
Abstract
Rare variants in the beta-glucocerebrosidase gene (GBA1) are common genetic risk factors for alpha synucleinopathy, which often manifests clinically as GBA-associated Parkinson’s disease (GBA-PD). Clinically, GBA-PD closely mimics idiopathic PD, but it may present at a younger age and often aggregates in families. Most carriers of GBA variants are, however, asymptomatic. Moreover, symptomatic PD patients without GBA variant have been reported in families with seemingly GBA-PD. These observations obscure the link between GBA variants and PD pathogenesis and point towards a role for unidentified additional genetic and/or environmental risk factors or second hits in GBA-PD. In this study, we explored whether rare genetic variants may be additional risk factors for PD in two families segregating the PD-associated GBA1 variants c.115+1G>A (ClinVar ID: 93445) and p.L444P (ClinVar ID: 4288). Our analysis identified rare genetic variants of the HSP70 co-chaperone DnaJ homolog subfamily B member 6 (DNAJB6) and lysosomal protein prosaposin (PSAP) as additional factors possibly influencing PD risk in the two families. In comparison to the wild-type proteins, variant DNAJB6 and PSAP proteins show altered functions in the context of cellular alpha-synuclein homeostasis when expressed in reporter cells. Furthermore, the segregation pattern of the rare variants in the genes encoding DNAJB6 and PSAP indicated a possible association with PD in the respective families. The occurrence of second hits or additional PD cosegregating rare variants has important implications for genetic counseling in PD families with GBA1 variant carriers and for the selection of PD patients for GBA targeted treatments.
Similar content being viewed by others

Introduction
Rare variants in the gene GBA1, which encodes beta-glucocerebrosidase (GBA, OMIM: #606463; EC 3.2.1.45), are considered as risk factors for developing alpha synucleinopathy manifesting clinically as Parkinson’s disease (PD)1,2. The GBA-associated PD (GBA-PD) often aggregates in families but the connection between GBA1 variants and PD pathogenesis remains ambiguous. Contributing to this ambiguity is an unknown molecular mechanism by which GBA variants translate into cellular alpha-synuclein mishandling. Genetic observations such as the occurrence of non-symptomatic GBA1 variant carriers (e.g. incomplete penetrance) and a high rate of PD symptomatic non-carriers (e.g. phenocopies of mostly unknown origin) in seemingly GBA-PD families further obscure the link between GBA and PD3,4,5. From a genetic point of view, reduced penetrance and phenocopies in GBA-PD families can be explained by interactions between GBA1 variants and unidentified additional genetic and/or environmental risk factors or second hits. For instance, additional risk factors may influence cellular alpha-synuclein homeostasis concurrently with GBA1 variants in an epistatic manner, thereby modifying the risk of alpha synucleinopathy attributed to GBA variants and hence their penetrance. Alternatively, additional risk factors may act independently of GBA variants and increase the PD risk non-epistatically of the GBA1 locus, thereby resulting in the occurrence of phenocopies in GBA-PD families.
The identification of additional factors such as second genetic hits can improve the clinical management of GBA-PD on many fronts. For instance, this knowledge will enable genetic risk management, counseling, and clinical surveillance in seemingly GBA-PD families, which is otherwise difficult to achieve solely based on GBA1 variants. Furthermore, GBA has recently become a prominent target for therapeutic development. Drugs such as GZ/SAR402671 (ClinicalTrials.gov Identifier: NCT02906020) and Ambroxol (ClinicalTrials.gov Identifier: NCT02941822 and NCT02914366) have been promoted for randomized interventional trials in PD patients with GBA1 variants. These drugs aim to augment lysosomal transport and function in GBA1 variant carriers thereby restoring healthy lysosomal function, which is required for cellular alpha-synuclein handling6,7,8. A major concern is that additional risk factors in GBA1 variant carriers may affect cellular alpha-synuclein homeostasis and thereby may confound the outcome of these trials.
Massively parallel sequencing, when combined with familial genetic studies, has the potential to reveal high-risk rare genetic variation in complex neurodegenerative disorders that show a substantially heritable component9. Application of sequencing technologies in PD genetics has recently led to the discovery of additional potentially pathogenic rare genetic variation in PD patients carrying primary pathogenic mutations, thus providing evidence for oligogenic inheritance and pathway interactions in PD10,11. In this study, we uncovered rare variants in DNAJB6 and PSAP as putative second hits in two PD families segregating GBA1 mutations c.115+1G>A and p.L444P, respectively.
Results
Clinical findings
Family A comprised four PD subjects, with a mean age of 58.5 years (range 45–67), mean age at onset of 46.5 years (range 40–51), and a mean disease duration of 12.5 years. Depression was noted in all PD patients of the family A. Patients with longer disease duration presented with cognitive decline (Supplementary Table 1 and Fig. 1, subjects IV:1 and IV:4). The four PD patients in family A were treated with dopaminergic medication with a good response. Two of the deceased individuals (Fig. 1A: III:5 and IV:3) of family A were described to have PD. Family B comprised four male PD subjects, with a mean age of 63.5 years (range 49–80), mean age at onset of 51.5 years (range 42–59), and a mean disease duration of 13 years (Supplementary Table 1 and Fig. 1C). Subjects II:4 and III:1 in family B presented with severe depression (Supplementary Table 1). The PD patients in family B were treated with carbidopa–levodopa (Sinemet) with a good response.

A The pedigree structure of families A and segregation of GBA: c.115+1G>A and DNAJB6; p.T193A (c.A577G) are shown. Affected individuals are shown as filled symbols and the arrow points to the index patient. The affection status of the family members of generation I and II could not be ascertained. The affection status of deceased individuals (diagonal lines) was reported by immediate family members and was confirmed by available medical records. Symbols with black lines (IV:6, V:4, and V:5) indicate asymptomatic GBA variant carriers. Asterisks mark the individuals for whom whole-genome sequencing and rare variant analysis was performed. B Representative Sanger sequence chromatograms showing GBA1: c.115+1G>A and DNAJB6; p.T193A (c.A577G) variant positions in subjects IV:4 and IV:7 of family A. Arrowhead points to heterozygous substitutions. C The pedigree structure of families B and segregation of GBA; p.L444P (c.1448T>C) and PSAP; p.N157S (c.A470G) variants is shown. Affected individuals are shown as filled symbols and the arrow points to the index patients. The symbol with a white circle indicates an individual with PD but without GBA variant (phenocopy PD patient, III:4). The PSAP; p.N157S (c.A470G) variant was found in all PD, and in addition in one healthy family member (IV:7). Asterisks mark the individuals for whom whole-genome sequencing and rare variant analysis was performed. D Representative sanger sequence chromatograms showing GBA; p.L444P (c.1448T>C) and PSAP; p.N157S (c.A470G) variant positions in subjects III:1 and III:2 of family B. Arrowhead points to heterozygous substitutions.
Identification and familial segregation of rare genetic variants in family A and B
The contribution of rare genetic variation to PD risk in family A and B was explored by whole-genome sequencing followed by rare variant filtering and segregation analysis. The summary of study design and data analysis is shown in Supplementary Fig. 1. Genome sequencing was performed for three PD patients in family A and one PD patient in family B (Fig. 1A, C; asterisks). Sequencing was also performed for one control individual from each family (Fig. 1A, C; asterisks), which helped to reduce the final list of candidate rare variants. The list of candidate rare variants identified in the two families is shown in Table 1.
A GBA1 splice-site variant (rs104886460-G/A; c.115+1G>A; ClinVar ID: 93445) was identified in the WGS data of family A. Segregation testing of this variant in family A revealed four PD subjects as heterozygous carriers of c.115+1G>A and in addition three asymptomatic carriers (Fig. 1A, symbols marked with thick lines; IV:6, V:4, and V:5). The younger asymptomatic carriers V:4 and V:5 (aged 41 and 40 years, respectively) may still develop PD or may already exhibit prodromal features of the disease as suggested previously12,13. We therefore assessed the three asymptomatic GBA1; c.115+1G>A carriers for olfactory (odor identification ability), visual and cognitive functions (Mini-Mental State Examination score), and depression (HAM-D score), which were all normal (data not shown). The occurrence of an asymptomatic carrier (IV:6, age 65 years) considerably older than the average age of PD onset in this family (i.e 46.5 years) suggests that the PD penetrance of the heterozygous variant GBA1; c.115+1G>A in family A is incomplete.
Another GBA1 gene variant (rs421016-T/C; c.1448T>C which encodes p.L444P, also called p.L483P; ClinVar ID: 4288), was identified by WGS of a PD patient (Fig. 1C, subject III:1) from family B. Segregation analysis of the p.L444P variant in family B revealed two additional PD patients as heterozygous carriers (Fig. 1C, subjects II:2 and II:4). No asymptomatic carrier of p.L444P was observed in this family. The PD patient III:4 in family B (Fig. 2C, male, black symbol with a white circle) did not segregate the GBA; p.L444P variant. He presented with action tremor, slight rigidity, and slowness of the movement in his upper right limb at the age of 49, which affected his daily activities, including writing, dressing, and his job as a restaurant worker. His motor symptoms worsened over a span of 2 years. Based on his medical history, a review of his signs and symptoms, and a neurological and physical examination he was formally diagnosed with PD at the age of 51 years (Supplementary Table 1). Due to his non-carrier status for the p.L444P variant in GBA1, which otherwise segregated with PD in family B, the patient III:4 was considered a phenocopy PD patient of unknown origin.

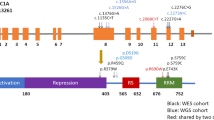
A Schematic diagram of the human DNAJB6 domain structure is shown. The approximate position of p.T193A is indicated. Alignment of a region of the human DNAJB6 protein sequence containing an S/T-rich motif with the corresponding polypeptide sequence from various species is shown below. Numbers indicate amino acid positions. Blue shading indicates the conservation status of the corresponding residues. Arrow points to the variant position, the S/T motif (SxSTST) is also highlighted. B The model structure of the DNAJB6 homodimer with the variant position (T193, red spheres) is shown. C Representative images showing the cellular distribution of alpha-synuclein-dsRed fusion protein in an alpha-synuclein aggregation reporter HEK293 cell line lacking endogenous DNAJB6 expression. Cells were transfected with plasmids expressing EGFP alone or as a fusion protein with wild type or the T193A variant containing DNAJB6. Focal and diffuse cytoplasmic distributions of alpha-synuclein-dsRed can be observed. D Quantification of focal cytoplasmic distribution of aggregated alpha-synuclein-dsRed (n = 3 transfections). Statistical analysis was performed by ANOVA. ***P < 0.001, *P < 0.05. Graphs represent mean and standard deviation.
In addition to the GBA1 splice variant c.115+1G>A segregating in family A, five single-nucleotide variants (SNVs) comprising two splice-site variants (Table 1: CDYL and TMEM209 genes) and two exonic variants (Table 1: DNAJB6 and TNR genes) were identified as candidates. In family B, which segregated GBA; p.L444P, the list of additional candidates comprised 11 exonic SNVs (Table 1: SUMF2, MAP4, MYLK, PSAP, ELF4G2, NAV2, ACACB, GIT2, FKBP10, ABCA7, and CDC25B genes). Besides a recently reported variant in the Tenascin-R gene (TNR) (rs61731112-A/C; c.A538C encoding p.N180H, ClinVAR ID 224863), which partially segregated with PD in family A, the list of additional candidates did not include exonic SNVs, CNVs, or small indels in known familial PD genes (SNCA, PKRN, PINK1, DJ1, LRRK2, ATP13A2, PLA2G6, FBX07, VPS35, DNAJC6, SYNJ1, DNAJC13, RAB39B)14. All additional candidate rare variants passed validation by bidirectional Sanger sequencing.
Exonic Rare variants c.A577G (p.T193A) in the DNAJB6 gene (NM_058246.4, rs770053224_A/G) and c.A470G (p.N157S) in the PSAP gene (NM_002778.4, rs756379007_T/C) segregated with PD in family A and B, respectively. All four PD patients (IV:1, IV:4, IV:5, and V:1) of family A were heterozygous carriers for the DNAJB6; p.T193A and GBA1; c.115+1G>A variants whereas the three non-penetrant carriers of GBA1; c.115+1G>A in family A (IV:6, V:4, and V:5) lacked this concurrence. This indicates that the DNAJB6; p.T193A variant contributes to PD risk in this family (Fig. 1A). In family B, segregation of PSAP; p.N157S in all four PD patients including the phenocopy PD patient III:4 (Fig. 1C, black symbol with a white circle) indicated that the rare genetic variation in the PSAP gene is an independent risk factor for PD in this family which complies with a previous report10.
The PD cosegregating variants in DNAJB6 and PSAP genes identified in this study are listed with extremely low frequencies (MAF < 0.0001) in the ethnically matched controls in GnomAD v2.1.1 (ref. 15) and were classified as the 1% most deleterious substitutions in the genome (CADD > 20)16 (Table 1). No other additional candidate rare variant segregated completely with PD in the two families.
Functional impact of the rare variant in DNAJB6
DNAJB6, a member of the HSP40 family of J-protein co-chaperones, acts as an aggregation suppressor preventing fibrillation of several aggregation-prone proteins17,18. Steady-state levels and cellular distribution profiles of the wild-type DNAJB6 and the p.T193A variant proteins expressed in HEK293 cells were not significantly different (data not shown). DNAJB6 dimer-/oligomerization was also not affected by p.T193A substitution (Supplementary Fig. 2), suggesting that p.T193A substitution exerts its influence by altering the cellular function of DNAJB6 protein. The p.T193A substitution in DNAJB6 protein identified in the current study is located in a highly conserved serine/threonine-rich motif (i.e SxSTST at residues 190 and 192–195), which lines the concave side of the DNAJB6 dimer near the peptide-binding cleft (Fig. 2A, B). This structural conformation is necessary for cellular functions of the DNAJB6 protein19,20, which includes alpha-synuclein homeostasis21. We expressed wild-type DNAJB6 or p.T193A variant containing DNAJB6 proteins in an alpha-synuclein reporter cell line lacking endogenous DNAJB6 (see “Methods”). Diffuse cytoplasmic distribution of alpha-synuclein-dsRed conjugate was observed in the majority of cells in case of wild-type DNAJB6 protein expression whereas the expression of DNAJB6; p.T193A only partially restored the diffuse cytoplasmic alpha-synuclein distribution. A significantly higher number of cells expressing DNAJB6; p.T193A variant displayed a focal cytoplasmic distribution of alpha-synuclein-dsRed compared to wild-type DNAJB6. Collectively, these data indicate altered cellular homeostasis of alpha-synuclein as a direct consequence of p.T193A variant expression in HEK293 cells (Fig. 2C, D).
Functional impact of the rare variant in PSAP
Prosaposin (PSAP) is a lysosomal protein, which in its native form or processed peptides (e.g. Saposins A–D and several prosaptides) plays distinct cellular roles22. The PD cosegregating PSAP variant p.N157S identified in family B (Fig. 2C, D) is located in the Intersaposin A–B region of the PSAP protein (Fig. 3A). Rare potentially pathogenic variants in other regions of PSAP protein have previously been reported to increase PD risk in a GBA-dependent manner10,23,24,25. To determine the functional impact of the p.N157S variant, we expressed wild type or p.N157S variant PSAP as Flag or EGFP fusion proteins in the HEK293 cells. EGFP-tagged PSAP; p.N157S displayed a lower co-localization with perinuclear lysosomal LAMP1-positive puncta compared to wild-type PSAP (Fig. 3C, D). The reduced lysosomal localization of the p.N157S variant may be due to an overall reduction in protein levels 48 h post-transfection (Fig. 3D, top panel), indicating that variant PSAP protein is less stable than that of wild-type protein in HEK293 cells. It has previously been shown that the intersaposin A–B region in the PSAP protein is necessary for cleavage of native PSAP into biologically active Saposin peptides26 (Fig. 3A, Saposin A–D). PSAP is a functional partner of lysosomal Cathepsin D (CTSD) and both proteins are mutually dependent for lysosomal trafficking. Moreover, PSAP–CTSD interaction is necessary for stimulation of the CTSD proteolytic activity, which mediates the processing of PSAP at the intersaposin A–B region26,27,28,29. Computational predictions indicated that p.N157S substitution in PSAP introduces a novel phosphorylation site (LESSKI, AA 153–158) in the intersaposin A–B region near a predicted CTSD-specific site (PSAP residues 176–179, LLL) (Fig. 3A, B and Supplementary Fig. 3A). We tested the influence of p.N157S substitution on the PSAP–CTSD interaction. Fluorescence resonance energy transfer (FRET) analysis of co-expressed PSAP and CTSD as FRET capable fusion proteins indicated that the p.N157S substitution reduces PSAP–CTSD interaction in the LAMP1+lysosomal perinuclear compartment (Supplementary Fig. 3B). Thus, reduced PSAP synthesis and reduced interaction with CTSD were identified as functional consequences of the p.N157S substitution. This may influence cellular alpha-synuclein homeostasis30,31,32,33 considering the role of PSAP in lysosomal CTSD delivery and activation27,28. We, therefore, tested the influence of the PSAP; p.N157S expression on lysosomal handling of alpha-synuclein in heterologous cells. To this end, we co-expressed mCherry-tagged PSAP or PSAP; p.N157S with an aggregation-prone alpha-synuclein-EGFP (see “Methods”). The number of LAMP1-positive alpha-synuclein-EGFP decorated inclusions per cell as well as the number of cells with such inclusions was higher when co-expressing PSAP; p.N157S than when co-expressing wild-type PSAP (Fig. 3E, F). Collectively, these results indicate that expression of p.N157S variant PSAP in HEK293 cells results in a lysosomal defect, which affects alpha-synuclein handling.

A The domain structure of human the PSAP protein is shown. The location of the p.N157S variant (red) and the predicted Cathepsin D site (L177–L179) in the intersaposin A–B region is indicated (green). The alignment of human PSAP with various species is shown. Numbers indicate the position of amino acid residues. Blue shading indicates the conservation status of the corresponding residues; arrow points to the variant position. The predicted Cathepsin D site, L177–L179, is indicated (underlined in green). B The structure of the human PSAP protein is shown. The variant positions (N157) in the intersaposin A–B region is marked in red and the predicted Cathepsin D site (L177–L179) is displayed in pink. C Representative images showing the distribution of LAMP1-mTurquoise2 and PSAP-EGFP fusion proteins in HEK293T cells. DAPI was used as a nuclear stain (blue). Arrowheads in the bottom right image point to the EGFP-PSAP puncta in the perinuclear region not overlapping with LAMP1-mTurquoise2. D Quantification of green (PSAP-EGFP fusion protein) signal intensity overlapping with turquoise signal (mTurquoise2-LAMP1 marking the perinuclear lysosomal compartment) is shown. Graphs represent mean and standard deviation ***P = 0.001. Also shown is the immunoblot image of wild type and N157S variant containing PSAP proteins from HEK293 cells (top panel). HEK293 cells were transfected with an equal amount of plasmids expressing the wild type or N157S variant PSAP proteins as Flag-tag conjugate. Full-length blots are presented in Supplementary Fig. 3C. All blots are derived from the same experiment and gels/blots were processed in parallel. E Representative images showing LAMP1-positive intracellular inclusions decorated with alpha-synuclein-EGFP. Cells were transfected with three plasmids: a plasmid-expressing EGFP-tagged aggregation-prone alpha-synuclein protein (see “Methods”), a plasmid-expressing mTurquoise2-LAMP1 expression protein as a lysosomal marker, and a plasmid-expressing wild type or p.PN157S variant containing PSAP. Arrowheads in the merged panel point to the EGFP-PSAP puncta in the perinuclear region overlapping with LAMP1-mTurquoise2 as well as alpha-syuclein-EGFP puncta indicating lysosomal aggregation. F The quantification of lysosomal alpha-synuclein-EGFP aggregates in transfected cells is shown (n = 3 transfections). Statistical analysis was performed by ANOVA. Graphs represent mean and standard deviation, **P < 0.01.
Discussion
We here report rare genetic variants in the HSP70 chaperone DNAJB6 and the lysosomal protein PSAP as likely second hits in addition to the GBA1 gene variants in two PD families. The segregation pattern of additional rare variants in both families (Fig. 1A, C) and their ability to manipulate cellular alpha-synuclein handling when expressed in reporter cells (Figs. 2C, D and 3E, F) lead to the hypothesis that they contribute to the PD pathophysiology.
The precise mechanism by which GBA1 variants increase the risk for developing alpha synucleinopathy in PD remains unknown. Cellular alpha-synuclein is intrinsically prone to aggregation and toxic fibrillation34,35. Thus, a major portion of alpha-synuclein is delivered to lysosomes for degradation via non-vesicular (chaperone-mediated) or vesicular (micro- and macro-autophagy) pathways36,37. It was proposed that GBA1 variants result in an abnormal lysosomal glycosphingolipid environment that leads to an impaired autophagy–lysosomal pathway and promotes alpha-synuclein aggregation38. However, genetic observations such as reduced PD penetrance5 of GBA1 variants in familial cases, the occurrence of phenocopies in GBA-PD families3,4, and comparable age‐specific PD risk in homo-and heterozygous GBA1 variant carriers39 indicate that GBA1 variants alone do not cause alpha synucleinopathy or PD in the heterozygous GBA1 variant carriers. This suggests that additional risk influence cellular alpha-synuclein homeostasis in GBA-PD.
DNAJB6 is a member of the HSP40 family of J-protein co-chaperones. Rare genetic variants in several members of this family have previously been associated with PD40,41,42,43,44. Our data support that a p.T193A substitution, which is located in a conserved serine-threonine-rich SxSTST motif (residues 190 and 192–195) of DNAJB6, affects its anti-aggregation properties with respect to cellar alpha-synuclein homeostasis as was described previously for other aggregation-prone proteins19,20. In our study, DNAJB6; p.T193A and GBA1; c.115+1G>A variants were found to segregate concurrently only in the PD patients of family A (Fig. 1A, B). Individuals lacking this concurrence did not show cardinal motor or prodromal signs of PD (Fig. 1A, subjects IV:6, IV:7, V:4, and V:5). The DNAJB6; p.T193A variant when expressed in HEK293 cells promotes alpha-synuclein aggregation (Fig. 2C, D). Taken together, our results indicate that DNAJB6; p.T193A is a rare genetic variant, which apparently acts independently in relation to GBA, to enhance PD risk in the GBA1; c.115+1G>A variant carriers of the family A.
PSAP is a multifunctional protein, which is secreted in its native form as ligand for the PD-associated neuroprotective receptor GPR37 (refs. 45,46). PSAP is intracellularly cleaved by lysosomal proteases such as Cathepsin D (CTSD)26 into mature monosaposins. One of the cleavage products, namely Saposin C, is essential cofactor for lysosomal GBA24,25. Short peptides derived from PSAP (e.g. Prosaptides) have protective effects on dopaminergic neurons in vitro and in models of PD in vivo47,48. At the molecular level, PSAP protein engages in reciprocal functional interaction with CTSD and this interaction affects lysosomal trafficking and function of both PSAP and CTSD27,28,29. Interestingly, PSAP mRNA is upregulated in the substantia nigra of PD patients49. This upregulation may be a protective mechanism against PD, given the neuroprotective functions of PSAP and our study, which indicates that a dysfunctional PSAP contributes to PD.
In family B, three PD patients were concurrent carriers of GBA; p.L444P and PSAP; p.N157S variants. However, patient III:4 carried only the PSAP; p.N157S variant. This patient was clinically indistinguishable from the three PD patients with both variants, indicating a similar underlying pathogenic mechanism (Supplementary Table 1, Family B). Indeed both GBA and PSAP proteins interact at the molecular level24,25. PD pathogenesis due to GBA; p.L444P variant involves accumulation of alpha-synuclein aggregates50,51. The PSAP; p.N157S variant when expressed in HEK293 cells also promoted alpha-synuclein aggregation (Fig. 3E, F). The exact molecular mechanism by which PSAP; p.N157S variant promotes alpha-synuclein is currently not known. One possibility is a reduced molecular interaction between PSAP; p.N157S variant and CTSD (Supplementary Fig. 3B), which may decrease lysosomal CTSD function27. Since CTSD is the main lysosomal protease clearing alpha-synuclein52,53, the PSAP; p.N157S variant may promote alpha-synuclein aggregation and PD risk independently of GBA; p.L444P variant. Thus, the PSAP; p.N157S variant may contribute to PD risk in all four PD patients of the family B independent of the GBA; p.L444P variant. We thus considered the subject III:4 of the family B to be a genetic phenocopy lacking the GBA; p.L444P variant.
Finally, we focused on rare genetic variation as additional risk factors for GBA-PD in a family-based design. Variants in the PSAP and DNAJB6 genes were selected from the list of background WGS candidates primarily based on segregation testing and the potential roles of DNAJB6 and PSAP proteins in the cellular alpha-synuclein handling. In this study, we tested in heterologous cells (HEK293 cells) whether the identified variants have disrupted functions compared to the wild-type DNAJB6 and PSAP proteins. Further investigation on patient-derived material such as iPSCs is required to definitely link the identified rare genetic variants in DNAJB6 and PSAP with PD pathogenesis. The prioritization of DNAJB6 and PSAP in this study does not ignore the role of other rare variants in the list of candidates (Table 1) that may in addition play a role in PD pathogenesis. For instance, a potential pathogenic rare variant in the Tenascin receptor gene (TNR: p.N180H; ClinVAR ID 224863) was found in family A (Table 1). This finding is an independent replication of a recent exome sequencing study involving familial PD patients from a distant population54. In our study, the TNR variant was detected in three of the four PD patients of family A but in none of the controls, which indicates that the variant may have a pathogenic effect in the segregating individuals. In addition, we did not consider common genetic variation at the loci associated with cellular alpha-synuclein homeostasis. Large-scale genetic association studies are required to uncover such interactions. Despite these limitations, findings from our study contribute to a deeper molecular understanding of GBA-PD. Such understanding will not only be helpful for the clinicians when providing genetic counseling to seemingly GBA-PD patients and families but also for selecting patients for novel interventional therapies aiming to treat PD by augmenting lysosomal GBA function.
Methods
Study participants and clinical evaluation
Our study followed the tenets of the Declaration of Helsinki. The Institutional Review Board of the Institute for biomedical and genetic engineering (IBGE, Islamabad, Pakistan) provided the approval (Ref. IBGE/SARK04/1201/2012). Written informed consent was obtained from all participants.
Medical records of the neurology department of Shaheed Zulfiqar Ali Bhutto Medical University in Islamabad were screened rigorously to identify PD patients with a positive family history (defined as having at least one first-degree relative with PD who was also registered in the same clinic). We identified two families (family A and B) with four PD patients each. There was no history of consanguinity in the two families. After obtaining written informed consent, members of family A and B underwent a detailed neurologic examination by at least one experienced movement disorders specialist. The diagnosis of the eight PD patients was verified based on the UK Parkinson’s disease society brain bank55 and Gelb criteria56. Clinical data were collected, which comprised assessment of motor disability scores (Unified Parkinson Disease Rating Motor subscale III, UPDRS-III, total score 108)57, PD severity assessment (Hoehn and Yahr scale, H-Y, stages 1–5)58, cognitive impairment rating (Mini-Mental State Examination, MMSE, cut-off 24/30)59, depression rating (7-item version of the Hamilton Depression Rating Scale; scores 0–8: normal, 9–16: mild, 17–24: moderate and ≥25: severe)60 and an assessment of olfactory and visual functions.
Genome sequencing and analysis
Genomic DNA was isolated from peripheral blood cells using a QIAamp DNA Blood Mini Kit. DNA libraries were prepared using the TruSeq Nano DNA PCR-free kit (Illumina San Diego CA USA). Paired-end sequencing (2 × 150 bp) was performed at >30× coverage per sample (Illumina HiSeq X TEN and HiSeq2500, Illumina San Diego CA). Raw reads were aligned to the human reference genome (version build GRCh37, version hs37d5) using bwa mem (version 0.7.8) with minimum score threshold set to zero [-T 0] and remaining settings left at default values61. Duplicates were marked using biobambam (version 0.0.148)62. SNVs were called using SAMtools (version 0.1.19)63 and short indels were called using Platypus (version 0.7.4)64. Functional classifications of the variants were done using ANNOVAR65 with gene model definitions from Gencode (version v19).
Variant filtering, candidate selection, and Sanger sequencing
Quality-filtered variants (QUAL score >20) with minimum coverage >10× were carried forward for filtering and candidate selection. A stringent filtering approach was used to prioritize rare pathogenic variants likely representing candidates worthy of functional validation. We only considered variants with minor allele frequency MAF < 0.01 in ExAC (version 0.3) since high-risk rare variants with large effect sizes tend to aggregate in families. All functional rare variants, including nonsynonymous SNVs, frameshift, and non-frameshift indels, stop-gain/loss and splice-site SNVs or indels were considered. Rare functional variants that were identified only in the control individuals but were absent in one or more of the affected individuals of the family were removed. Deleteriousness prediction was carried out using a set of prediction tools, including CADD16, PolyPhen-2 (ref. 66), SIFT67, LRT68, Mutation Taster69, FATHMM70, PROVEAN71, and MetaSVM and MetaLR72. Benign variants were filtered out using the mutation significance cut‐off (MSC) tool, a gene‐level‐specific cut‐off for CADD/PolyPhen-2/SIFT scores as described previously73,74. Variants in genes with high brain expression in the GTEx Portal75 were prioritized. The SmallPedigree-WGS module of the Canvas program (version 1.40.0.1613) (https://academic.oup.com/bioinformatics/article/32/15/2375/1743834) was applied to the WGS data of two families separately to detect CNVs.
Commercially available M13-tailed primer pairs (Table 1; Thermo Fisher Scientific) were used for the amplification of candidate genomic regions. The genomic segment covering the position of p.L444P variant in GBA1 (rs421016 position) was amplified using forward (TGGGTGCGTAACTTTGTCGACAG) and reverse (CCACAGCAGGATCCTTGATGGTAA) primers, as described previously76. The genomic segment covering a novel stop-gain variant in the ABCA7 gene (corresponding to p.Q1062X) was amplified using M13-tailed forward (TACTACCTGACGCTGGTGAAGG) and reverse (AGTTAAGGCACAGCCACCCCACTG) primers (M13 sequence not shown). Thermocycling conditions used were as follows: 3 min at 94 °C, 30 cycles of 94 °C for 30 s, 60 °C for 30 s, and 72 °C for 45 s, and a final extension for 5 min at 72 °C. PCR products were resolved on 1.5% agarose gel and purified using the MinElute Gel Extraction Kit (Qiagen, Germany). Bidirectional Sanger sequencing was performed at StarSEQ GmbH, Mainz, Germany.
In silico analysis and protein structure predictions
Online tools PROSITE My Domains77, ClustalOmega78, and Jalview79 were used for drawing protein domain features, sequence alignment, and visualization, respectively. N-glycosylation sites were predicted using the GlycoEP and NetNGlyc web-servers80,81. Analysis of post-translational modification sites was performed with the Eukaryotic Linear Motif web-server82. Three-dimensional structure of human PSAP protein (Uniprot ID: P07602) was predicted using the I-TASSER web-server83. The ModLoop web-server84 was used to repair broken peptide bonds. Previously, Sun et al.85 showed that Cathepsin D (CTSD) specific sites are leucine rich. Based on this observation, we scanned the PSAP sequence, which predicted residues 176–179 (LLL) in the intersaposin region A–B of the PSAP as a potential CTSD-specific site. The three-dimensional structure of the human DNAJB6 homodimer was described previously by Söderberg et al.19 and was kindly provided by Professor Cecilia Emanuelsson (Lund University, Sweden). Figures of the PSAP and DNAJB6 structures were generated with PyMOL Molecular Graphics System (version 1.8).
Plasmids, cell culture, and transfection
Full-length cDNA encoding wild-type human PSAP cloned in pCMV3 with a C-terminal Flag tag was obtained from Sino Biological (HG16224-CF) and subcloned into pEGFP-N1 with HindIII and AgeI to obtain pEGFP-N1-PSAP (WT) plasmid. A fragment of PSAP cDNA encoding PSAP amino acid residues 1–320 encoding N157S variant was synthesized via GeneArt synthesis (Thermo Fisher Scientific) and inserted in pEGFP-N1-PSAP (WT) via HindIII and EcoRI, replacing the corresponding wild-type sequence to generate the pEGFP-N1-PSAP (N157S) plasmid. PSAP cDNA sequences encoding the wild type and the p.N157S variant were similarly cloned in pmCherry-N1. The cDNA sequence encoding human Cathepsin D (CTSD) was amplified from a human brain cDNA library and inserted into pmCherry-N1 using HindIII and AgeI restriction sites. The pcDNA3.1 plasmid-expressing wild-type DNAJB6 as a fluorescent fusion protein was previously published86 and was kindly provided by Professor Conrad Chris Weihl (Washington University School of Medicine). A wild-type DNAJB6 cDNA clone was modified by PCR to introduce the c.A577G/p.T193A variant in the coding sequence using the following primers: CAAATCGATATCAGCTTCAAC (forward primer; variant position is underlined) and CAACAGATGGCTGGCAACTAG (reverse primer). An alpha-synuclein-dsRed aggregation reporter cell line lacking endogenous DNAJB6 (ref. 21) was kindly provided by Dr. Cristian Hansen, Lund University, Sweden. A bicistronic plasmid encoding aggregation-prone alpha-synuclein (A53T) cDNA tagged with PDZ-binding motif (HSTTRV) of neuroligin-1 and PDZ1 domain of synaptic scaffolding molecule (S-SCAM) fused to mCherry has previously been described87 and was kindly provided by Professor Björn H. Falkenburger (University Hospital Carl Gustav Carus at the Dresden University of Technology, Germany). This plasmid was modified to replace mCherry with the EGFP coding sequence. A plasmid encoding Lamp1-mTurquoise2 was obtained from Addgene (Plasmid # 98828). Sanger sequencing verified the sequence integrity of all the above-mentioned plasmids.
Human embryonic kidney 293 cells (HEK293) were cultured in high glucose Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin–streptomycin (Gibco). For transient transfections, HEK293 cells were grown on circular glass coverslips (13 mm diameter) coated before for 1 h with 300 μl of 0.1% (w/v) poly-l-lysine solution (Sigma). Cells were transfected with plasmid DNA (0.25 µg each) using Lipofectamine 2000 reagent in OPTIMEM medium (Thermo Fisher Scientific).
Immunoblotting, cell imaging, and quantification
For immunoblotting, HEK293 cells transfected with plasmids expressing Flag-tagged proteins were washed and lysed. SDS-PAGE was performed with 10% acrylamide gels. Proteins were transferred to polyvinylidene difluoride (PVDF) membrane, which was blocked in 10% skimmed milk for 1 h. The membranes were incubated with mouse anti-Flag antibody (1:1000, F1804, Sigma) overnight at 4 °C, followed by incubation with peroxidase-linked secondary antibody (1:10,000, P0161, Dako) for 1 h. Protein bands were detected on a ChemiDoc XRS + imaging system (Bio-Rad). After Flag tag detection, the PVDF membrane was stripped and beta-actin expression was detected on the same membrane using a rabbit anti-actin antibody (1:1000, ab8227, Abcam).
For cell imaging, cells transfected with appropriate plasmids were washed three times with phosphate buffer saline (PBS) solution and were fixed in 4% paraformaldehyde for 10 min. Nuclei were stained with 4′,6′-diamidino-2-phenylindole (Sigma, St Louis, MO, USA). Coverslips were mounted onto slides with ProLong Gold anti-fade mountant (Thermo Fisher Scientific). Images were obtained with a ×63/1.4 oil objective on a TCS SP5 confocal imaging system (Leica Microsystems, Heidelberg GmbH). For the analysis of lysosomal localization of PSAP variants, Fiji software was used to quantify the integrated intensity of EGFP fluorescence (i.e PSAP-EGFP fusion) overlapping with turquoise signal (i.e mTurquoise2-LAMP1 fusion). At least 30 cells for each condition from triplicate coverslips were analyzed. Quantification of alpha-synuclein aggregation was performed as described previously21,87. Briefly, whole-field images were obtained from three coverslips for each condition. Alpha-synuclein aggregation was defined as a cellular region with markedly increased corresponding immunofluorescence signal compared with surrounding cytoplasmic areas of the same cell. An investigator blinded to the experimental conditions classified at least 100 cells per coverslip as positive or negative for cytoplasmic alpha-synuclein aggregates (Fig. 2), or cells with no, single, or multiple lysosomal aggregates (Fig. 3). All experiments were repeated at least three times.
FRET analysis
For the FRET experiments, cells were transfected with plasmids expressing EGFP and mCherry as a donor–acceptor FRET pair alone as controls or as fusion proteins. A construct expressing EGFP and mCherry as a tandem dimer separated by a spacer was used as FRET standard. Twenty-four hours after transfection, cells were fixed and mounted as described above. FRET measurements were performed by using the FRET acceptor photobleaching wizard in the Leica Application Suite on a Leica scanning confocal microscope (TCS SP5 Leica Microsystems, Heidelberg GmbH, Germany). FRET measurements were performed for at least three independent transfections for each condition. FRET efficiency was defined as FRETeff = (donorpost − donorpre)/donorpost, where donorpost describes the fluorescence intensity after, and donorpre the intensity before acceptor photobleaching. FRETeff was calculated by the FRET wizard of the Leica Application Suite.
Statistical analysis
We used QtiPlot software (version 4.8.7) for statistical analysis and data presentation. Graphs represent mean ± standard deviation. The statistical tests and number of independent experiments for each analysis are noted in the figure legends. P < 0.05 was considered significant.
MAÖ and RCW gratefully acknowledge the support of the Klaus Tschira Foundation and the European Union’s Horizon 2020 Framework Programme for Research and Innovation under the Specific Grant Agreement Nos. 720270 and 785907 (Human Brain Project SGA1 and SGA2). A.U. and S.N. gratefully acknowledge the support from the German Academic Exchange Service (DAAD Scholarship) and International Research Support Initiative Program of the higher education commission of Pakistan respectively.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The datasets generated during and/or analyzed during the current study are available in the EGA repository under the following accessions: Study: EGAS00001004777 and Dataset: EGAD00001006561.
Code availability
The custom scripts used to annotate, parse, and filter the variants are made available in the following public repositories:
https://github.com/NagaComBio/PlatypusGermlinePipeline, https://github.com/NagaComBio/GermlineSmallVariantAnalysis.
Following tools were used to map, de-duplicate, and call variants from the sample: bwa mem (version 0.7.8, parameter: -T 0), biobambam (version 0.0.148), SAMtools (version 0.1.19), Platypus (version 0.7.4). The following tools were used to annotate the variants: ANNOVAR, Gencode (version v19), CADD (version 1.3), dbNSFP (version 2.9). The germline CNVs were called used the SmallPedigree-WGS module of the Canvas program (version 1.40.0.1613).
The ClinVar accession numbers of the deposited variants are as follows: SCV001244288 and SCV001244287.
References
Choi, J. H. et al. Aggregation of α-synuclein in brain samples from subjects with glucocerebrosidase mutations. Mol. Genet. Metab. https://doi.org/10.1016/j.ymgme.2011.06.008 (2011).
Nichols, W. C. et al. Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 72, 310–316 (2009).
Lesage, S. et al. Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum. Mol. Genet. https://doi.org/10.1093/hmg/ddq454 (2011).
Klein, C., Chuang, R., Marras, C. & Lang, A. E. The curious case of phenocopies in families with genetic Parkinson’s disease. Mov. Disord. https://doi.org/10.1002/mds.23853 (2011).
Anheim, M. et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78, 417–420 (2012).
Luan, Z. et al. The chaperone activity and toxicity of ambroxol on Gaucher cells and normal mice. Brain Dev. https://doi.org/10.1016/j.braindev.2012.05.008 (2013).
Migdalska-Richards, A., Daly, L., Bezard, E. & Schapira, A. H. V. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann. Neurol. https://doi.org/10.1002/ana.24790 (2016).
Sardi, S. P. et al. Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc. Natl Acad. Sci. USA. https://doi.org/10.1073/pnas.1616152114 (2017)
Van Blitterswijk, M. et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. https://doi.org/10.1093/hmg/dds199 (2012).
Robak, L. A. et al. Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain. https://doi.org/10.1093/brain/awx285 (2017).
Lubbe, S. J. et al. Additional rare variant analysis in Parkinson’s disease cases with and without known pathogenic mutations: evidence for oligogenic inheritance. Hum. Mol. Genet. https://doi.org/10.1093/hmg/ddw348 (2016).
Mcneill, A. et al. Hyposmia and cognitive impairment in Gaucher disease patients and carriers. Mov. Disord. https://doi.org/10.1002/mds.24945 (2012).
Beavan, M. et al. Evolution of prodromal clinical markers of parkinson disease in a GBA mutation-positive cohort. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2014.2950 (2015).
U.S. National Library of Medicine. Genes—Genetics Home Reference—NIH (2018).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. https://doi.org/10.1038/nature19057 (2016).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
Gillis, J. et al. The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J. Biol. Chem. https://doi.org/10.1074/jbc.M112.421685 (2013).
Månsson, C. et al. DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones. https://doi.org/10.1007/s12192-013-0448-5 (2014).
Söderberg, C. A. G. et al. Structural modelling of the DNAJB6 oligomeric chaperone shows a peptide-binding cleft lined with conserved S/T-residues at the dimer interface. Sci. Rep. https://doi.org/10.1038/s41598-018-23035-9 (2018).
Kakkar, V. et al. The S/T-Rich Motif in the DNAJB6 Chaperone Delays Polyglutamine Aggregation and the Onset of Disease in a Mouse Model. Mol. Cell. https://doi.org/10.1016/j.molcel.2016.03.017 (2016).
Aprile, F. A. et al. The molecular chaperones DNAJB6 and Hsp70 cooperate to suppress α-synuclein aggregation. Sci. Rep. https://doi.org/10.1038/s41598-017-08324-z (2017).
Meyer, R. C., Giddens, M. M., Coleman, B. M. & Hall, R. A. The protective role of prosaposin and its receptors in the nervous system. Brain Res. 1585, 1–12 (2014).
Ouled Amar Bencheikh, B. et al. Sequencing of the GBA coactivator, Saposin C, in Parkinson disease. Neurobiol. Aging. https://doi.org/10.1016/j.neurobiolaging.2018.06.034 (2018).
Yap, T. L., Gruschus, J. M., Velayati, A., Sidransky, E. & Lee, J. C. Saposin C protects glucocerebrosidase against α-synuclein inhibition. Biochemistry. https://doi.org/10.1021/bi401191v (2013).
Sun, Y., Qi, X. & Grabowski, G. A. Saposin C is required for normal resistance of acid β-glucosidase to proteolytic degradation. J. Biol. Chem. https://doi.org/10.1074/jbc.M302752200 (2003).
Hiraiwa, M. et al. Lysosomal proteolysis of prosaposin, the precursor of saposins (sphingolipid activator proteins): Its mechanism and inhibition by ganglioside. Arch. Biochem. Biophys. https://doi.org/10.1006/abbi.1997.9958 (1997).
Gopalakrishnan, M. M. et al. Purified recombinant human prosaposin forms oligomers that bind procathepsin D and affect its autoactivation. Biochem. J. 383, 507–515 (2004).
Zhu, Y. & Conner, G. E. Intermolecular association of lysosomal protein precursors during biosynthesis. J. Biol. Chem. 269, 3846–3851 (1994).
Grässel, S. & Hasilik, A. Human cathepsin D precursor is associated with a 60 kDa glycosylated polypeptide. Biochem. Biophys. Res. Commun. https://doi.org/10.1016/S0006-291X(05)80141-X (1992).
Sevlever, D., Jiang, P. & Yen, S. H. C. Cathepsin D is the main lysosomal enzyme involved in the degradation of??-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47, 9678–9687 (2008).
Bae, E. J. et al. Haploinsufficiency of cathepsin D leads to lysosomal dysfunction and promotes cell-to-cell transmission of α-synuclein aggregates. Cell Death Dis. https://doi.org/10.1038/cddis.2015.283 (2015).
Cullen, V. et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol. Brain. https://doi.org/10.1186/1756-6606-2-5 (2009).
Qiao, L. et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol. Brain. https://doi.org/10.1186/1756-6606-1-17 (2008).
Mantsyzov, A. B. et al. A maximum entropy approach to the study of residue-specific backbone angle distributions in α-synuclein, an intrinsically disordered protein. Protein Sci. https://doi.org/10.1002/pro.2511 (2014).
Coelho-Cerqueira, E., Carmo-Gonçalves, P., Sá Pinheiro, A., Cortines, J. & Follmer, C. α-Synuclein as an intrinsically disordered monomer—fact or artefact? FEBS J. https://doi.org/10.1111/febs.12471 (2013).
Cuervo, A. M., Stafanis, L., Fredenburg, R., Lansbury, P. T. & Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science. https://doi.org/10.1126/science.1101738 (2004).
Cao, Y. L. et al. A role of BAG3 in regulating SNCA/α-synuclein clearance via selective macroautophagy. Neurobiol. Aging. https://doi.org/10.1016/j.neurobiolaging.2017.08.023 (2017).
Magalhaes, J. et al. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum. Mol. Genet. 25, 3432–3445 (2015).
Alcal––ay, R. N. et al. Comparison of parkinson risk in ashkenazi jewish patients with gaucher disease and gba heterozygotes. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2014.313 (2014).
Vilariño-Güell, C. et al. DNAJC13 mutations in Parkinson disease. Hum. Mol. Genet. https://doi.org/10.1093/hmg/ddt570 (2014).
Lorenzo-Betancor, O. et al. DNAJC13 p.Asn855Ser mutation screening in Parkinson’s disease and pathologically confirmed Lewy body disease patients. Eur. J. Neurol. https://doi.org/10.1111/ene.12770 (2015).
Elsayed, L. E. O. et al. A novel nonsense mutation in DNAJC6 expands the phenotype of autosomal-recessive juvenile-onset Parkinson’s disease. Ann. Neurol. https://doi.org/10.1002/ana.24591 (2016).
Yuan, L. et al. Systematic analysis of genetic variants in Han Chinese patients with sporadic Parkinson’s disease. Sci. Rep. https://doi.org/10.1038/srep33850 (2016).
Durrenberger, P. F. et al. DnaJB6 is present in the core of Lewy bodies and is highly up-regulated in Parkinsonian astrocytes. J. Neurosci. Res. https://doi.org/10.1002/jnr.21819 (2009).
Meyer, R. C., Giddens, M. M., Schaefer, S. A. & Hall, R. A. GPR37 and GPR37L1 are receptors for the neuroprotective and glioprotective factors prosaptide and prosaposin. Proc. Natl. Acad. Sci. USA. https://doi.org/10.1073/pnas.1219004110 (2013).
Leinartaité, L. & Svenningsson, P. Folding underlies bidirectional role of GPR37/Pael-R in Parkinson disease. Trends Pharmacol. Sci. https://doi.org/10.1016/j.tips.2017.05.006 (2017).
Liu, J., Wang, C. Y. & O’Brien, J. S. Prosaptide D5, a retro-inverso 11-mer peptidomimetic, rescued dopaminergic neurons in a model of Parkinson’s disease. FASEB J. 15, 1080–1082 (2001).
Gao, H. L. et al. Attenuation of MPTP/MPP+ toxicity in vivo and in vitro by an 18-mer peptide derived from prosaposin. Neuroscience. https://doi.org/10.1016/j.neuroscience.2013.01.007 (2013).
Miller, R. M. et al. Robust dysregulation of gene expression in substantia nigra and striatum in Parkinson’s disease. Neurobiol. Dis. https://doi.org/10.1016/j.nbd.2005.07.010 (2006).
Yun, S. P. et al. α-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol. Neurodegener. https://doi.org/10.1186/s13024-017-0233-5 (2018).
Migdalska-Richards, A. et al. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain. https://doi.org/10.1093/brain/awx221 (2017).
Rosenbloom, B. et al. The incidence of Parkinsonism in patients with type 1 Gaucher disease: data from the ICGG Gaucher Registry. Blood Cells Mol. Dis. https://doi.org/10.1016/j.bcmd.2010.10.006 (2011).
Massano, J. & Bhatia, K. P. Clinical approach to Parkinson’s disease: features, diagnosis, and principles of management. Cold Spring Harb. Perspect. Med. https://doi.org/10.1101/cshperspect.a008870 (2012).
Farlow, J. L. et al. Whole-exome sequencing in familial Parkinson disease. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2015.3266 (2016).
Hughes, A. J., Daniel, S. E., Kilford, L. & Lees, A. J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 55, 181–184 (1992).
Gelb, D. J., Oliver, E. & Gilman, S. Diagnostic criteria for Parkinson disease. JAMA Neurol. 56, 33–39 (1999).
Fahn, S., Elton, R. L. & UPDRS program members. Unified Parkinsons Disease Rating Scale. In: Fahn, S., Marsden, C. D., Goldstein, M., Calne, D. B., editors. Recent developments in Parkinsons disease, vol 2. Florham Park, NJ: Macmillan Healthcare Information, p 153–163 (1987).
Goetz, C. G. et al. The Movement Disorder Society Task Force on rating scales for Parkinson’s disease. Movement Disorder Society Task Force report on the Hoehn and Yahr staging scale: status and recommendations. Mov. Disord. 19, 1020–1028 https://doi.org/10.1002/mds.20213 (2004).
Folstein, M. F., Folstein, S. E. & McHugh, P. R. ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. https://doi.org/10.1016/0022-3956(75)90026-6 (1975).
HAMILTON, M. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry. 23, 56–62 https://doi.org/10.1136/jnnp.23.1.56 (1960).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Tischler, G. & Leonard, S. Biobambam: Tools for read pair collation based algorithms on BAM files. Source Code Biol. Med. https://doi.org/10.1186/1751-0473-9-13 (2014).
Li, H. et al. The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Rimmer, A. et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 46, 1–9 (2014).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat. Methods. https://doi.org/10.1038/nmeth0410-248 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. https://doi.org/10.1038/nprot.2009.86 (2009).
Chun, S. & Fay, J. C. Identification of deleterious mutations within three human genomes. https://doi.org/10.1101/gr.092619.109.2001 (2009).
Schwarz, J. M., Rödelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. https://doi.org/10.1038/nmeth0810-575 (2010).
Shihab, H. A. et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum. Mutat. 34, 57–65 https://doi.org/10.1002/humu.22225 (2013). Epub 2 Nov 2012.
Choi, Y. & Chan, A. P. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. https://doi.org/10.1093/bioinformatics/btv195 (2015).
Dong, C. et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. https://doi.org/10.1093/hmg/ddu733 (2015).
Itan, Y. et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat. Methods. https://doi.org/10.1038/nmeth.3739 (2016).
Parlato, M. et al. Human ALPI deficiency causes inflammatory bowel disease and highlights a key mechanism of gut homeostasis. EMBO Mol. Med. https://doi.org/10.15252/emmm.201708483 (2018).
Carithers, L. J. & Moore, H. M. The Genotype-Tissue Expression (GTEx) Project. Biopreserv. Biobank. https://doi.org/10.1089/bio.2015.29031.hmm (2015).
Brown, J. T., Lahey, C., Laosinchai-Wolf, W. & Hadd, A. G. Polymorphisms in the glucocerebrosidase gene and pseudogene urge caution in clinical analysis of Gaucher disease allele c.1448T>C (L444P). BMC Med. Genet. https://doi.org/10.1186/1471-2350-7-69 (2006).
Hulo, N. The PROSITE database. Nucleic Acids Res. https://doi.org/10.1093/nar/gkj063 (2006).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. https://doi.org/10.1093/nar/gkz268 (2019).
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M. & Barton, G. J. Jalview Version 2-A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Chauhan, J. S., Rao, A. & Raghava, G. P. S. In silico platform for prediction of N-, O- and C-glycosites in eukaryotic protein sequences. PLoS ONE 8, e67008 https://doi.org/10.1371/journal.pone.0067008 (2013).
Gupta, R. & Brunak, S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pacific Symposium on Biocomputing. 310–322 (2002) Center for Biological Sequence Analysis, Bldg-208, Bio-Centrum Technical University of Denmark, DK-2800 Lyngby, Denmark.
Dinkel, H. et al. ELM 2016—data update and new functionality of the eukaryotic linear motif resource. Nucleic Acids Res. 44, D294–D300 (2016).
Yang, J. et al. The I-TASSER suite: protein structure and function prediction. Nat. Methods. https://doi.org/10.1038/nmeth.3213 (2014).
Fiser, A. & Sali, A. ModLoop: automated modeling of loops in protein structures. Bioinformatics. https://doi.org/10.1093/bioinformatics/btg362 (2003).
Sun, H. et al. Proteolytic characteristics of Cathepsin D related to the recognition and cleavage of its target proteins. PLoS ONE. https://doi.org/10.1371/journal.pone.0065733 (2013).
Bengoechea, R., Pittman, S. K., Tuck, E. P., True, H. L. & Weihl, C. C. Myofibrillar disruption and RNA-binding protein aggregation in a mouse model of limb-girdle muscular dystrophy 1D. Hum. Mol. Genet. https://doi.org/10.1093/hmg/ddv363 (2015).
Dinter, E. et al. Rab7 induces clearance of α-synuclein aggregates. J. Neurochem. https://doi.org/10.1111/jnc.13712 (2016).
Acknowledgements
We thank Professor Conrad Chris Weihl (Washington University School of Medicine), Dr. Cristian Hansen and Professor Cecilia Emanuelsson (Lund University, Sweden), and Professor Björn H. Falkenburger (University Hospital Carl Gustav Carus at the Dresden University of Technology, Germany) for providing material used in this study. We thank the High-Throughput Sequencing Unit of the Genome and Proteome Core Facility of the German Cancer Research Center (DKFZ, Heidelberg) and Microscopy Core Facility of the Institute for Molecular Biology (IMB, Mainz) for excellent technical support.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
M.A., N.A., J.v.E., and M.S. conceived of the presented study; M.A. and J.v.E. designed the experiments and supervised the study; M.A., A.U., N.K., and S.N. performed all experiments; N.A. recruited the families; N.P., M.S., M.A., and M.I. analyzed genome sequencing data; A.U. and N.A. interviewed the family; M.B. examined clinical records and the patients; and M.A. and J.v.E. wrote the manuscript. All authors provided critical feedback and helped shape the research, analysis, and manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aslam, M., Kandasamy, N., Ullah, A. et al. Putative second hit rare genetic variants in families with seemingly GBA-associated Parkinson’s disease. npj Genom. Med. 6, 2 (2021). https://doi.org/10.1038/s41525-020-00163-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41525-020-00163-8
This article is cited by
-
Prosaposin maintains lipid homeostasis in dopamine neurons and counteracts experimental parkinsonism in rodents
Nature Communications (2023)
-
GBA-associated Parkinson’s disease in Hungary: clinical features and genetic insights
Neurological Sciences (2023)
-
Who is at Risk of Parkinson Disease? Refining the Preclinical Phase of GBA1 and LRRK2 Variant Carriers: a Clinical, Biochemical, and Imaging Approach
Current Neurology and Neuroscience Reports (2023)
-
PRINCESS: comprehensive detection of haplotype resolved SNVs, SVs, and methylation
Genome Biology (2021)
