growth when monitored with this end-point approach: Log phase growth occurred between 0 and 6 h, and both cultures were in early stationary phase at 8 h growth, followed by entry into late stationary phase/death phase between 24 and 48 h growth. Interestingly, differences in macroscopic cell aggregation were consistently observed between the simulated microgravity and normal gravity HARV conditions when using the end-point growth analysis. Specifically, the simulated microgravity condition tended to form round/compact cellular structures, whereas the aggregates in the normal gravity growth condition tended to form less compact structures (Fig. 2).

End-point growth curve analysis of S. mutans simulated microgravity and normal gravity HARV cultures. Simulated microgravity (dark gray bars) and normal gravity (light gray bars) HARV end-point growth was measured by serial dilution and CFU/ml plating at t = 0 (time of inoculation; average of n = 4 independent experiments), and t = 4, 6, 8, 12, 24, and 48 h postinoculation (average of n = 2 independent experiments per time point per growth condition), as described in “Materials and methods”. The vertical dotted line on the graph indicates that the t = 24 and 48 h data were obtained in a separate experimental run from the t = 4, 6, 8, and 12 h time points. Error bars = standard error of the mean (SEM)

Representative S. mutans end-point HARV cultures grown for 8 h in biofilm media under simulated microgravity (a) and normal gravity (b) conditions. Note the structural differences of the self-aggregating bacterial cells under each condition (indicated by arrows)
Metabolomics analysis of “end-point” HARV cultures
To monitor potential differences in the metabolome of simulated microgravity and normal gravity HARV cultures, cell pellets and culture supernatants were harvested from a second end-point HARV culture experiment (n = 3 independent experiments per time point per growth condition) at t = 4, t = 8, and t = 24 h samples, corresponding to log-phase, early stationary phase, and late stationary/death phase, respectively (Supplemental Fig. S2 ). Given that macroscopic differences in the self-aggregating clusters of S. mutans cells were consistently observed at the t = 8 h time point (Fig. 2), RNA-seq analysis was also performed on the t = 8 h samples. Principle component analysis (PCA) of metabolomics data generated from t = 4, 8, and 24 h HARV cultures (Supplemental Files S2 and S3) revealed a strong relationship between the time of growth (t = 4, 8, and 24 h) and metabolite profile for each of the simulated microgravity and normal gravity cultures (Fig. 3). However, a much weaker “time-treatment” relationship existed when comparing the metabolite profile between simulated microgravity vs. normal gravity cultures at each time point. Welch’s two sample t tests revealed 8, 14, and 10 statistically significant differences (p ≤ 0.05) in cellular metabolite production between cells grown under normal gravity and microgravity conditions at the 4, 8, and 24 h time points, respectively (Supplemental File S2). Likewise, 21, 21, and 23 statistically significant differences were observed between the corresponding culture supernatant samples at these time points (Supplemental File S3). Keeping in mind the caveat that the number of significant differences at each time point were close to that expected by random chance alone (5% error rate), the significant changes of cell-associated and supernatant metabolites of the 8 h HARV cultures are summarized in Table 1, and their potential relationship to the corresponding 8 h RNA-seq data are discussed below.

Principle component analysis (PCA) of cellular (a) and supernatant (b) metabolites. Values on X and Y axis labels = % of the total eigen values
RNA-seq analysis
Because macroscopic structural differences between the S. mutans simulated microgravity and normal gravity HARV cultures were consistently observed at 8 h growth (early stationary phase), we chose to perform RNA-seq analysis on n = 3 independent biological samples per growth condition (simulated microgravity and normal gravity) isolated from this time point. Compared to the metabolomics data, differential expression analysis of the 8 h RNA-seq data revealed a large number of statistically significant gene expression changes in normal gravity relative to simulated microgravity growth (Supplemental File S4). Specifically, 94 genes were up-regulated (log2 fold-change ≥1.0) and 153 genes were down-regulated (log2 fold-change ≤−1.0) in normal gravity cultures relative to simulated microgravity cultures. These patterns in gene expression were confirmed by qPCR for a subset of genes (Supplemental Table S1). Functional classification (Fig. 4) revealed that expression of a large number of genes associated with transposon sequences, as well as several genes encoding predicted phage proteins, were down-regulated in normal gravity cultures relative to simulated microgravity growth. A second trend of interest was the significant down-regulated expression of genes involved in carbohydrate metabolism, including various phosphotransferase (PTS) systems, the carbon catabolite protein regulator ccpA, and the redox sensitive transcriptional regulator rex, in the 8 h normal gravity cultures relative to the simulated microgravity cultures (Fig. 4 and Supplemental File S4). Increased expression of genes encoding ribosomal proteins, tRNAs and other aspects of protein translation was also observed in the normal gravity cultures compared to simulated microgravity growth, whereas expression of a large number of genes encoding transcriptional regulators was decreased in the normal gravity cultures (Fig. 4). As well, altered expression of stress genes was also observed in the normal gravity HARV cultures, including decreased expression of the toxin–antitoxin (TA) systems mazEF (SMU_172/173) and relBE (SMU_895/896), decreased expression of the holin-like lrgA, decreased expression of the peroxide resistance gene dpr, and increased expression of genes encoding glutathione S-transferase (SMU_1296) and glutaredoxin (SMU_669c) (Fig. 4).

Distribution of gene functional categories differentially expressed during normal gravity growth relative to simulated microgravity. RNA isolated from 8 h normal gravity and simulated microgravity S. mutans HARV cultures (n = 3 independent experiments per growth condition) was subjected to RNA-seq transcriptome profiling as described in “Materials and methods”. Differential expression and statistical analysis was carried out using a cutoff FDR-adjusted p value of 0.05. Of this dataset, those genes with a log2 (fold-change) value of at least ±1.0 were grouped by functional classification based on DAVID gene functional categorization analysis, NCBI gene annotations and KEGG pathway analysis. Total number of up-regulated genes (log2 fold-change ≥1.0, black bars) = 94, total number of down-regulated genes (log2 fold-change ≤ −1.0, gray bars) = 153
DAVID bioinformatics resource22, 23 was used to further probe the RNA-seq dataset for functional annotation groups that were enriched in the genes that displayed a log2 fold change expression of at least ±1.0 in the normal gravity vs. simulated microgravity HARV cultures (Supplemental Table S2). This analysis revealed the presence of four significant (enrichment scores ≥1.3) functional annotation clusters in the down-regulated normal gravity genes, which included genes involved in carbohydrate metabolism and transport (cluster I), transcriptional regulators (clusters II and III), and carbohydrate transporters/other membrane proteins (cluster IV). As well, functional annotation clusters involved in arginine metabolism (cluster V) and nucleoside/tRNA metabolism (cluster VI) were significantly enriched (enrichment scores ≥1.3) in the up-regulated normal gravity genes. To identify metabolic pathways potentially altered under the simulated microgravity growth condition, all statistically-significant changes in 8 h growth gene expression from the RNA-seq differential expression analysis with a minimum log2 fold-change of at least ±0.6 were also analyzed using the bioinformatics program Metacyc.24. The top metabolic pathways identified by this analysis were mixed acid fermentation (seven genes), arginine biosynthesis (six genes), and pyrimidine deoxyribonucleotide biosynthesis (five genes) (Supplemental Fig. S3). Specifically, genes involved in the conversion of acetyl-CoA to 2-oxoglutarate (a TCA cycle intermediate), arginine biosynthesis, and dCDP/dTMP biosynthesis were upregulated in the normal gravity HARV cultures, whereas genes involved in ethanol and acetate fermentation were down-regulated in normal gravity HARV cultures. Interestingly, when comparing these metabolic pathways to statistically-significant changes in cellular metabolites at 8 h growth (Table 1), several matched to the corresponding metabolic pathway changes (Supplemental Fig. S3) identified in the RNA-seq data by Metacyc, including 2-oxogluarate (alpha-ketoglutarate; mixed acid fermentation), pyruvate (mixed acid fermentation), argininosuccinate (arginine biosynthesis), uridine monophosphate and uracil (pyrmidine deoxynucleotide de novo biosynthesis). The apparent increased sensitivity of the RNA-seq data in detecting more changes in S. mutans metabolism relative to the corresponding metabolomics data may reflect a suboptimal component of the metabolomics sample collection method and/or processing time.
Assessment of oxidative stress and competence phenotypes
S. mutans encounters significant oxidative stress in the oral cavity in the form of H2O2 production by competing non-cariogenic oral streptococci,25,26 as well as from dental care products (toothpastes, mouthwash) that contain H2O2. Furthermore, expression of several oxidative stress-related genes (dpr, rex, SMU_1296, SMU_669c) was altered in the 8 h simulated microgravity HARV cultures. Therefore, the ability of S. mutans to survive exogenous H2O2 treatment was compared in cells isolated from exponential (t = 4), early-stationary (t = 8) and late stationary (t = 16) simulated microgravity and normal gravity HARV cultures (Fig. 5). Although no significant differences in oxidative stress resistance were observed between normal gravity and simulated microgravity cells isolated from 4 and 8 h growth, the H2O2-treated 4 h cells (Fig. 5a) displayed a greater overall loss of viability (~1% survival after 90 min of treatment) compared to the 8 h cells (Fig. 5b; ~50% survival after 90 min of treatment). Interestingly, when this experiment was repeated on cells from normal gravity and simulated microgravity cultures harvested at 16 h growth (Fig. 5c), the simulated microgravity cells displayed significantly (P < 0.05, one-tailed T test) increased H2O2 killing (~22% survival after 60 min treatment) compared to the normal gravity cells (~43% survival after 60 min treatment).

Percent survival of S. mutans post-treatment with 5 mM H2O2. Cells from t = 4 (a), t = 8 (b), or t = 16 (c) hour simulated microgravity (triangles) and normal gravity (circles) HARV cultures (n = 3 independent experiments per time point per growth condition) were harvested by centrifugation and resuspended in sterile HBSS. Samples were withdrawn to determine initial CFU/ml (“t = 0”), and then H2O2 was added to a final concentration of 5 mM. CFU/ml of each cell suspension was sampled at 20, 40, 60, 90, and 120 min post-H2O2 addition, and CFU/ml were determined. % Survival was calculated by 100 × [(t = final CFU/ml)/(t = 0 CFU/ml)]. Error bars = SEM. Asterisk denotes statistical significance, one-tailed t test (p < 0.05)
Because previous S. mutans studies have implicated a complex link between oxidative stress, biofilm formation, and natural competence [reviewed in27], we were curious if competence was also subject to regulation by the low-shear modeled microgravity (LSMMG) environment. Therefore early-exponential phase (growth phase at which S. mutans is most competent during standard in vitro growth) simulated microgravity and normal gravity HARV cultures were tested for their ability to uptake plasmid DNA. However, transformation efficiency (as measured by uptake of a plasmid conferring erythromycin resistance) was nearly identical between simulated microgravity and normal gravity HARV cultures (Fig. 6). The transformation efficiency was also not affected by HARV growth itself, as the HARV transformation efficiencies were similar to those measured in a parallel non-rotating culture of S. mutans (Fig. 6).

S. mutans competence phenotype assessed in simulated microgravity and normal gravity HARV cultures. Simulated microgravity and normal gravity HARV cultures, as well as a parallel static Falcon tube control culture, were each grown for 1.5 h in biofilm media prior to addition of 2 µg pOri23 plasmid DNA. All three cultures were then grown for 2.5 more hours followed by harvest and serial dilution plating on BHI agar ± 10 µg/ml erythromycin (selective antibiotic to monitor plasmid transformation). Transformation efficiency was calculated as the percentage of transformants (CFU/ml on BHI + erythromycin) among total viable cells (CFU/ml on BHI). Data represent the average of n = 3 independent experiments. Error bars = SEM
Discussion
Although the effects of both spaceflight and microgravity analog growth on virulence-related phenotypes of bacterial pathogens such as Salmonella enterica serovar Typhimurium, Escherichia coli, Pseudomonas aeruginosa, and Staphylococcus aureus have been well-described,28–38 comparatively little research has been dedicated to studying the specific effects of space flight and simulated microgravity on virulence attributes of oral bacteria such as S. mutans. In the Skylab missions conducted in the 1970s, the effects of space flight and diet on the oral cavity and oral microbiota of astronauts were documented using culture-dependent methods. These studies demonstrated that increased counts of specific oral bacteria (i.e. Bacteriodes, Veillonella, Fusobacterium, Neisseria, S. sanguinus, and S. mutans) were recovered from the astronauts in response to space flight.16 Furthermore, the astronauts in these studies exhibited decreased salivary lysozyme and increased dental calculus and gingival inflammation.16 More recently, the effect of simulated microgravity on the physiology and biofilm structure of S. mutans was also assessed using a superconducting magnet capable of generating a large gradient high magnetic field (LG-HMF),39 which mimics the space-like gravity environment by achieving stable levitation of diamagnetic materials such as biological cells and tissues.40 The results of this study suggested that growth of S. mutans under simulated microgravity enhanced its acid tolerance, altered its biofilm architecture, and increased its proportion within dual-species biofilms co-cultured with S. sanguinis, an H2O2-producing early colonizer of dental plaque biofilm.39 Collectively, these previous studies suggest that both space flight and the microgravity analog environment have the ability to alter the cariogenic potential of S. mutans.
In our current work, HARVs rotated on an axis perpendicular to the Earth’s gravitational vector were used to simulate a low-shear modeled microgravity (LSMMG) growth environment. This ground-based model of microgravity has been used to study the response of a variety of microorganisms to “weightlessness” that would be encountered during space flight [reviewed in19, 41, 42]. However, it is important to keep in mind that the S. mutans LSMMG-associated metabolic and physiological changes (i.e. alterations in cell aggregation, transcriptome, and sensitivity to oxidative stress) presented here could represent the bacteria’s response to either simulated microgravity and/or low-fluid shear (<1 dyn/cm2) specific environmental signals, the latter of which may be relevant to the fluid shear of saliva flow (<0.8 dyn/cm2) experienced by S. mutans in the oral cavity.19,21 Therefore, the data obtained in this study have the potential to shed light on S. mutans physiology and virulence properties relevant to environmental signals experienced in the host, and will hopefully provide a springboard for planning future space flight studies of oral bacteria. It is also important to note that all of the S. mutans experiments presented in this study were conducted in semi-defined culture medium containing 11 mM glucose and 10 mM sucrose, previously shown to promote in vitro S. mutans biofilm formation.43 The addition of sucrose, a host dietary sugar to which S. mutans is exposed to in the oral cavity, promotes production of extracellular glucan polymers by S. mutans, important biofilm matrix components that promote intracellular adhesion.7 In reality, the in vivo plaque biofilm environment of S. mutans is much more complex, including stress conditions such as interspecies competition for nutrients and space within the biofilm, as well as alternating periods of “feast and famine” with respect to dietary carbohydrate consumption. Therefore, the growth condition used in this study should be considered a simple representation of caries-promoting “feast” conditions experienced by S. mutans in the oral cavity. With these caveats in mind, the following discussion points represent interesting patterns/conclusions from the data obtained in this study:
1. S. mutans displays an altered early stationary-phase metabolism under simulated microgravity conditions: As indicated in the “Results” section and Fig. 1, S. mutans “end-point” simulated microgravity and normal gravity cultures appear to enter stationary phase at the same point in growth (6 h) compared to normal gravity cultures, and there were no apparent differences in CFU/ml of each culture at each time point. This being said, the 8 h (early stationary phase) metabolomics and RNA-seq data suggest that S. mutans simulated microgravity metabolism at this point is altered relative to normal gravity growth. For example, simulated microgravity cultures experienced a ten-fold decrease in supernatant glucose levels (Table 1 and Supplemental File S3) at 8 h growth relative to the normal gravity cultures. Furthermore, expression of PTS system genes involved in transport of carbohydrates (glucose, mannitol, trehalose, mannose and cellobiose) was significantly upregulated during simulated microgravity growth, whereas expression of genes encoding ribosomal proteins, tRNAs, and other aspects of protein translation was decreased in the microgravity cultures at this time point (Fig. 4 and Supplemental File S4 ). Bioinformatics analysis also revealed patterns of decreased arginine metabolism and decreased pyrimidine biosynthesis (Supplemental Table S2 and Supplemental Fig. S3) in the 8 h simulated microgravity cultures. Finally, expression of genes encoding the central metabolic transcriptional regulators CcpA and Rex were also both upregulated (2.2-fold and 2.8-fold, respectively) at 8 h growth in the simulated microgravity cultures. CcpA has been shown to regulate S. mutans catabolite repression and the ability to utilize non-preferred carbohydrates, acid tolerance, acid production, and biofilm formation,44,45,46 whereas Rex regulates expression of S. mutans genes involved with carbohydrate fermentation, biofilm formation, and oxidative stress resistance in response to cellular redox.47,48 Interestingly, a role for Rex in the transcriptional response of Staphylococcus aureus to LSMMG was also previously implicated by the altered expression of a number of Rex-regulated genes under this growth condition.29 Although great care was taken in each experiment to vigorously vortex all samples prior to and during serial dilution plating, we cannot exclude the possibility that sucrose-dependent cell clumping may have masked more apparent growth differences between the simulated microgravity and normal gravity end-point HARV cultures in these studies.
2. S. mutans late stationary-phase simulated microgravity cultures appear to be subject to increased stress: A striking phenotypic property observed in S. mutans simulated microgravity cultures was the greatly increased sensitivity of cells from late stationary phase (16 h) to H2O2 treatment (Fig. 5c) relative to cells from normal gravity cultures. However, no apparent differences in oxidative stress resistance were observed between the simulated microgravity and normal gravity cells harvested at earlier time points (log phase and early stationary phase, Fig. 5a, b, respectively). This result correlated with the LG-HMF S. mutans microgravity study by Cheng and co-workers,39 which found that mid-exponential phase S. mutans microgravity and normal gravity cells did not differ in their sensitivity to H2O2. In previous studies, late stationary-phase (20–24 h) cultures of S. typhimurium and P. aeruginosa were both shown to be more resistant to oxidative stress when grown in simulated microgravity,30,49 whereas S. typhimurium simulated microgravity cultures grown to an earlier phase of growth (10 h) displayed increased sensitivity to H2O2 treatment.31 Furthermore, the sensitivity of stationary phase simulated microgravity cultures of adherent-invasive E. coli to oxidative stress was shown to be strain-dependent.33 The susceptibility of S. aureus to oxidative stress when grown in LSMMG has also been previously investigated: compared to normal gravity growth, S. aureus cells isolated from simulated microgravity cultures displayed increased sensitivity to challenge with H2O2, which was demonstrated to be related to decreased carotenoid (yellow membrane pigment) production under this growth condition.29 These previously-published studies, in conjunction with the S. mutans results presented here, clearly demonstrate that the microgravity model used, growth phase at which oxidative stress is tested, and/or other inherent species and strain-dependent differences, make it difficult to discern broad conclusions regarding the effect of the LSMMG on oxidative stress resistance.
Although direct comparisons cannot be extrapolated due to time point differences between the 8 h RNA-seq data (Fig. 4 and Supplemental File S4) and 16 h oxidative stress data (Fig. 5c), it is interesting to note that the 8 h simulated microgravity cultures displayed increased expression of genes previously-shown to be involved in the response to oxidative stress in S. mutans. These included lrgA, which encodes a predicted holin-like protein,50,51 dpr, encoding an iron-binding protein that reduces the intracellular free iron pool during oxidative stress,52,53 and rex.48 As well, S. mutans simulated microgravity cultures displayed increased expression of genes encoding phosphotransacetylase (Pta; SMU_1043c) and two putative acetate kinase (Act) genes (ackA and SMU_1299c), involved in the conversion of acetyl-CoA to acetyl phosphate and acetyl phosphate to acetate, respectively.54 In S. mutans, aspects of the Pta-Ack pathway have been shown to influence the cellular response to oxidative stress resistance.54 These results suggest that the simulated microgravity HARV cultures may be experiencing an oxidative-like stress response in early stationary phase (8 h growth), and the altered expression of these oxidative-stress related genes and metabolic pathways is perhaps occurring in response to this stress. Further support for the idea that the S. mutans simulated microgravity cultures may be experiencing more inherent stress relative to the normal gravity cultures stems from the observation that TA system genes mazEF and relBE were significantly upregulated during simulated microgravity growth (Fig. 4 and Supplemental File S4). MazF and RelE have been shown in several bacteria to selectively inhibit translation by cleaving specific mRNA recognition sequences in response to nutritional stress [reviewed in55,56]. These data collectively suggest that S. mutans simulated microgravity HARV cultures display characteristics associated with stationary phase cellular stress. However, it is not clear from this data whether these cultures are experiencing overlapping simultaneous stresses (such as oxidative and starvation stress), or if the sum of these phenotype and gene expression changes represent a stress response specific to LSMMG.
3. S. mutans displays an altered cell aggregation phenotype under simulated microgravity conditions: Micro-organisms such as Micrococcus luteus,57 E. coli,58 P. aeruginosa,34,35,59 S. aureus,29 S. typhimurium,36 and Candida albicans 60,61 have all displayed increased biofilm growth when grown in either ground-based simulated and/or space flight microgravity conditions. In some instances, this biofilm increase was paralleled by documented increases in expression of biofilm-promoting regulatory genes or adhesion factors.29,34,35,36,49,58,61 Given these observations in other micro-organisms, we predicted that S. mutans would also display an increase in biofilm formation when grown in HARV microgravity cultures. Although biofilms attached to a solid surface were not assessed in these studies, macroscopic differences in cell–cell aggregation were typically observed in simulated microgravity compared to normal gravity HARV cultures at stationary phase (8 h growth; Fig. 2), suggesting that cell–cell adhesion and/or biofilm-like properties of this bacterium were altered in response to simulated microgravity growth. Although cell aggregation was observed in both the normal gravity and simulated microgravity HARV cultures grown in biofilm-promoting media containing sucrose, the simulated microgravity cell aggregates were more compact and more difficult to disrupt relative to the aggregates formed during normal gravity growth. Follow-up studies will be required to better characterize the structural properties of these cell aggregates (i.e. production of sucrose-dependent exopolysaccharide) and whether they are actually similar to biofilms that form on hydroxyapatite in vitro and/or the tooth surface in vivo. However, it is noteworthy that altered cell aggregation and/or “attachment-independent” phenotypes have also been observed in other biofilm-forming microbes in both LSMMG29,34 and during spaceflight.61
In summary, the results from our experiments suggest that S. mutans undergoes changes in metabolism, physiology, and global gene expression during stationary-phase simulated microgravity growth that impact cell aggregation and oxidative stress resistance. Defining the exact regulatory circuits and contributions of these differentially regulated genes to the ability of S. mutans to survive and adapt to LSMMG and spaceflight environments will help in the development of treatment and control strategies to combat the caries potential of this pathogen. Furthermore, these studies may provide insight into the response of S. mutans to low-fluid shear encountered in the oral cavity. Our future research efforts will focus on assessing S. mutans biofilm formation during simulated microgravity growth using culture conditions more closely mimicking the oral cavity (i.e., artificial saliva media and periodic pulsing of sucrose addition), as well as growth of S. mutans as part of a mixed-species biofilm consortium.
Materials and methods
Bacterial strain and growth conditions
S. mutans UA15962 was used for all of the experiments described below. All S. mutans cultures (including HARV vessels) were grown at 37 °C in a 5% CO2 incubator. For each experiment, S. mutans was freshly streaked from a frozen 30% (vol vol−1) glycerol stock onto Brain heart infusion (BHI) agar and grown for 48 h. A single colony was then inoculated into 30 ml BHI broth, and grown as a static culture for 16–18 h prior to inoculation of HARVs. Disposable vessel rotary cell culture systems (Synthecon) with vertical and horizontal 4-station rotator bases were used to simulate microgravity (HARV rotated on axis perpendicular to gravitational vector) and “normal” (1 × g) gravity (HARV rotated on axis parallel to gravitational vector), respectively (Supplemental Figure S4). All HARV manipulations were performed under a Class II A2 biosafety cabinet (Labconco). HARV vessels were filled with sterile media according to manufacturer’s recommendations, and incubated at room temperature for 18 h prior to each experiment. This media was then aseptically removed, and 20× dilute S. mutans overnight culture (approximate OD600 = 0.06/ml) in room-temperature semi-defined biofilm media (BM)43 containing 11 mM glucose and 10 mM sucrose, was used to fill each HARV. Prior to HARV rotation and incubation, sterile 5-ml luer lock syringes were used to remove air bubbles according to manufacturer’s protocols.
Continuous HARV growth curves
For each experiment (n = 3 independent experiments per growth condition), overnight S. mutans cultures were used to inoculate 120 ml of sterile BM containing 11 mM glucose and 10 mM sucrose. This inoculum was mixed well by vigorous swirling and vortexing, and 1 ml withdrawn to measure the time-of-inoculation (t = 0) CFU/ml. The remaining inoculum volume was then subjected again to vigorous swirling and vortexing and immediately used to fill 2 × 50 ml disposable HARVs as described above. Each HARV was rotated either vertically (“simulated microgravity”) or horizontally (“normal gravity”) at 37 °C, 5% CO2 for 24 h. HARVs were rotated at 30 RPM for the first 8 h of growth, and the rotation speed of each reactor was increased to 32 RPM after 8 h of growth to ensure that S. mutans cell aggregates did not settle to the bottom of the simulated microgravity HARV. At 2, 4, 6, 8, 12, and 24 h postinoculation, each HARV was removed from the incubator, vigorously agitated to remove adherent cell clumps from the walls and membrane of the HARV reactor and evenly dispersing cell clumps, and 1 ml culture was immediately removed for CFU/ml assessment. The same volume of sterile media was then added back to each HARV and air bubbles removed as described above prior to re-initiation of simulated microgravity or normal gravity HARV growth. For CFU/ml quantification, each 1 ml sample was vortexed vigorously for at least 30 s to resuspend cell clumps, followed immediately by serial dilution (each tube vortexed vigorously for at least 15 s between each dilution) and CFU/ml determination using the track plating method.63
HARV “end-point” growth curves
Overnight S. mutans cultures were used to inoculate 500 ml sterile BM containing 11 mM glucose and 10 mM sucrose to an approximate OD600 = 0.06/ml as described above. This inoculum was mixed well by vigorous swirling and vortexing, and 1 ml was withdrawn to measure the t = 0 OD600, pH, and CFU/ml. The remaining inoculum volume was then immediately used to fill 8 × 50 ml disposable HARVs (inoculum volume was subjected to vigorous swirling and vortexing before addition to each HARV). For end-point growth curve analysis at t = 4, 6, 8, and 12 h (n = 2 independent experiments total per time point per growth condition), four simulated microgravity and four normal gravity HARV cultures were grown in parallel at 37 °C, 5% CO2. HARVs were rotated at 30 RPM for the first 8 h of growth, and the rotation speed of each reactor was increased to 32 RPM after 8 h of growth to ensure that S. mutans cell aggregates did not settle to the bottom of the simulated microgravity cultures. At each time point, one simulated microgravity and one normal gravity HARV were each harvested as follows: 25 ml of HARV culture was transferred to a sterile 50 ml Falcon tube. The HARV was then closed and shaken vigorously for approximately 5 s (until there were no remaining visible clumps of bacteria stuck to the inside walls and membrane of the HARV). The reactor was then immediately uncapped and the remaining culture transferred to the 50 ml Falcon tube, which was vortexed at high speed for 30 s prior to removing 100 µl for CFU counts. The tube was vortexed again for 10 s, and 900 µl immediately removed for measuring OD600 and pH readings. OD600 readings were obtained using a Genesys 10 Bio spectrophotometer (Thermo Scientific), culture pH measured with colorpHast indicator strips (EMD), and CFU/ml calculated by serial dilution plating as described above. For the end-point growth curve analysis at t = 24 and t = 48 h, a separate experiment (n = 2 biological replicates for each time point) was performed as described above.
HARV end-point growth for metabolomic and RNA-seq analyses
Overnight S. mutans cultures were used to inoculate 350 ml sterile BM containing 11 mM glucose and 10 mM sucrose to an approximate OD600 = 0.06/ml as described above. This inoculum was mixed well by vigorous swirling and vortexing, and 1 ml was withdrawn to measure the t = 0 OD600, pH, and CFU/ml. The remaining inoculum volume was then immediately used to fill 6 × 50 ml disposable HARVs (inoculum volume was subjected to vigorous swirling and vortexing before addition to each HARV). A sterile media control was also collected at the beginning of each experiment and stored at −80 °C for metabolomics analysis (see below). For each experiment (n = 3 independent experiments total), three simulated microgravity and three normal gravity HARV cultures were grown in parallel at 37 °C, 5% CO2 as described above for the end-point growth curve analysis. For each experiment, one simulated microgravity and one normal gravity HARV were each harvested at t = 4, t = 8 h, and t = 24 h growth, as described above for the end-point growth curve analysis (for a total of n = 3 independent samples per time point per growth condition). HARVs were photographed prior to culture disruption and harvest to document macroscopic growth observations. After harvesting, 34 ml of each t = 4, 8, and 24 h HARV culture was centrifuged at 3000 RPM, 4 °C for 20 min and immediately placed on ice. 1 ml of culture supernatant was removed from each centrifuged sample, transferred to 1.5 ml tubes, and immediately frozen at −80 °C. The remaining culture supernatants were decanted and cell pellets immediately frozen at −80 °C. For RNA-seq samples (t = 8 h only), a 10-ml aliquot of each HARV culture was also centrifuged at 3000 RPM, 4 °C for 20 min and immediately placed on ice. Culture supernatants were decanted and 1 ml RNAlater (Ambion) was added to each cell pellet prior to immediate storage at −80 °C.
Metabolite extraction and metabolomics analysis
Extraction of metabolites (n = 3 independent biological samples per time point and growth condition) and all metabolomics analyses (including statistics) were performed by Metabolon, Inc. (Durham, NC) following previously-published methods.64–68 Details specific to this experiment can be found in Supplemental File S1 (Supplemental Materials and Methods).
RNA isolation and RNA-seq analysis
RNA was isolated from t = 8 h cell pellets stored at −80 °C in RNAlater (collected from n = 3 independent experiments each for simulated microgravity and normal gravity HARV cultures) with the RNeasy Kit (Qiagen) and FASTPREP lysing matrix B tubes (MP Biomedical) using previously-described methods.50,69 Each RNA sample was then subjected to a second DNAse treatment using the TURBO DNA-free™ Kit (Thermo Fisher Scientific) per the manufacturer’s protocols. Lack of contaminating genomic DNA in each RNA sample was then determined using quantitative real-time PCR (i.e. no cDNA synthesis step) and S. mutans-specific primers (1547-F/R; Supplemental Table S3) prior to sending ≥5 µg of each RNA sample to Seqwright Genomic Services (Houston, TX), who performed all steps of the RNA-seq workflow, data curation, differential expression, and corresponding statistical analysis. In brief, each RNA sample was subjected to an mRNA enrichment step and generation of a library of cDNA template molecules of known strand origin using the TruSeq Total mRNA Sample Preparation Kit (Illumina). Sequencing was performed using the Illumina HiSeq 2500 platform, generating 2 × 100 bp read lengths for a total of 2 × 10 M reads per sample. Illumina CASAVA software was used to create genomic builds and count reads, whereas TopHat-2.0.8b and Cufflinks-2.0.170 were used to align reads to the S. mutans UA159 genome (Genbank accession # NC_004350.2). All RNA-seq data have been deposited to NCBI's Gene Expression Omnibus and are accessible through GEO Series accession number GSE90166.
Hydrogen peroxide (H2O2) assay
For each experiment (n = 3 independent biological samples each for simulated microgravity and normal gravity HARV cultures), two S. mutans 10-ml HARV cultures were inoculated as described above, and each was grown for either 4, 8, or 16 h. For the 4 and 8 h experiments, the HARV rotation speed was 30 RPM. For the 16 h experiment, the HARV rotation speed was 30 RPM for the first 8 h of growth, and was then increased to 35 RPM to counteract sedimentation of cellular aggregates. S. mutans cultures were then harvested from each HARV vessel, transferred to a 50 ml Falcon tube, and centrifuged for 10 min at 4000 RPM. Cell pellets were resuspended in 5 ml sterile 1× Hanks Buffered Salt Solution (HBSS), and 100 µl was withdrawn from each tube and subjected to serial dilution plating as described above to enumerate CFU/ml of each cell suspension at “t = 0” (prior to H2O2 addition). H2O2 was added to each remaining 4.9 ml cell suspension to a final concentration of 5 mM, and incubated at 37 °C, 5% CO2. Samples of each cell suspension were removed at 15, 30, 45, 60, and 120 min post-H2O2 addition to enumerate CFU/ml, and % survival for each sample was calculated by 100× (t = × CFU/ml/t = 0 CFU/ml).
Competence assay
The ability of S. mutans to take up exogenous DNA was assessed in microgravity and normal gravity HARV cultures based on previously-described methods.50,71 For each experiment (n = 3 independent biological samples each for simulated microgravity and normal gravity HARV cultures), S. mutans 10-ml micro-gravity and normal gravity HARV cultures were grown in BM containing 11 mM glucose and 10 mM sucrose for 90 min [corresponding to 2.3 × 106 CFU/ml (±3.0 × 105 SEM) for normal gravity and 3.1 × 106 CFU/ml (±1.5 × 105 SEM) for simulated microgravity] at 30 RPM. HARVS were removed from rotator bases and 2 µg of unmethylated pOri23 plasmid DNA was added to each reactor. Microgravity and normal gravity HARVs were then incubated for 2.5 h at 30 RPM prior to serial dilution and plating of each culture on BHI agar ±10 µg/ml erythromycin (Erm; selectable antibiotic for pOri23). CFU/ml were determined after 48 h plate growth, and transformation efficiencies were calculated as the percentage of transformants (BHI-Erm10 CFU/ml) among the total viable cells (BHI CFU/ml). For each experiment, a non-rotating control culture (15-ml Falcon tube containing 15 ml of S. mutans culture) was grown in parallel and assessed for competence as described above.
Bioinformatics
The DAVID Bioinformatics Resource22,23 was used to aid in functional categorization of the RNA-seq differential expression data and to perform functional annotation clustering. Only genes with statistically-significant log2-fold changes of at least ±1.0 were included in these analyses. The lowest classification stringency was used for functional categorization. For functional annotation clustering analysis, the default analysis parameters were used: Medium classification stringency, similarity threshold = 0.5, enrichment threshold (EASE score) = 1.0. As recommended previously,22 an enrichment score cutoff of ≥1.3 was applied to the functional annotation clustering analysis. For identifying trends in metabolic pathway changes, all statistically-significant RNA-seq differential expression data with a log2-fold change of at least ±0.6 were also analyzed using Metacyc.24
Statistics
For metabolomics, statistical analyses were performed in ArrayStudio on log transformed data. For those analyses not standard in ArrayStudio, the programs R (http://cran.r-project.org/) or JMP (SAS, http://www.jmp.com) were used as previously described.72 Two-way ANOVA was used to compare the effects of treatment (simulated microgravity vs. normal gravity growth) and growth time (t = 4, 8, and 24 h) on metabolite differences. Multiple comparisons were accounted for with the false discovery rate (FDR) method, and each FDR was estimated by q-values. PCA was also applied to the metabolomics data. For RNA-seq, differential expression analysis was performed by Seqwright Genomic Services using FPKM (fragments per kilobase per million mapped reads) values and fold-change was expressed as log2 (FPKM normal gravity/FPKM simulated microgravity). For all other data, statistical analyses were performed using Sigmaplot version 12.5 (Build 12.5.0.38, Systat Software, Inc.). Data was tested for normality and equal variance, followed by a T-test or Rank-sum test, as appropriate.
References
Marchant, S., Brailsford, S. R., Twomey, A. C., Roberts, G. J. & Beighton, D. The predominant microflora of nursing caries lesions. Caries Res. 35, 397–406 (2001). PubMed PMID: 11799279.
Fitzgerald, R. J., Jordan, H. V. & Stanley, H. R. Experimental caries and gingival pathologic changes in the gnotobiotic rat. J. Dent. Res. 39, 923–935 (1960). PubMed PMID: 13700062.
Okada, M. et al. Longitudinal study of dental caries incidence associated with Streptococcus mutans and Streptococcus sobrinus in pre-school children. J. Med. Microbiol. 54, 661–665 (2005). PubMed PMID: 15947431.
Rozier, R. G. Effectiveness of methods used by dental professionals for the primary prevention of dental caries. J. Dent. Educ. 65, 1063–1072 (2001). PubMed PMID: 11699978.
U.S. Department of Health and Human Services. Oral Health in America: a Report of the Surgeon General. Rockville, MD: US Department of Health and Human Services, National Institute of Dental and Craniofacial Research, National Institutes of Health (2000).
Nakano, K. et al. Detection of cariogenic Streptococcus mutans in extirpated heart valve and atheromatous plaque specimens. J. Clin. Microbiol. 44, 3313–3317 (2006). PubMed PMID: 16954266.
Ooshima, T. et al. Contributions of three glycosyltransferases to sucrose-dependent adherence of Streptococcus mutans. J. Dent. Res. 80, 1672–1677 (2001). PubMed PMID: 11597030.
Petersen, F. C., Assev, S., van der Mei, H. C., Busscher, H. J. & Scheie, A. A. Functional variation of the antigen I/II surface protein in Streptococcus mutans and Streptococcus intermedius. Infect. Immun. 70, 249–256 (2002). PubMed PMID: 11748190.
Zhu, L. et al. Functional characterization of cell-wall-associated protein WapA in Streptococcus mutans. Microbiology. 152, 2395–2404 (2006). PubMed PMID: 16849803.
Ahn, S. J. & Burne, R. A. The atlA operon of Streptococcus mutans: role in autolysin maturation and cell surface biogenesis. J. Bacteriol. 188, 6877–6888 (2006). Pubmed Central PMCID: 1595523.
Senadheera, D. & Cvitkovitch, D. G. Quorum sensing and biofilm formation by Streptococcus mutans. Adv. Exp. Med. Biol. 631, 178–188 (2008). PubMed PMID: 18792689.
Das, T., Sharma, P. K., Krom, B. P., van der Mei, H. C. & Busscher, H. J. Role of eDNA on the adhesion forces between Streptococcus mutans and substratum surfaces: influence of ionic strength and substratum hydrophobicity. Langmuir. 27, 10113–10118 (2011). PubMed PMID: 21740034.
Rai, B., Kaur, J. & Catalina, M. Bone mineral density, bone mineral content, gingival crevicular fluid (matrix metalloproteinases, cathepsin K, osteocalcin), and salivary and serum osteocalcin levels in human mandible and alveolar bone under conditions of simulated microgravity. J. Oral. Sci. 52, 385–390 (2010). PubMed PMID: 20881330.
Rai, B., Kaur, J. & Foing, B. H. Evaluation by an aeronautic dentist on the adverse effects of a six-week period of microgravity on the oral cavity. Int. J. Dent 2011, 548068 (2011). PubMed PMID: 22190932. Pubmed Central PMCID: 3235462.
Rai, B. & Kaur, J. The history and importance of aeronautic dentistry. J. Oral. Sci. 53, 143–146 (2011). PubMed PMID: 21712617.
Brown, L.R., Frome, W. J., Handler, S., Wheatcroft, M. G. & Rider, L. J. Skylab oral health studies in Biomedical Results from Skylab National Aeronautics and Space Administration (Johnston, R. S. & Dietlein, L. F.) (Johnson Space Center, 1977). pp. 35–44.
Brown, L. R., Wheatcroft, M. G., Frome, W. J. & Rider, L. J. Effects of a simulated skylab mission on the oral health of astronauts. J. Dent. Res. 53, 1268–1275 (1974). PubMed PMID: 4153662.
Volozhin, A. I., Tsarev, V. N., Malneva, N. S., Sashkina, T. I. & Saldusova, I. V. Interaction peculiarities between microbial cenosis and local immunity of periodontium of humans under extreme conditions. Acta Astronaut. 49, 53–57 (2001). PubMed PMID: 11858254.
Nickerson, C. A., Ott, C. M., Wilson, J. W., Ramamurthy, R. & Pierson, D. L. Microbial responses to microgravity and other low-shear environments. Microbiol. Mol. Biol. Rev. 68, 345–361 (2004). PubMed PMID: 15187188. Pubmed Central PMCID: 419922.
Nickerson, C. A. et al. Low-shear modeled microgravity: a global environmental regulatory signal affecting bacterial gene expression, physiology, and pathogenesis. J. Microbiol. Methods 54, 1–11 (2003). PubMed PMID: 12732416.
Prakobphol, A., Burdsal, C. A. & Fisher, S. J. Quantifying the strength of bacterial adhesive interactions with salivary glycoproteins. J. Dent. Res. 74, 1212–1218 (1995). PubMed PMID: 7790599.
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). PubMed PMID: 19131956.
Huang da, W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009). Pubmed Central PMCID: 2615629.
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids. Res. 44, D471–D480 (2016). PubMed PMID: 26527732. Pubmed Central PMCID: 4702838.
Kreth, J., Zhang, Y. & Herzberg, M. C. Streptococcal antagonism in oral biofilms: Streptococcus sanguinis and Streptococcus gordonii interference with Streptococcus mutans. J. Bacteriol. 190, 4632–4640 (2008). PubMed PMID: 18441055. Pubmed Central PMCID: 2446780.
Zheng, X. et al. Involvement of gshAB in the interspecies competition within oral biofilm. J. Dent. Res. 92, 819–824 (2013). PubMed PMID: 23872989.
Lemos, J. A., Quivey, R. G. Jr., Koo, H. & Abranches, J. Streptococcus mutans: a new Gram-positive paradigm?. Microbiology. 159, 436–445 (2013). PubMed PMID: 23393147. Pubmed Central PMCID: 4083656.
Tucker, D. L. et al. Characterization of Escherichia coli MG1655 grown in a low-shear modeled microgravity environment. BMC Microbiol. 7, 15 (2007). Pubmed Central PMCID: 1852313.
Castro, S. L., Nelman-Gonzalez, M., Nickerson, C. A. & Ott, C. M. Induction of attachment-independent biofilm formation and repression of Hfq expression by low-fluid-shear culture of Staphylococcus aureus. Appl. Environ. Microbiol. 77, 6368–6378 (2011). Pubmed Central PMCID: 3187170.
Pacello, F., Rotilio, G. & Battistoni, A. Low-shear modeled microgravity enhances Salmonella enterica resistance to hydrogen peroxide through a mechanism involving katG and katN. Open Microbiol. J. 6, 53–64 (2012). PubMed PMID: 22888375. Pubmed Central PMCID: 3414715.
Wilson, J. W. et al. Low-shear modeled microgravity alters the Salmonella enterica serovar typhimurium stress response in an RpoS-independent manner. Appl. Environ. Microbiol. 68, 5408–5416 (2002). PubMed PMID: 12406731. Pubmed Central PMCID: 129924.
Nickerson, C. A. et al. Microgravity as a novel environmental signal affecting Salmonella enterica serovar Typhimurium virulence. Infect. Immun. 68, 3147–3152 (2000). PubMed PMID: 10816456. Pubmed Central PMCID: 97548.
Allen, C. A., Niesel, D. W. & Torres, A. G. The effects of low-shear stress on adherent-invasive Escherichia coli. Environ. Microbiol. 10, 1512–1525, doi:10.111/j.462-2920.008.01567.x (2008).
Crabbe, A. et al. Use of the rotating wall vessel technology to study the effect of shear stress on growth behaviour of Pseudomonas aeruginosa PA01. Environ. Microbiol. 10, 2098–2110, doi:10.1111/j.462-2920.008.01631.x (2008).
Kim, W. et al. Spaceflight promotes biofilm formation by Pseudomonas aeruginosa. PLoS One 8, e62437, doi:10.1371/journal.pone.0062437 (2013).
Wilson, J. W. et al. Space flight alters bacterial gene expression and virulence and reveals a role for global regulator Hfq. Proc. Natl Acad. Sci. USA 104, 16299–16304 (2007).
Wilson, J. W. et al. Media ion composition controls regulatory and virulence response of Salmonella in spaceflight. PLoS One 3, e3923, doi:10.1371/journal.pone.0003923 (2008).
Wilson, J. W. et al. Microarray analysis identifies Salmonella genes belonging to the low-shear modeled microgravity regulon. Proc. Natl Acad. Sci. USA 99, 13807–13812 (2002). NASA Discipline Cell Biology Non-NASA Center.
Cheng, X. et al. Effects of simulated microgravity on Streptococcus mutans physiology and biofilm structure. FEMS Microbiol. Lett. 359, 94–101 (2014). PubMed PMID: 25109245.
Qian, A. R. et al. Large gradient high magnetic fields affect osteoblast ultrastructure and function by disrupting collagen I or fibronectin/alphabeta1 integrin. PLoS One 8, e51036 (2013). PubMed PMID: 23382804. Pubmed Central PMCID: 3558520.
Rosenzweig, J. A. et al. Spaceflight and modeled microgravity effects on microbial growth and virulence. Appl. Microbiol. Biotechnol. 85, 885–891, doi:10.1007/s00253-009-2237-8 (2010).
Rosenzweig, J. A., Ahmed, S., Eunson, J. Jr. & Chopra, A. K. Low-shear force associated with modeled microgravity and spaceflight does not similarly impact the virulence of notable bacterial pathogens. Appl. Microbiol. Biotechnol. 98, 8797–8807, doi:10.1007/s00253-014-6025-8 (2014).
Biswas, I., Drake, L. & Biswas, S. Regulation of gbpC expression in Streptococcus mutans. J. Bacteriol. 189, 6521–6531 (2007). PubMed PMID: 17616585. Pubmed Central PMCID: 2045159.
Abranches, J. et al. CcpA regulates central metabolism and virulence gene expression in Streptococcus mutans. J. Bacteriol. 190, 2340–2349, doi:10.1128/JB.01237-07 (2008).
Zeng, L. & Burne, R. A. Multiple sugar: phosphotransferase system permeases participate in catabolite modification of gene expression in Streptococcus mutans. Mol. Microbiol. 70, 197–208, doi:10.1111/j.365-2958.008.06403.x (2008).
Wen, Z. T. & Burne, R. A. Functional genomics approach to identifying genes required for biofilm development by Streptococcus mutans. Appl. Environ. Microbiol. 68, 1196–1203 (2002). PubMed PMID: 11872468. Pubmed Central PMCID: 123778.
Bitoun, J. P., Nguyen, A. H., Fan, Y., Burne, R. A. & Wen, Z. T. Transcriptional repressor Rex is involved in regulation of oxidative stress response and biofilm formation by Streptococcus mutans. FEMS Microbiol. Lett. 320, 110–117, doi:10.1111/j.574-6968.2011.02293.x (2011).
Bitoun, J. P., Liao, S., Yao, X., Xie, G. G. & Wen, Z. T. The redox-sensing regulator Rex modulates central carbon metabolism, stress tolerance response and biofilm formation by Streptococcus mutans. PLoS One 7, e44766, doi:10.1371/journal.pone.0044766 (2012).
Crabbe, A. et al. Response of Pseudomonas aeruginosa PAO1 to low shear modelled microgravity involves AlgU regulation. Environ. Microbiol. 12, 1545–1564 (2010). PubMed PMID: 20236169.
Ahn, S. J., Qu, M. D., Roberts, E., Burne, R. A. & Rice, K. C. Identification of the Streptococcus mutans LytST two-component regulon reveals its contribution to oxidative stress tolerance. BMC. Microbiol. 12, 187 (2012). PubMed PMID: 22937869.
Ahn, S. J., Rice, K. C., Oleas, J., Bayles, K. W. & Burne, R. A. The Streptococcus mutans Cid and Lrg systems modulate virulence traits in response to multiple environmental signals. Microbiology. 156, 3136–3147, doi:10.1099/mic.0.039586-0 (2010).
Yamamoto, Y. et al. Regulation of the intracellular free iron pool by Dpr provides oxygen tolerance to Streptococcus mutans. J. Bacteriol. 186, 5997–6002 (2004).
Yamamoto, Y., Poole, L. B., Hantgan, R. R. & Kamio, Y. An iron-binding protein, Dpr, from Streptococcus mutans prevents iron-dependent hydroxyl radical formation in vitro. J. Bacteriol. 184, 2931–2939 (2002).
Kim, J. N., Ahn, S. J. & Burne, R. A. Genetics and physiology of acetate metabolism by the Pta-Ack pathway of Streptococcus mutans. Appl. Environ. Microbiol. 81, 5015–5025, doi:10.1128/AEM.01160-15 (2015).
Cook, G. M. et al. Ribonucleases in bacterial toxin-antitoxin systems. Biochim. Biophys. Acta. 1829, 523–531 (2013). PubMed PMID: 23454553.
Gerdes, K., Christensen, S. K. & Lobner-Olesen, A. Prokaryotic toxin–antitoxin stress response loci. Nat. Rev. Microbiol. 3, 371–382 (2005). PubMed PMID: 15864262.
Mauclaire, L. & Egli, M. Effect of simulated microgravity on growth and production of exopolymeric substances of Micrococcus luteus space and earth isolates. FEMS Immunol. Med. Microbiol. 59, 350–356 (2010). PubMed PMID: 20482631.
Lynch, S. V., Mukundakrishnan, K., Benoit, M. R., Ayyaswamy, P. S. & Matin, A. Escherichia coli biofilms formed under low-shear modeled microgravity in a ground-based system. Appl. Environ. Microbiol. 72, 7701–7710 (2006). PubMed PMID: 17028231. Pubmed Central PMCID: 1694224.
McLean, R. J., Cassanto, J. M., Barnes, M. B. & Koo, J. H. Bacterial biofilm formation under microgravity conditions. FEMS Microbiol. Lett. 195, 115–119 (2001). PubMed PMID: 11179638.
Searles, S. C., Woolley, C. M., Petersen, R. A., Hyman, L. E. & Nielsen-Preiss, S. M. Modeled microgravity increases filamentation, biofilm formation, phenotypic switching, and antimicrobial resistance in Candida albicans. Astrobiology. 11, 825–836 (2011). PubMed PMID: 21936634.
Crabbe, A. et al. Spaceflight enhances cell aggregation and random budding in Candida albicans. PLoS One 8, e80677 (2013). PubMed PMID: 24324620. Pubmed Central PMCID: 3851762.
Shah, G. R. & Caufield, P. W. Enhanced transformation of Streptococcus mutans by modifications in culture conditions. Anal. Biochem. 214, 343–346 (1993). PubMed PMID: 8250246.
Jett, B. D., Hatter, K. L., Huycke, M. M. & Gilmore, M. S. Simplified agar plate method for quantifying viable bacteria. BioTechniques. 23, 648–650 (1997). PubMed PMID: 9343684.
Ohta, T. et al. Untargeted metabolomic profiling as an evaluative tool of fenofibrate-induced toxicology in Fischer 344 male rats. Toxicol. Pathol. 37, 521–535 (2009). PubMed PMID: 19458390.
Evans, A. M., DeHaven, C. D., Barrett, T., Mitchell, M. & Milgram, E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 81, 6656–6667 (2009). PubMed PMID: 19624122.
Evans, C. R. et al. Untargeted LC-MS metabolomics of bronchoalveolar lavage fluid differentiates acute respiratory distress syndrome from health. J. Proteome Res. 13, 640–649 (2014). PubMed PMID: 24289193. Pubmed Central PMCID: 4068805.
Dehaven, C. D., Evans, A. M., Dai, H. & Lawton, K. A. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J. Cheminform. 2, 9 (2010). Pubmed Central PMCID: 2984397.
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl Acad. Sci. USA. 100, 9440–9445 (2003). PubMed PMID: 12883005. Pubmed Central PMCID: 170937.
Patton, T. G., Rice, K. C., Foster, M. K. & Bayles, K. W. The Staphylococcus aureus cidC gene encodes a pyruvate oxidase that affects acetate metabolism and cell death in stationary phase. Mol. Microbiol. 56, 1664–1674 (2005). PubMed PMID: 15916614.
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012). PubMed PMID: 22383036. Pubmed Central PMCID: 3334321.
Seaton, K., Ahn, S. J., Sagstetter, A. M. & Burne, R. A. A transcriptional regulator and ABC transporters link stress tolerance, (p)ppGpp, and genetic competence in Streptococcus mutans. J. Bacteriol. 193, 862–874 (2011). PubMed PMID: 21148727. Pubmed Central PMCID: 3028664.
Yobi, A. et al. Comparative metabolic profiling between desiccation-sensitive and desiccation-tolerant species of Selaginella reveals insights into the resurrection trait. Plant J. 72, 983–999 (2012). PubMed PMID: 23061970.
Acknowledgements
This work was funded in part by a Florida Education Fund McKnight Doctoral Fellowship to S.S.O, NASA grant NNX13AM09G to K.C.R., and NIH grant R01 AI118999 to K.C.R.
Author Contributions
S.S.O. and A.M.L. contributed to experimental design, performed experiments, analyzed data, and assisted in the writing and editing of the manuscript. K.C.R. performed experiments, directed the overall experimental design, analyzed data, and wrote the manuscript.
Competing interests
The authors declare no competing financial interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Orsini, S.S., Lewis, A.M. & Rice, K.C. Investigation of simulated microgravity effects on Streptococcus mutans physiology and global gene expression. npj Microgravity 3, 4 (2017). https://doi.org/10.1038/s41526-016-0006-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41526-016-0006-4
This article is cited by
-
Whole transcriptome analysis highlights nutrient limitation of nitrogen cycle bacteria in simulated microgravity
npj Microgravity (2024)
-
Enhanced assembly of bacteriophage T7 produced in cell-free reactions under simulated microgravity
npj Microgravity (2024)
-
Metabolic modeling of the International Space Station microbiome reveals key microbial interactions
Microbiome (2022)
-
Adaptation to simulated microgravity in Streptococcus mutans
npj Microgravity (2022)
-
Molecular response of Deinococcus radiodurans to simulated microgravity explored by proteometabolomic approach
Scientific Reports (2019)
