
Abstract
Deep learning (DL) has indeed emerged as a powerful tool for rapidly and accurately predicting materials properties from big data, such as the design of current commercial Li-ion batteries. However, its practical utility for multivalent metal-ion batteries (MIBs), the most promising future solution of large-scale energy storage, is limited due to scarce MIB data availability and poor DL model interpretability. Here, we develop an interpretable DL model as an effective and accurate method for learning electrode voltages of multivalent MIBs (divalent magnesium, calcium, zinc, and trivalent aluminum) at small dataset limits (150–500). Using the experimental results as validation, our model is much more accurate than machine-learning models, which usually are better than DL in the small dataset regime. Besides the high accuracy, our feature-engineering-free DL model is explainable, which automatically extracts the atom covalent radius as the most important feature for the voltage learning by visualizing vectors from the layers of the neural network. The presented model potentially accelerates the design and optimization of multivalent MIB materials with fewer data and less domain-knowledge restriction and is implemented into a publicly available online tool kit in http://batteries.2dmatpedia.org/ for the battery community.
Similar content being viewed by others

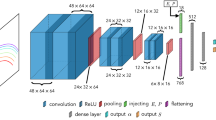

Introduction
Lithium-ion batteries (LIBs) have shown great success as the major power source for transportation and as an energy storage solution for grid applications1,2,3. Nowadays, the relatively low-energy density and the scarcity of Li raw materials are the main issues of LIBs for large-scale applications4,5. These issues call for high density, cheaper and sustainable alternatives to present LIB technologies. Multivalent metal-ion batteries (MIBs), including Mg2+, Ca2+, Zn2+, Al3+, have the potential to meet this purpose, due to the relatively high abundance of these elements in the Earth’s crust and high-energy density6.
Within the arena of big data, deep learning (DL) has emerged as a game-changing technique very recently, enabling numerous scientific applications in chemistry7, mathematics8, physics9, and biology10. In materials science, several DL models11,12,13 have also been developed, such as AtomSets14, SchNet15, Material Optimal Descriptor Network (MODNet)16, Compositionally Restricted Attention-Based Network (CrabNet)17,18, Crystal Graph Convolutional Neural Network (CGCNN)19,20,21,22,23,24. They have achieved great success in the applications, for instance, identifying degradation patterns of LIBs25, designing solid-state LIBs26, learning properties from multi-fidelity data27, discovering stable lead-free hybrid organic–inorganic perovskites28, screening 2D ferromagnets29, discovering and designing electrocatalysts30,31, mapping the crystal-structure phase32, and designing material microstructures33. It has been widely demonstrated that DL has much lower model errors than conventional machine-learning (ML) models, such as support vector regression (SVR) and kernel ridge regression (KRR), when dealing with big data34. For example, the reported mean absolute error (MAE) of the deep-neural network (DNN) model is lower than conventional ML in predicting the volume change and voltage of LIB electrodes35,36. However, DL still cannot solve many problems in the field of batteries, especially the MIBs beyond lithium, due to insufficient data available. For example, Joshi et al. predicted the voltages of Na-ion battery electrodes with a DNN model, but MAE is much higher than that of LIBs35. Moreover, due to being highly complex, DNN takes a long training time and is not explainable, which may not outperform shallow learning in solving some battery problems, especially for the design of batteries, in which explanations are in great demand. Therefore, the high-performance and interpretable DL model is of high need for predicting a variety of properties of multivalent MIBs from small data and then designing high-performing multivalent MIBs.
In this work, we take the voltage of multivalent MIBs as an example to demonstrate how an interpretable deep transfer-learning (TL) model, (Fig. 1) can be used for exploration and design of electrode materials for battery applications, addressing the conventional ML issues (low prediction accuracy and heavy feature-engineering dependence) and DL limitations (low interpretability and high big-data demand). It is worth noting that high-voltage electrode materials can enhance the voltage platform of batteries, which is the key component for high-energy density MIBs and is generally used in the performance prediction of materials of battery electrodes35,36,37. We firstly train our DL models with relatively large data of the electrode voltage of LIBs (2000+ data) from Materials Project (MP)10,38. It is found that MAE of LIBs is only 0.32 V. Nevertheless, MAE of multivalent MIBs is significantly high (up to 2.14 V) using the same method because of small datasets of multivalent MIBs (as low as 149). We, thus, integrate the TL technique39, widely used to address less data restriction40,41, into our DL models. It greatly reduces the MAEs for Zn-, Ca-, Mg-, and Al-ion batteries, for example, from 2.14 V down to only 0.47 V for the Zn-ion battery. To interpret our DL models, we perform the visualization of the similarity between the elements and local environments in different layers of the deep-neural network. The DL models can automatically hierarchically extract key features and explain the different contributions of element groups in the periodic table to the corresponding electrode voltages (Supplementary Fig. 1). Our results show that the highly accurate and interpretable deep model could accelerate the discovery and design of electrode materials for multivalent MIBs and the development of the large-scale battery industry.

A crystal is converted and characterized by atomic and bonding vectors. The elemental and local environment representations for visualization of the model are constructed from the embedding and the convolutional layers, respectively. The weights in the hidden layer and output layer are trained during transfer learning.
Results and discussion
Deep learning of Li-ion batteries
In all, 2190 samples of LIB electrode materials are used for model training and assessment after excluding the outliers and inconsistencies. The data (target data) firstly is randomly split into training, validation, and test parts with the ratio of 0.8:0.1:0.1, respectively. The three parts show similar distributions (Fig. 2a, top), indicating a reasonable split. Then, we train and supervise our DL model (Fig. 1) with training and validation data, respectively. The mean squared errors (MSEs) are chosen to be the loss function and MAEs are the evaluation metric. After fully training, MAE of our DL model for predicting the voltages of LIBs is only 0.32 V, which is lower than the conventional ML results (0.40 V). Our results are quite similar to the previous reports for LIBs35,36. The predicted voltages have similar distributions (Fig. 2a, right) with the target voltage (Fig. 2a, top) for all the training, validation, and test sets. The data points of predicted voltages plotted against the target voltages are around the dashed straight line of y = x (Fig. 2a, dash line), which also indicates the high accuracy of our model. However, our DL model developed from LIBs (named Li-model) cannot be directly used for multivalent MIBs (quite large MAEs Fig. 2b–e) because of the different properties between LIBs and multivalent MIBs. It’s also inadvisable to train DL models from small data of multivalent MIBs. Furthermore, it is found that the MAEs of conventional ML are similar to the Li-model (Fig. 2b–e). Thus, neither DL nor conventional ML is suitable for multivalent MIBs. This critical problem will be addressed in the late part of this paper.

a The data points are predicted with the deep-learning model and those in b–e are predicted with the transfer-learning model. The model errors of the Li-, Ca-, Mg-, Zn-, and Al-ion battery are shown in the right-down corner. The histograms at the top and right show the distributions of predicted and target voltages, respectively. The black dashed line is the identity line (y = x) for reference. The TL, CML, and Li-model mean the transfer learning, conventional machine learning, and the model trained on LIB database only.
Interpretability and visualization of the DL model
Although the state-of-the-art DL models outperform conventional ML models in the large dataset regime, they are generally viewed as black-box models due to the high complexity and are often achieved at the cost of interpretability. This is a major drawback of DL models for applications in which the interpretability of decisions is a critical prerequisite, such as new materials discovery and design20,42,43. Here, we visualize the embedding and convolutional layers of the deep-neural network, separately20 (see in Fig. 1) to interpret the contribution of underlying features to the electrode-voltage prediction. Taking the LIB as a proof of concept, we first project the 64-dimensional elemental representation vectors on a two-dimensional (2D) plane constituted by the first two principal components, dimension 1 and 2, using the principal component analysis (PCA)44,45. Figure 3a shows the visualized element features automatically extracted from the embedded layers (Fig. 1). As can be seen, the elements can be clearly clustered into three groups according to the location of the corresponding elements in the periodic table. For instance, the first two group elements, alkali, and alkaline-earth elements, locate at the upper right corner of the plot; the early transition metal (TM) elements locate at the left side; and the rest elements mainly distribute at the lower right side. Such a distribution indicates that the voltage of a crystal has a strong correlation with the group number of the constituent elements. Interestingly, we can observe a linear relationship between the covalent radius of the elements and the second principal component (dimension 2) (Fig. 3a inset). Thus, the covalent radius of the constituent elements of electrodes should be one of the important features for crystal voltage prediction. A conventional ML-based random forest regression (RFR) model with 273 materials features46,47 is also trained for analyzing the Gini importance factor by the weight of features. The covalent radius is also found to be the most important feature with the importance ratio as high as 20% (Supplementary Fig. 2). Such a result indicates the validation of the visualization analysis of our interpretable DL model.

a, b The visualization of the two principal dimensions with PCA for the element representations and the local oxygen-coordination-environment representations, respectively. Dimension 1 and 2 in the plot constitute a plane in the corresponding vector space that can approximate the representations best. The points in a are colored according to their elemental groups. c the median value of the learned local voltages for each element in the oxygen-coordination environments depicted in the elemental parodic table. The elements in b and c are labeled according to the type of center atoms in oxygen-coordination environments and the colors are coded with learned local voltages. The gray arrow in b indicates the change of local voltages.
Besides the visualization of the element representations in the neural network, the interpretable model can get a clear picture of the local environment from the electrode-voltage prediction. We next visualize the local environment (see more details in the Methods section), and take the oxygen coordination, at least two oxygen atoms around the central element, as the local environment representation because of the large number of oxides for battery electrodes. There are 23,022 local oxygen-coordination environments in total in our dataset. We first project all the oxygen local environments into a 2D plane using the PCA (Fig. 3b), and the elements are colored according to their local voltage, which is the voltage for each atom in the corresponding crystal and is calculated using Equation (2) in Methods. It shows that the local voltage color changes along the diagonal direction (gray arrow) in the dimension 1- dimension 2 plot. Such a phenomenon indicates that both dimension 1 and dimension 2 have linear relationships with the local voltage, implying the local voltage information has been learned by our model. To surface such a relationship, we visualize the oxygen-coordination-environments with the t-distributed stochastic neighbor embedding (t-SNE) algorithm48 (Supplementary Fig. 3). We find that oxygen environments can be approximately grouped into three large groups according to the t-SNE distribution: (i) the IA group with low voltages, (ii) the main group with high voltages, and (iii) the TM group with both low and high voltages. In order to validate such voltage performance extracted from the neural network, we calculate and project the median voltage (Vi) for each element (under the oxygen-coordination environment) on the elemental periodic table as shown in Fig. 3c. As can be seen, it is quite similar to the t-SNE analysis, but with more details. The elements in the main group have high-median local voltage except for the alkali metal elements, which have low voltages. For the TMs, the early TMs mainly have relatively low-median local voltages, while the late TMs perform reversely. Such a relationship of local voltages with elements probably provides a promising strategy to design electrodes of batteries with a high voltage through the elemental replacement, such as to replace an early transition metal, Ti, by a late transition metal, Cu, which can significantly increase the cathode voltage from 1.32 V of NaTiPO4F to 5.52 V of NaCuPO4F for Na-ion batteries49. Although, a high-voltage cathode often experiences a poor reversibility in the experiment, which should be taken into consideration in actual selection of cathode materials in the experiment.
Interestingly, the PCA of the elemental features (Fig. 3a) also performs similar element-voltage relations with that of the local voltage of oxygen-coordination environment analysis. For example, the elements with the low local voltages occupy the left and right parts (two regions shaded ion blue in Fig. 3a), while the middle area shaded in orange is mostly occupied by the elements with the high local voltages. This means that instead of extracting from the late convolutional layers (Fig. 1), the voltage information has already been learned in the early layer in our neural work. The oxygen-coordination environment and the covalent radius shows a similar trend with respect to the element (Supplementary Fig. 4), in line with the previous element representation analysis. Moreover, the local voltage of each element (Fig. 3c) is not only connected with the elemental properties, such as the electronegativity, but also affected by its oxygen-coordination environment, which are not available from conventional ML.
The p-orbital elements and late transition metal elements prefer to accept electrons, resulting in the reduction reaction. These characters make crystals with these elements have large chemical potentials and thus high voltages. The alkali metals and the early transition metals are easy to lose their electrons and get oxidation. Thus, electrode materials with these elements under the oxygen environment would have small chemical potentials and low voltages. The alkaline-earth metal ions prefer to attract electrons because of the electrostatic force between the cation and the electrons, and thus, the existence of these bivalent cations would make the cathodes have large chemical potentials and high voltages.
Transfer learning of multivalent metal-ion batteries
In this section, we will address the aforementioned problem that it is hard to get high-performance DL models for multivalent MIBs because of data scarcity, for example, only 149 Al-ion samples. Multivalent MIBs have the similar mechanism with LIBs, which allows us to reuse the pre-trained DL model on LIBs to multivalent MIBs. Here, we integrate the layer-freezing TL method45, which would only fine tune the last two fully connected layers, into our deep-neural network (see in Fig. 1) to predict the voltages of Mg-, Ca-, Zn-, and Al-ion battery electrodes. For comparison, two conventional ML models and one DL model trained from LIBs (named Li-model) are also used on multivalent MIBs. Thus, the DL model, CGCNN, and the two conventional ML models are not only used for the Li-ion battery voltage prediction, but also the multivalent MIBs, while the TL model is only used for the multivalent MIBs. The overview of the architecture for these models are shown in Supplementary Fig. 1. Figure 2b–e show the model error, MAE, for voltage prediction using the conventional ML model, Li-model, and TL model. As can be seen, the Li-model and conventional ML models have very similar MAEs, which are much larger than those predicted by the TL model. It demonstrates that the prediction accuracy from small data has really been largely improved by the TL technique, especially for the Zn and Al-ion battery electrodes, whose voltage predictions have been significantly improved by 1.67 V and 1.03 V, respectively. Moreover, Fig. 2b–e show that the predicted and the corresponding target voltage datasets have similar normal distributions, indicating that the datasets are reasonable, and the predictions are accurate. It is worth noting that the Li-model can provide a relatively good prediction for Na- and K-ion batteries (Supplementary Table 2) because Li-, Na-, and K-ions are all monovalent and having similar performances in electrodes35. However, our TL model still can further improve the performance in predicting monovalent MIBs (Supplementary Table 2). The MODNet16 and CrabNet17,18 are another two neural network models commonly used for the small dataset. We train these two models on the electrode voltages of Al- and Mg-ion battery datasets for comparation. Our results show that the MODNet has higher MAEs than the CGCNN TL with the MAE of 0.63 and 0.47 V for Al- and Mg-ion batteries, respectively. It is found that the CrabNet is highly overfitting for predicting voltages of electrode materials. It is because the MAE of the training set for Mg-ion batteries is only 0.13 V, but 0.36 V for the validation set. Besides, the CrabNet model is time consuming compared with TL because it is an attention-based model.
We validate our model by comparing it with the machine learning and available experimental results as shown in Fig. 4. As can be seen, our model outperforms the conventional ML model in predicting the electrode voltage for multivalent Mg-, Ca-, Zn-, and Al-ion batteries. Therefore, the voltages predicted by the TL model are more reliable to guide experiments in finding appropriate electrode materials for improving the performance of future multivalent MIBs. To help the battery community, we thus implemented our model into a publicly available online tool kit that can be used for quickly pre-checking and screening the voltage of any electrode with the crystal structure only.

Finally, we predict the voltages of 1637 materials for Al-ion (613), Ca-ion (278), Mg-ion (369), and Zn-ion (377) battery electrodes in Supplementary Tables 3–6, which are not available in Materials Project database. Within the 1637 materials, 113/613 of Al-ion, 128/278 of Ca-ion, 107/369 of Mg-ion, and 4/377 of Zn-ion battery electrode materials have an average voltage higher than 3.0 V as the potential candidates. It is worth noting that only around 1% cathode materials for Zn-ion batteries have the average voltage of 3.0 V or above. It means that finding a high-voltage electrode material for Zn-ion batteries would be a challenge. As a general design rule, our results of local voltages of each element (Fig. 3a) indicate the relationship between crystal elements and corresponding electrode voltages. Such element-voltage relationship may provide a promising strategy to design high electrode voltages by the elemental replacement for Zn-ion batteries.
Online artificial intelligence tool kit for voltage prediction
A web tool, provided for the voltage prediction for both multivalent and monovalent MIB electrodes, is publicly available online (http://batteries.2dmatpedia.org/). The voltage of a crystal can be obtained within a few seconds, with the crystal structure or the material ID in the MP and type of metal ion as inputs. The voltages for LIBs are predicted by the interpretable DL model and others are predicted by the deep TL model.
In summary, we develop an interpretable TL model to accurately predict the electrode voltages for MIBs (especially the multivalent batteries with very small data) and explain the underlying physical pictures as the important features for the voltage prediction by visualizing the vectors in layers in the neural network. The prediction with our model is much accurate in comparison with the conventional ML models and validated by the reported experimental results. The high-performing and interpretable DL models with the booming growth of battery materials data would greatly benefit the battery community that can be used for AI-enabled high-speed materials screening and rational design of electrodes of MIBs, such as substituting high local voltage elements (Fig. 3c) in a crystal to improve its voltage and energy density.
Methods
Training datasets
The data are extracted from the MP database38,50 accessed through the application programming interface (API) implemented in pymatgen51. The database contains a total number of 4401 intercalation-based battery electrode materials, in which, 2291 entries are electrode materials for LIBs, and 393, 484, 385, 149, 328, and 125 instances for the Mg-, Ca-, Zn-, Al-, Na-, and K-ion battery, respectively (Supplementary Table 1). Before training, outliers and inconsistent data are removed, and 2190, 387, 471, 378, 149, 287 and 125 instances are kept to train the model for Li-, Mg-, Ca-, Zn-, Al-, Na-, and K-ion battery electrode materials, respectively.
Machine-learning model
The conventional support vector regression (SVR) and kernel ridge regression (KRR) models are used in this work to predict the electrode voltages. They produce quite similar voltages for both multivalent and monovalent MIBs (Supplementary Fig. 5).
Deep-learning model
Our DL models are mainly based on the CGCNN4. The CGCNN model presents a periodic crystal structure into a multigraph G. Each atom in a structure is represented by a node i in G, which is represented by the atomic feature vector vi. The vector is then transformed into a 64-dimensional vector in the embedding layer. The nearest 12 neighbors for each atom are also considered in the CGCNN model and the chemical bond of the neighboring atom i and j is expressed as an edge (i, j)k in G, which is represented by vector u(i, j)k. The subscript k indicates the kth edge between note i and j because of the periodicity of the crystal19. Then, the atom and bond features are input of convolutional layers using a convolution function designed in the following equation.
where \({{{\boldsymbol{z}}}}_{(i,j)_k}^{(t)} = {{{\boldsymbol{v}}}}_i^{(t)} \oplus {{{\boldsymbol{v}}}}_j^{(t)} \oplus {{{\boldsymbol{u}}}}_{\left( {i,j} \right)_k}\) is a neighbor feature vector, and \(\oplus\) denotes the concatenation of vectors of the atomic and bonding feature of neighboring atoms of the ith atom. The \(\odot\) denotes element-wise multiplication, σ is a sigmoid function, and g is a non-linear activation function. W and b denote weights and biases of the neural networks, respectively. Behind three convolutional layers, the output vectors are then fitted into a pooling layer to create an overall feature vector. We, then, fully connect the resulting vector through a hidden layer, which is followed by a linear transformation to scalar values.
In the CGCNN model, only elemental and structural features of the electrode crystals are used, because our training datasets retrieved from Materials Project database are on the basis of DFT calculations. The average voltage in Materials Project is calculated by Supplementary Equation (3). Only the internal energy difference between the crystals before and after discharge is used to obtain the average voltage as an approximation, the details of which can be found in SI. It has been reported that the calculated voltages with such approximation are comparable to the experimental results. For example, the calculated average voltage of olivine LiFePO4 and LiNiPO4 is about 3.2 and 4.9 V, respectively, which are consistent with the experimental results of 3.4 and 5.1 V, respectively52,53,54,55. The calculated voltage curves for RuO2, SnO2, and SnS2 have the similar voltage curve change trend as those measured in experiments56.
Interpretability and visualization of deep-neural network
Ante-hoc and post-hoc are two commonly used interpretation models. Ante-hoc model would open the black-box and to gauge how a certain neural network is about to contribute to its predictions. Post-hoc only pries about the model from the outside of the model57. In the CGCNN network4, the output vectors of each layer are available for public, the ante-hoc could be used to probe the whole model. In this work, the element and the local environment representations, who are endured from embedding and the convolutional layers in the model respectively (Fig. 1), are mapped by two dimensions for visualizing the feature vectors in the layers. The local voltages are also explored to evaluate the contribution of each atom to the crystal voltage.
Element representations completely depend on the type of elements in the crystal. In the CGCNN model, the ith atom is a node i in the multigraph representation G. The node stores a 92-dimensional vector vi, which include all the elemental information, for instance atom covalent radius, valence electrons in each atom orbital. Then, the vector vi is put into the embedding layer and converted into a 64-dimensional vector \({{{\boldsymbol{v}}}}_i^{(0)}\). In order to visualize the features in the embedding layer, the elemental representation vector \({{{\boldsymbol{v}}}}_i^{(0)}\) are dragged out from the model. However, it cannot be directly visualized due to the large dimension of the vector. Thus, we adopt the PCA analysis to reduce the elemental representation into a two-dimensional plane.
Local environment representations depend on both the element type and their neighbors in crystal. After the three convolutional layers in the CGCNN model, a 64-dimensional vector \({{{\boldsymbol{v}}}}_i^{(3)}\), who represents the local environments of atom i and contains the information of both the atom and their neighbors, is obtained. In our work, only the vectors with the local oxygen-coordination environments, in which the working atom must have at least two oxygen atoms as neighbors, are dragged out for visualization. It is because cooperation among the same environment is more reasonable, and the oxygen-coordination environment is the commonest in the electrode materials dataset of MIBs. The local oxygen environment vector \({{{\boldsymbol{v}}}}_i^{(3)}\) is then reduced into two dimensions with the PCA and t-SNE method in order to have a clear visualization about what information has been learned in the convolutional layer.
Local voltage representation is derived from the local oxygen-coordination environment vectors and represents the contributions of each atom to the voltages of the crystal. A linear transformation is performed to map \({{{\boldsymbol{v}}}}_i^{(3)}\) to the local voltage Vi,
where Wl and bl denote weights and biases. These two parameter vectors are trained solely during the model visualization process. The voltage of the crystal is predicted using the average of the local voltage \(V = \frac{1}{n}\mathop {\sum }\limits_i V_i\) where n is the number of atoms in the crystal. The large local voltage of an atom means that electrode materials with such atom may have a larger voltage. The local voltage of elements in the oxygen-coordination environment is a specific local voltage for the atom, having at least two nearest neighboring oxygen atoms in the crystal. The oxygen-coordination environment is considered specially in this work because there are a large number of oxides in the electrode materials, which may make our models more explainable from such local environment.
Transfer learning
TL is an effective DL model for the prediction with insufficient datasets and is expected to speed up and improve the performance of the convolution neural network. There are two major TL scenarios for loading parameters from previous neural networks. One is to fine tune the convolution network. Herein, the parameters of the target network are initially loaded with a pre-trained network. Thereafter, all the parameters are optimized just as usual. The other one is to set the convolutional network as fixed feature extractor. This method freezes the weights of the earlier layers and only the last few fully connected layers are trained. In this study, the second TL method is used for the voltage predictions of multivalent MIBs and monovalent Na- and K-ion batteries. Only parameters in the last two fully connected layers in the CNN structure are optimized in the TL model, which are the hidden layer and the output layer (Fig. 1 orange box).
Data availability
All battery data and experimental data are available in Supplemental Materials and GitHub. The data for all figures and extended data figures are available in Source Data.
Code availability
Models and codes are available at https://github.com/mpeshel/Interpretable_AI_for_battery.git.
References
Armand, M. & Tarascon, J. M. Building better batteries. Nature 451, 652–657 (2008).
Dunn, B., Kamath, H. & Tarascon, J.-M. Electrical energy storage for the grid: a battery of choices. Science 334, 928–935 (2011).
Tarascon, J.-M. & Armand, M. In Materials for Sustainable Energy: A Collection of Peer-Reviewed Research and Review Articles from Nature Publishing Group (ed. Dusastre, V.) 171–179 (World Scientific Publishing Co., 2010).
Chu, S., Cui, Y. & Liu, N. The path towards sustainable energy. Nat. Mater. 16, 16–22 (2017).
Scrosati, B. & Garche, J. Lithium batteries: status, prospects and future. J. Power Sources 195, 2419–2430 (2010).
Taylor, S. R. Abundance of chemical elements in the continental crust: a new table. Geochim. Cosmochim. Acta 28, 1273–1285 (1964).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Davies, A. et al. Advancing mathematics by guiding human intuition with AI. Nature 600, 70–74 (2021).
Kirkpatrick, J. et al. Pushing the frontiers of density functionals by solving the fractional electron problem. Science 374, 1385–1389 (2021).
Zhou, F., Cococcioni, M., Marianetti, C. A., Morgan, D. & Ceder, G. First-principles prediction of redox potentials in transition-metal compounds with LDA+U. Phys. Rev. B Condens. Matter Mater. Phys. 70, 235121 (2004).
Chen, C., Ye, W., Zuo, Y., Zheng, C. & Ong, S. P. Graph networks as a universal machine learning framework for molecules and crystals. Chem. Mater. 31, 3564–3572 (2019).
Choudhary, K. & DeCost, B. Atomistic line graph neural network for improved materials property predictions. NPJ Comput. Mater. 7, 185 (2021).
Hu, J. et al. MaterialsAtlas.org: a materials informatics web app platform for materials discovery and survey of state-of-the-art. NPJ Comput. Mater. 8, 65 (2022).
Chen, C. & Ong, S. P. AtomSets as a hierarchical transfer learning framework for small and large materials datasets. NPJ Comput. Mater. 7, 173 (2021).
Schutt, K. T., Sauceda, H. E., Kindermans, P. J., Tkatchenko, A. & Muller, K. R. SchNet-a deep learning architecture for molecules and materials. J. Chem. Phys. 148, 241722 (2018).
De Breuck, P.-P., Hautier, G. & Rignanese, G.-M. Materials property prediction for limited datasets enabled by feature selection and joint learning with MODNet. NPJ Comput. Mater. 7, 83 (2021).
Wang, A. Y.-T., Kauwe, S. K., Murdock, R. J. & Sparks, T. D. Compositionally restricted attention-based network for materials property predictions. NPJ Comput. Mater. 7, 77 (2021).
Wang, A. Y.-T., Mahmoud, M. S., Czasny, M. & Gurlo, A. CrabNet for explainable deep learning in materials science: bridging the gap between academia and industry. Integr. Mater. Manuf. Innov. 11, 41–56 (2022).
Xie, T. & Grossman, J. C. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Xie, T. & Grossman, J. C. Hierarchical visualization of materials space with graph convolutional neural networks. J. Chem. Phys. 149, 174111 (2018).
Back, S. et al. Convolutional neural network of atomic surface structures to predict binding energies for high-throughput screening of catalysts. J. Phys. Chem. Lett. 10, 4401–4408 (2019).
Karamad, M. et al. Orbital graph convolutional neural network for material property prediction. Phys. Rev. Mater. 4, 093801 (2020).
Lam Pham, T. et al. Machine learning reveals orbital interaction in materials. Sci. Technol. Adv. Mater. 18, 756–765 (2017).
Park, C. W. & Wolverton, C. Developing an improved crystal graph convolutional neural network framework for accelerated materials discovery. Phys. Rev. Mater. 4, 063801 (2020).
Zhang, Y. et al. Identifying degradation patterns of lithium ion batteries from impedance spectroscopy using machine learning. Nat. Commun. 11, 1706 (2020).
Swift, M. W., Swift, J. W. & Qi, Y. Modeling the electrical double layer at solid-state electrochemical interfaces. Nat. Comput. Sci. 1, 212–220 (2021).
Chen, C., Zuo, Y., Ye, W., Li, X. & Ong, S. P. Learning properties of ordered and disordered materials from multi-fidelity data. Nat. Comput. Sci. 1, 46–53 (2021).
Lu, S. et al. Accelerated discovery of stable lead-free hybrid organic-inorganic perovskites via machine learning. Nat. Commun. 9, 3405 (2018).
Lu, S., Zhou, Q., Guo, Y. & Wang, J. On-the-fly interpretable machine learning for rapid discovery of two-dimensional ferromagnets with high Curie temperature. Chem 8, 769–783 (2022).
Tran, K. & Ulissi, Z. W. Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution. Nat. Catal. 1, 696–703 (2018).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
Chen, D. et al. Automating crystal-structure phase mapping by combining deep learning with constraint reasoning. Nat. Mach. Intell. 3, 812–822 (2021).
Lee, X. Y. et al. Fast inverse design of microstructures via generative invariance networks. Nat. Comp. Sci. 1, 229–238 (2021).
Dunn, A., Wang, Q., Ganose, A., Dopp, D. & Jain, A. Benchmarking materials property prediction methods: the Matbench test set and Automatminer reference algorithm. NPJ Comput. Mater. 6, 138 (2020).
Joshi, R. P. et al. Machine learning the voltage of electrode materials in metal-ion batteries. ACS Appl. Mater. Inter. 11, 18494–18503 (2019).
Moses, I. A. et al. Machine learning screening of metal-ion battery electrode materials. ACS Appl. Mater. Inter. 13, 53355–53362 (2021).
Sarkar, T., Sharma, A., Das, A. K., Deodhare, D. & Bharadwaj, M. D. A neural network based approach to predict high voltage Li-ion battery cathode materials. In 2014 2nd International Conference on Devices, Circuits and Systems (ICDCS) 1–3 (IEEE, 2014).
Jain, A. et al. Commentary: the materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Shao, L., Zhu, F. & Li, X. Transfer learning for visual categorization: a survey. IEEE T. Neur. Net. Lear. 26, 1019–1034 (2015).
Schmidt, J., Marques, M. R. G., Botti, S. & Marques, M. A. L. Recent advances and applications of machine learning in solid-state materials science. NPJ Comput. Mater. 5, 83 (2019).
Lee, J. & Asahi, R. Transfer learning for materials informatics using crystal graph convolutional neural network. Comp. Mater. Sci. 190, 110314 (2021).
Iten, R., Metger, T., Wilming, H., Del Rio, L. & Renner, R. Discovering physical concepts with neural networks. Phys. Rev. Lett. 124, 010508 (2020).
Zeiler, M. D. & Fergus, R. Visualizing and understanding convolutional networks. In European Conference on Computer Vision (ECCV) 818–833 (Springer International Publishing, 2014).
Hotelling, H. Relations between two sets of variates. Biometrika 28, 321–377 (1936).
Pearson, K. On lines and planes of closest fit to systems of points in space. Philos. Mag. 2, 559–572 (1901).
Ward, L., Agrawal, A., Choudhary, A. & Wolverton, C. A general-purpose machine learning framework for predicting properties of inorganic materials. NPJ Comput. Mater. 2, 16028 (2016).
Ward, L. et al. Including crystal structure attributes in machine learning models of formation energies via Voronoi tessellations. Phys. Rev. B Condens. Matter Mater. Phys. 96, 024104 (2017).
Maaten, L. V. D. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Louis, S. Y. et al. Accurate prediction of voltage of battery electrode materials using attention-based graph neural networks. ACS Appl. Mater. Inter. 14, 26587–26594 (2022).
Jain, A. et al. A high-throughput infrastructure for density functional theory calculations. Comp. Mater. Sci. 50, 2295–2310 (2011).
Ong, S. P. et al. The Materials Application Programming Interface (API): a simple, flexible and efficient API for materials data based on REpresentational State Transfer (REST) principles. Comp. Mater. Sci. 97, 209–215 (2015).
Dong, G., Wei, J., Zhang, C. & Chen, Z. Online state of charge estimation and open circuit voltage hysteresis modeling of LiFePO4 battery using invariant imbedding method. Appl. Energy 162, 163–171 (2016).
Panchal, S., Dincer, I., Agelin-Chaab, M., Fraser, R. & Fowler, M. Experimental and simulated temperature variations in a LiFePO4-20 Ah battery during discharge process. Appl. Energy 180, 504–515 (2016).
Nasir, M. H., Janjua, N. K. & Santoki, J. Electrochemical performance of carbon modified LiNiPO4 as Li-Ion battery cathode: a combined experimental and theoretical study. J. Electrochem. Soc. 167, 130526 (2020).
Zhang, Y., Pan, Y., Liu, J., Wang, G. & Cao, D. Synthesis and electrochemical studies of carbon-modified LiNiPO4 as the cathode material of Li-ion batteries. Chem. Res. Chin. Univ. 31, 117–122 (2015).
Hassan, A. S., Moyer, K., Ramachandran, B. R. & Wick, C. D. Comparison of storage mechanisms in RuO2, SnO2, and SnS2 for lithium-ion battery anode materials. J. Phys. Chem. C. 120, 2036–2046 (2016).
Buhrmester, V., Münch, D. & Arens, M. Analysis of explainers of black box deep neural networks for computer vision: a survey. Mach. Learn. Knowl. Extr. 3, 966–989 (2021).
Yokozaki, R., Kobayashi, H. & Honma, I. Reductive solvothermal synthesis of MgMn2O4 spinel nanoparticles for Mg-ion battery cathodes. Ceram. Int. 47, 10236–10241 (2021).
Orikasa, Y. et al. High energy density rechargeable magnesium battery using earth-abundant and non-toxic elements. Sci. Rep. 4, 5622 (2014).
Gu, Y., Katsura, Y., Yoshino, T., Takagi, H. & Taniguchi, K. Rechargeable magnesium-ion battery based on a TiSe2-cathode with d-p orbital hybridized electronic structure. Sci. Rep. 5, 12486 (2015).
Sun, X. et al. A high capacity thiospinel cathode for Mg batteries. Energy Environ. Sci. 9, 2273–2277 (2016).
Park, H., Cui, Y., Kim, S., Vaughey, J. T. & Zapol, P. Ca cobaltites as potential cathode materials for rechargeable Ca-ion batteries: theory and experiment. J. Phys. Chem. C. 124, 5902–5909 (2020).
Shiga, T., Kondo, H., Kato, Y. & Inoue, M. Insertion of calcium ion into prussian blue analogue in nonaqueous solutions and its application to a rechargeable battery with dual carriers. J. Phys. Chem. C. 119, 27946–27953 (2015).
Shoji, T. & Yamamoto, T. Charging and discharging behavior of zinc—manganese dioxide galvanic cells using zinc sulfate as electrolyte. J. Electroanal. Chem. 362, 153–157 (1993).
Lin, M. C. et al. An ultrafast rechargeable aluminium-ion battery. Nature 520, 325–328 (2015).
Geng, L., Lv, G., Xing, X. & Guo, J. Reversible electrochemical intercalation of aluminum in Mo6S8. Chem. Mater. 27, 4926–4929 (2015).
Acknowledgements
The authors thank Dr. Zeng Minggang and Dr. Xu Lei for their helpful discussion. This work was supported by MOE, Singapore Ministry of Education (No. MOE2019-T2-2-030, No. R-723-000-029-112, and No. R265-000-691-114), conducted at the National University of Singapore. The authors gratefully acknowledge the Center of Advanced 2D Materials (NUS), NSCC (Singapore) for providing computational resources, and the China Scholarship Council for financial support.
Author information
Authors and Affiliations
Contributions
L.S. and X.Z. conceived the work. X.Z. designed the model, conducted the experiment, and took the lead in writing the manuscript with input from all authors. J.Z. and J.L. gave scientific and technical advice throughout the project and helped with interpretation of data. The project was supervised by L.S. All authors provided feedback and contributed to the discussion and analyses of the data.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, X., Zhou, J., Lu, J. et al. Interpretable learning of voltage for electrode design of multivalent metal-ion batteries. npj Comput Mater 8, 175 (2022). https://doi.org/10.1038/s41524-022-00858-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-022-00858-9
