
Abstract
Batteries are of paramount importance for the energy storage, consumption, and transportation in the current and future society. Recently machine learning (ML) has demonstrated success for improving lithium-ion technologies and beyond. This in-depth review aims to provide state-of-art achievements in the interdisciplinary field of ML and battery research and engineering, the battery informatics. We highlight a crucial hurdle in battery informatics, the availability of battery data, and explain the mitigation of the data scarcity challenge with a detailed review of recent achievements. This review is concluded with a perspective in this new but exciting field.
Similar content being viewed by others


Introduction
The continuous growth of economics and global energy consumption has increased the CO2 emission by 45% from 2000 to 20191. To meet the goal of carbon neutrality, replacing current reliability on fossil fuel with cleaner and renewable energy resources is urged. Rechargeable batteries play a vital role in a green society for energy storage, consumption, and transportation. The market size for Li-ion batteries was at 36.7 billion dollars in 2019, and is projected at 128.3 billion by 2027 with a compounded annual growth rate estimated at 18% from 2020 to 2027, driven mostly by the shift from combustion engine vehicles to hybrid and electric transportation2. In the past decade, the desire to meet the demanded large-scale applications with higher energy density and power density, larger capacity, longer durability, and better safety has motivated tremendous research efforts to improve current Li-ion technology as well as developing new battery chemistries.
A battery is a complex electrochemical ensemble of multiple components of cathode, anode, electrolyte, separator, current collectors, and housing materials. The complicated electrochemically coupled transport processes across a wide range of time and length scales haunts quantitative understanding of the relationship among the performance, materials, design, and operation of a battery. The traditional simulation and experiment methods in battery research usually require large research resources in combination with sophisticated domain knowledge or experience to enhance the effectiveness of trial-and-error approaches. In recent years, data-driven techniques have emerged as the fourth paradigm of materials research in parallel to empirical, model-based, and computation-based science3,4,5,6. Machine learning (ML) has been flourishing in materials representation7,8,9, accelerating atomic simulations10,11,12, reaction network13 and synthesizability network analysis14, experimental design15,16,17, and the discovery of numerous functional candidates with an unprecedented rate18,19,20,21,22,23,24,25,26. Integrating ML into conventional experimental and computational techniques has achieved success in various aspects of battery research. From 2010 to 2020, the number of publications in the interdisciplinary field of battery informatics has increased by ~20 times, matching well to the growing interest of ML in other materials domains.
This review is devoted to summarizing the achievements of battery informatics in the past years. Herein, the battery informatics is defined as the research that utilizes machine learning as the main technique or relies on machine learning as a major tool for data analysis and interpretation. The employment of ML offers the surrogate function of observables to circumvent the challenge to understand the underlying mechanism of the complex battery systems in conventional approaches. There are several excellent reviews in the literature covering the fundamental mathematics of ML as well as the application in materials domains3,4,5,6. In battery informatics, the work in Liu et al. reviewed the application of ML in the design and discovery of novel battery materials27. The work of Chen et al. summarized the application of ML in energy storage materials22. For batteries materials, they reviewed the ML prediction of diffusion, mechanical properties as well as developing interatomic potential for dynamical simulations of battery materials. The work of Guo et al. reviewed the application of ML to accelerate first-principles calculations and facilitate the modeling of battery materials28. The work of Liu et al. summarized the discovery of solid-state electrolyte through ML29. These reviews have highlighted the progress and achievements in certain subareas of battery studies. Amid the broad range of battery research from fundamental materials development to system-level operation and optimization, a more comprehensive review is desired for better summary of the state-of-art work as well as providing instructive guidance into future research. The structure of the remainder of this paper is illustrated as follows. In the section “Data for battery informatics” we review available data source of battery research and explain the data scarcity challenge for battery informatics. In the section “Circumvent the data scarity challenge through algorithm development” we briefly discuss how the data scarcity challenge can be mitigated through appropriate ML algorithms. In the section “Application of machine learning in battery research”, we summarize applications of ML in various aspects of battery research in detail and highlight several exciting achievements of ML in battery engineering in the section “Machine learning in battery engineering”. A concluding remark is provided in the last section.
Data for battery informatics
Data scarcity challenge
Machine learning is a data-centered technique to generalize trends observed from existing examples to make decisions without explicating programming to achieve so. Among many factors determining the success of ML, data are central to the task as the availability of good quality data in a large quantity allows more accurate detecting of underlying patterns and eventually better prediction of unknown scenario. For example, in the computation vision field, the standardized dataset of the Modified National Institute of Standards and Technology database of hand-written digits includes 70 thousand images of hand-written digits for each number30. For speech recognition, the Chime-5 challenge recorded a total of over 50 h of conversation composed of 98,448 utterances31. Although the requirement of data volume necessary for good ML performance varies with the choice of model algorithm, data processing pipeline, and the latent dimension of the target problem, in general, higher data availability will lead to better ML modeling. In addition, these large, standardized, and well-organized datasets provide excellent platforms that algorithms and technologies can be developed, compared and advanced.
The materials community, however, have not fully enjoyed such luxury in informatics enterprise. Only a number of materials properties have been organized in good quality and high quantity. The lack of data availability presents a significant challenge towards generalizing ML as a standard tool in materials research. Table 1 summarizes different types of datasets available for the battery informatics research. Based on the method used to generate and collect the data, we categorize the data into the computational database, experimental database, high-throughput experimentation data, and database through text mining techniques and discuss accordingly.
Computational databases for battery informatics
Computational databases use sophisticated pipelines of simulation to calculate and store the thermodynamic, electronic, and structural information for several tens of thousands of inorganic compounds at the level of density functional theory32,33,34,35,36,37,38. The large volume and good quality of data in these highly curated computational materials databases has promoted a significant portion of materials informatics research. The modeling of formation energies, for example, serves as one of the first few examples that demonstrated the potential capability of leveraging statistical data technique in materials research and is continuously employed for testing and improvement of new ML approaches for feature engineering and pattern mining of materials properties39,40,41,42,43,44,45.
The data from computational materials databases allows the estimation of many thermodynamic properties of battery materials. The open-circuit voltages of electrode materials, for example, can be obtained once the phases in discharge and charge states are both included in the dataset46. Materials Project includes the calculated voltages for 4730 intercalation-type and 16,128 conversion-type electrode materials dated to May 202132. Using the data from Materials Project, the voltage trends of oxide-based cathode candidates for Li-ion battery were statistically analyzed to unveil the effects of polyanion group, redox metal, and the ratio of oxygen to counter cation on voltage and O2 release temperature47. Taking advantage of the data abundance, general rules for designing safe cathode systems were summarized. Another example of materials properties that can be directly estimated from the data in the computational materials database is the stability of interfaces between the electrode and solid-state electrolyte48. Utilizing the computational data from OQMD, Aykol et al. screened more than 130,000 oxygen-bearing materials with high phase stability, electrochemical stability, and hydrofluoric-acid resistance to serve as cathode coating layers49. They identified optimal hydrofluoric-acid scavengers of Li2SrSiO4, Li2CaSiO4, and CaIn2O4 for the layered LiCoO2, and Li2GeO3, Li4NiTeO6, and Li2MnO3 for the spinel LiMn2O4 cathodes. Xiao et al. screened 104,082 Li-containing compounds to find coating materials with high phase stability, electrochemical stability, and chemical compatibility with Li3PS4 solid-state electrolyte and LiNi1/3Co1/3Mn1/3O2 cathode50. After a detailed analysis of stability and conductivity, three oxide candidates, LiH2PO4, LiTi2(PO4)3, and LiPO3 were identified for cathode coating. The large amount of good quality data stored in computational materials databases enables these studies to screen a board compositional space for materials with specific functionality without the necessary ML participation.
Properties that can be calculated with reasonable computational resources only compose a small portion of targets of interest in battery research. Rate capability, cycling behavior, degradation, and performance at the cell level are all examples of crucial properties that are not straightforwardly simulated using computational techniques. Even properties that can be calculated in well-established computational methods may face high computational cost when the pipeline of exploration is extended to a large and highly diverse configurational space. One representative example is the ionic transport properties in solid-state materials. The energy barriers for the solid-state diffusion of charge carriers can be calculated using the nudge elastic band method (NEB)51. Ab initio molecular dynamics (AIMD) provides an additional means to estimate the diffusivity in comparable agreement with experimental measurements52. However, both NEB and AIMD methods are much more computationally extensive than structure relaxation. It restricts the availability of diffusion data when the ML approach is attempted. With the leverage of modern information technology infrastructure and software tools, the assessment of alkali superionic conductors was facilitated at the rate of about 200 compositions within the space of two years using relatively modest computational resources53. This rate of exploration, however, is much slower compared to the calculation of thermodynamic properties.
Experimental database
In parallel to the computational database, experimentally based, large, and structured materials property datasets have been pursued. Inorganic crystal structure database (ICSD) stores the crystalline structure information of inorganic substances published since 191354. As of December 2020, ISCD contains over 210,000 entries and is updated twice a year. Crystallography open database (COD) contains more than 150,000 structures and offers the searching and downloading possibilities55. As of January 2019, Pauling files stores 51,974 entries of experimental and computational temperature-composition phase diagrams, 357,612 entries of crystalline structure information and 156,274 records of a broad range of intrinsic physical properties of inorganic solids from the processing of 23,876, 113,556, and 56,219 publications, respectively56. The construction of such a large database requires inputs from the entire community and necessitates good quality control on the targeted information. For individual researchers, a common practice is to apply a standard procedure to a parameter space and augment the data from discrete measurements. Several databases have been publicly available through the standard experimentation such as three battery datasets accessible from NASA portfolio57,58,59 and the electrochemical performance of commercial 18650 cells at a variety of temperatures and discharge currents60. Due to the standardized protocols for data collection, the data quality is usually consistent and well-controlled. However, data collection through single and discrete experiments requires considerable experimental resources focusing on the measurement of specific properties, making the large-scale accumulation expensive and time-consuming for individual researchers.
High-throughput experimentation
In recent years, the advancement in experimental automation techniques has reached the level that executes a large number of experiments can be executed in parallel and result in a wealth of experimental data for better technical decisions. In biology and pharmaceutical industry, high-throughput experimentation (HTE) has matured to the point that experiments are now routinely executed for the screening of drug libraries61,62. For battery research, the experimentation involves several steps including synthesis, characterization, cell fabrication, electrochemical testing, and other performance evaluations. In the past decade, HTE has gradually extended its territory to these fields with the successful implementation of materials synthesis and cell fabrication, electrochemical property measurement and multiple materials characterization techniques in the pipeline63,64,65,66. HTE offers the direct examination of candidates in a combination of external tunable parameters, yielding better electrochemical functionalities for the compositional screening of Li-ion battery cathode67,68,69,70,71, Na-ion battery cathode72, liquid electrolyte73, solid-state electrolyte74, cathode-electrolyte interlayer75, electrolyte additive76 as well as evaluating cell design parameters63.
The integration of data-driven techniques with HTE could eventually close the loop of automated materials discovery, design, and optimization (Fig. 1). In the close-loop approach, a ML engine receives the data from HTE and make decisions for the next step. The experimental engine then receives the direction from ML engine and perform the experiments accordingly. The data are augmented to start the next loop of collaboration between these two engines. In the real-time operation, a ML agent can narrow down the chemical space to be examined prior to the execution of combinatorial chemistry. Matsubara et al. used ML to predict the O2− conductivity in 13,384 oxides materials and identified the system of Bi, Nb, Ta, and alkaline earth metals (Ca, Sr, and Ba) for the subsequent combinatorial experiments77. Implementing high-throughput conductivity measurements and high-throughput XRD increased the total experimental throughput to chemical space not included in the informatics screening. In the ideal situation, the close-loop strategy should be executed in the fully automated manner with robotics carrying out serial experiments and deposit the data directly to the ML domain. An example of close-loop exploration was the exploration of a new aqueous electrolyte. Whitcare et al. built the robotic platform of Otto for the automated measurement of pH, conductivity, and voltage stability of liquid electrolytes78,79. By connecting Otto to a Bayesian optimizer, the machine-learning model directed the experimental execution on the basis of measurement feedback in real time to optimize the electrochemical window of aqueous sodium electrolyte in the design space of mixtures of NaNO3, NaClO4, Na2SO4, and NaBr and mixtures of LiNO3, LiClO4, and Li2SO480. The automation examined 140 electrolyte formulas in 40 h of experimentation and discovered a blend receipt with more resistance to oxygen evolution reaction on platinum than high-concentration NaClO4 electrolyte.

ML could assist the pre-selection of candidate before HTE execution, or guide the sampling in the HTE.
Although HTE provides high-quality data in an unprecedented rate compared to conventional experimentation, the high capital cost is still the main hurdle for its implementation in general battery research community. HTE is usually carried out in homogenous environments and thus lacks the flexibility to optimize the performance through process engineering. This is particularly important in battery research, because many macroscopic properties of battery materials strongly depend on the synthesis, processing and even measurement techniques. Lifting the restriction of HTE to include the processing space as variables of exploration thus deserves attention.
Collect unstructured data from literature
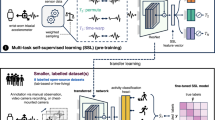
Given the much desired needs to mine knowledge directly from experimental outputs, the information presented as numerical text or image-based information in publications, patents, and other text archives composes an invaluable source of data in unstructured format. Identifying and harvesting them from documents through text mining presents an avenue to collect the mass volume of materials data for subsequent ML tasks. Due to the presence of specialized vernacular, terminology, and chemical semantics, generic natural langrage processing tools is not performing well in the materials science domain. In recent years, several materials-specific text mining tools have been developed to harvest information from materials literature following the general overflow of acquiring text content, recognizing entities of interest, collecting and storing the entity information and performing post analysis and modeling (Fig. 2a)81,82,83. The usage of these tools generates libraries of information to explore, which forms the foundation for the designing and performing next phase research. For example, text mining has been used to extract the synthesis conditions of inorganic compounds84. The data were then fueled to predict the appropriate conditions to synthesize titania nanotubes via hydrothermal routes and clarifying the procedures to synthesize inorganic materials85. He et al. trained a two-step bi-long-short-term-memory model to distinguish precursors and targets in the inorganic synthesis reaction in 86,544 literature papers, which allowed the subsequent meta-analysis on the similarities and differences between precursors86. Tshitoyan and coauthors showed the knowledge extracted data from text mining provided implicit relevance of compounds to a new application26. The lateral structure–property relationships led to the discovery of new thermoelectric materials several years before their discovery as a case of demonstration.

a Illustration of the workflow of text mining process for materials database. b Temperatures of processing temperatures for solid-state lithium-ion electrolytes. The middle and right figures show the temperatures of processing garnet LLZO reported in literature. Reproduced with permission from ref. 84. Copyright Elsevier 2020. c Distribution of battery capacity and conductivity from text mining the literature. Reproduced with permission from ref. 88. Copyright Springer Nature 2020.
One of the primary goals of text mining is to construct the structured database to prompt the subsequent data-driven discoveries. Mahbub et al. collected the processing temperature for solid-state electrolytes of Li2S-P2S5, Li7P3S11, β-Li3PS4, Li10GeP2S12 (LGPS), and garnet Li7La3Zr2O12 (LLZO) oxides from published reports (Fig. 2b)87. The processing temperature can be further broken down, for example, for garnet LLZO, to investigate the temperature regime of specific processing steps (drying, annealing, calcination, and sintering) and shed lights on efforts towards low-temperature processing of solid-state LLZO electrolytes. As shown in Fig. 2c, A battery database based on text mined information was recently published by Huang and Cole88. Using the software of ChemDataExtractor version 1.5 to mine 229,061 academic papers, they collected 292,313 data records, with 214,617 unique chemical-property data relations between 17,354 unique chemicals and up to five material properties: capacity, voltage, conductivity, Coulombic efficiency and energy. The data were deposited in both relational and non-relational formats of database shared at figshare.
Data fidelity
The precious value of materials data naturally motivates efforts to maximize the efficiency of data utilization by consolidating data from different resources for the modeling of the same property. It should be, however, cautious that data from different sources is likely to have varied degree of uncertainties. A similar issue of fidelity control presents when data from different levels of theory are mixed for the computational database in the open repository. Without clarifying the fidelities of different datasets, the high-quality data will be polluted by the presence of low-quality data in the modeling. Appropriate inclusion of the fidelity information in the modeling could, on the other hand, enhance the model quality. One strategy is to distinguish the low- and high-fidelity data as input feature and output properties, respectively. Despite its less accuracy, the crude estimation of targeted property usually has strong correlation with ground truth value; hence the inclusion of this specific feature adds knowledge to improve the inference of target and mitigate the data requirement for modeling (Fig. 3a)23,89,90. A multi-fidelity graph network to encode the data fidelity level to a trainable fidelity embedding matrix was proposed by Chen et al (Fig. 3b)91. They demonstrated that the inclusion of low-fidelity Perdew–Burke–Ernzerhof band gaps reduced the error of experimental band gap predictions by 22–45% and offered an approach to model disordered materials. Fujimura et al. used the high temperature (1600 K) diffusivity of LISICON compounds obtained from AIMD simulation (D1600) to predict the more precious experimentally measured conductivities at 373 K (σ373)92. These studies all suggest the data from different sources can be effectively utilized once the labeling of uncertainties and fidelities can be appropriately addressed.

a Use the crude estimation of target property as feature to improve the modeling accuracy. Reproduced with permission from ref. 90. Copyright Springer Nature 2017. b Improving the model accuracy by encoding the fidelity in multi-fidelity graph network. Reproduced with permission from ref. 91. Copyright Springer Nature 2021.
Data bias and anthropogenic bias
Because of the complex interplays among the electronic, structural, and microstructural degree of freedom, the macroscopic properties of battery materials are affected by factors across a broad range of length scales. Taking the conductivity of solid electrolyte as an example, in the atomic scale, the conduction is affected by the crystalline structure and chemical composition of the electrolyte. Beyond the atomic scale, the conductivity is affected by the microstructures of electrolyte such as particle morphology, size, and packing. On the cell level, the conductivity is further affected by the reaction between electrolyte and electrode and the corresponding interface layer formed in between93. These factors in combination causes a large variance of measured conductivities even for materials with the same composition, resulting in bias of collected data. For instance, depending on the synthesis methods and temperatures, the conductivity of garnet Li5La3Ta2O12 varied two orders of magnitude between 10−6 and 10−4 S cm−1 94. Such complexity raises the importance of labeling the data beyond the level of the materials to include information about the synthesis, processing, and characterization. This is a vast challenge: not all data contain every necessary characterization of materials. Even for information well presented in all publications, correctly pairing the materials properties-characterization is still challenging due to the requirement to scan a large portion of the article. This distant co- or cross-referencing is a significant problem to move from human-readable to machine-readable contents. Recently, a canonical ontology for materials synthesis consisting of a controlled vocabulary with restricted relations between concepts was proposed95. It still takes time for the community to digest and transit to improve the communication of materials synthesis, extend the impact of the insights contained in each published synthesis method and contribute toward a global body of unified materials synthesis knowledge.
Another type of data bias is the anthropogenic bias unconsciously presented in the sampling procedure. Scientists lean to explore a system with the highest confidence of success and prefer to select the most salient results for showcasing scientific points. It leads to both the overpopulation in a local domain and the absence of negative examples in the published literature. A survey of lithium-containing compounds in inorganic crystal database clearly reveals these biases. Among 2,986 compounds, 80 (2.7%) compounds are in the family of spinel Li4Ti5O12, 86 (3.0%) has with the formula of Li3x−1La1−xTiO3 and 30 (1.0%) belongs to the garnet family of compounds. The heavy population in a few families of compounds is easily understood. Li-containing compounds are famous for their potential usages in battery and these over-populated samples are known with promising properties for battery applications. Spinel Li4Ti5O12 is a popular anode material, while perovskite Li3x−1La1−xTiO3 and garnet compounds are good candidates of solid-state electrolyte. On the other hand, the application of other Li-containing materials for battery and other potential applications have either not been attempted, or the negative results have discouraged the researcher from publishing the data.
From a realistic viewpoint, materials exhibiting a special functionality should only compose a small portion of the entire materials space. Negative data not considered to deserve publication benefits ML models for a trustful exploration of unknown domains96. Sampling skewed by the anthropogenic bias ignores the abundance of negative data and will not reflect the true data distribution. Compared machine-learning models trained on the complete set of human-selected (biased) reactions to models trained on randomly generated and unbiased reactions for the synthesis of amine-templated metal oxides, correcting anthropogenic bias improved machine-learning models and led to faster discovery of new materials97.
Avoiding the contamination of model performance by data bias and anthropogenic bias requires the complete transparency of data quantity and quality. It should be cautious that the quantity and quality of datasets are not always straightforward to assess and often subjective, depending on the choice of ML algorithms and the intended applications. Therefore, the data quantity and quality should not be regarded as judgement criteria when reporting and evaluating ML research. A more important step is to disclose the data collection and pre-processing procedure in addition to the encouraged open access of published data. In recent work, Artrith et al. outline a set of guidelines when reporting machine learning models composed of listing all data sources, documenting the strategy for data selection, including access dates or version numbers, describing data cleaning procedure, and evaluating the extent of data pre-processing98. Their work provides the checklist for reporting and evaluating machine learning models towards the standard of a high data reporting protocol in the materials domain.
Circumvent the data scarity challenge through algorithm development
The field of ML includes a vast number of algorithms ranging from simple linear regression to complex methods such as convolutional neural network and generative adversarial network. We note here that our intention is not to generalize the best algorithms for battery informatics, although the performance comparison for different algorithms on the same task and the same dataset is important and necessary. The no-free-lunch theorem states that the computational cost of finding a solution is the same for all solution methods when averaged on all problems in the class99. No solution therefore offers a better capability on all problems. Therefore, our review will not be restricted to any specific algorithm-related topics. Instead, we aim to discuss the mitigation of data scarcity challenge through appropriate algorithms in battery informatics. Reviews of the mathematical foundation of ML algorithms would be beyond the scope of this review and the readers of interest are encouraged to statistical and ML textbooks as well as several excellent reviews covering this topic4,5,22.
Regression and classification in supervised learning
Supervised learning utilizes labeled data to make decisions by seeking patterns in the labeled features for an analytics process. The data used in supervised learning is labeled to make the task that a direct relationship between the input variable and output properties can be constructed. In battery informatics, supervised learning is the most adopted type of methods and finds broad applications to predict materials properties, discover new materials and forecast future behaviors. A main goal of supervised learning is to reduce the expensive experiments by providing guidance for the next step of experimentation. From this aspect, the supervised learning is often partnered with high-throughput simulation in a way that the simulation fuels supervised learning with necessary data for training, while the supervised learning accelerates the throughput rate by rapid screening of chemical space unseen in the simulation.
The typical tasks in supervised learning are regression and classification. In a classification task, the observable is labeled to a set of categories and the goal is to identify whether a new observation belongs to a specific class in a yes or no manner. In a regression task, the model seeks to map a real-valued numerical output to independent variables. Regression analysis is widely adapted for prediction and forecasting while classification is mostly used for grouping and boundary detection. However, it is not necessarily the natural reasoning to consider which is the most suitable for the specific materials problem. For example, to predict which materials will be the most promising candidates for an application, one may naturally consider it as a regression problem and contract models to predict the performance of a list of candidates. The selection can be made by sorting the predicted functionalities and choose the one with the best-predicted value. Alternatively, the regression task can be converted to classification with the use of proper thresholds to define “promising” and “non-promising”. Sendek et al applied this strategy in their exploration of solid-state ionic conductors100. They defined material is conductive if the room temperature conductivity is higher than 10−4 S cm−1 and nonconductive vice versa. The conversion from a regression task to the classification was believed to mitigate the shortage of data availability, resulting in a prediction of logistic classification on 40 data points. Liu et al. trained a support vector machine model to classify whether a doped LLZO compound is stable against the reaction with metallic lithium101. Trained on 100 data points, their model discovered a clear boundary between stable and unstable doped phases. The output of the classification model is the probability that material belongs to a specific pre-defined class and should not be treated as the indication of values of true property. Due to this limitation, the estimation of true property can only be obtained through the regression model, or from the subsequent experiments or highly accurate simulations.
Utilize unlabeled data through unsupervised learning
Unsupervised learning performs learning on dataset without labels. Taking the advantage of more abundancy of unlabeled data, unsupervised learning usually enjoys more data availability compared to the supervise learning models. Typical tasks of unsupervised learning include grouping and clustering, data visualization, dimension reduction, and feature extraction. In materials informatics, unsupervised learning is widely used to visualize materials in latent space to explore underlying relation among different materials groups102,103,104,105. Our recent work revealed the previously shadowed potential of unsupervised learning in the task of materials discovery25. Built on the premise that the Li-ion conduction in a solid is tightly connected to the crystalline lattice, we deviated from the supervised prediction of the conductivity property to unsupervised grouping of all Li-containing compounds based on their crystalline structural features. Compared to supervised learning, the capability to utilize Li-compounds without conduction property circumvented the challenge brought by the scarcity of conductivity data, resulting good clustering of Li-conductive and nonconductive materials in separated groups. Our unsupervised learning scheme provides a powerful alternative to the most widely adapted supervised approach for the discovery of other functional materials, especially under conditions of scarce materials data.
Enhance sampling efficiency through active learning and Bayesian optimization
Active learning is a type of learning strategy that requires the interaction between the learning agent and a domain expert. In active learning, the learning algorithm iteratively chooses unlabeled examples and query the domain expert for new labeling. Because the learner decides the examples, selection strategies can be taken to suggest what examples most deserves to be labeled, thus reducing the cost of expensive and time-consuming labeling process while keeping the performance comparable to supervised learners. A commonly used active learning approach is Bayesian optimization (BO)106. In BO, the learner agent uses the posterior for the black box target function conditioned on the past evaluations to construct an acquisition function; then determines the next point to label through maximizing the acquisition function. Several choices for the acquisition function are available, such as upper confidence bound107, entropy-based methods108, probability of improvement109, expected improvement110, top-two expected improvement111, knowledge gradient112, and Thompson sampling113. Similarly, the prior for the target function can be a variety of ML models such as neural network114 and random forest115, while the most popular choice is the Gaussian process for the simultaneous prediction of the value of targeted function and uncertainty116. These two parameters control the exploration and exploitation strategy in the acquiring function. For the acquiring focusing on the target value, the model encourages an exploitation to query regions where we have more confidence to find better targets, while focusing on the uncertainty encourages an exploratory strategy to explore regions we have yet queried.
Both reinforcement learning and active learning are relatively new to battery informatics. But their promising potential has already been demonstrated in several studies. Bayesian optimization is demonstrated with faster speed to optimize materials properties compared to the search of random sampling. Homma used BO to find the composition ratio of ternary Li3PO4–Li3BO3–Li2SO4 for optimized Li-ion conductivity117. Harada et al. examined the efficiency of BO in finding the composition from 49 compounds in the family of NASICON-type Li1+x+2yZr2–x–yYxCay(PO4)3 solid electrolyte for the highest conductivity at 30 °C118. BO found the optimal after 16 trials with the average failure rate of <0.1%, which was about three times faster than the random search. Nakayama et al. calculated the migration energies in ~400 Li- and Zn-containing oxides of varied crystal structures using the bond valance force field method119. Based on the calculated data, they demonstrated better search performance of BO approach than random sampling. On average, the BO approach required ~15% of the total dataset to discover the material with highest conductivity. We note, however, that the optimization of the conventional materials is commonly guided by domain knowledge and/or empirical mathematical analysis and should not be regarded as random sampling. In the work of Harada et al., multi-object BO was carried out to find the Pareto frontiers of the relative density for mechanical properties and ionic conductivity118. The Pareto frontier is defined as a set of points where one property cannot be improved without sacrificing any other properties. The multi-object BO was more efficient to find the Pareto frontiers than multi-objective optimization approach based on the non-dominated sorting genetic algorithm II.
Active learning is particularly attractive to connect the intelligence agent with the simulation or experiment agent to close the loop of automated materials discovery and optimization as discussed in the previous section. This is because the simulation and experimentation in close-loop strategy is the natural step of labeling new samples while the recommendation from ML serves as the learner to select samples for labeling. Therefore, the architecture of active learning excellently matches the framework of close-loop materials informatics. Dave et al. connected the robotic HTE platform of Otto to the BO software of Dragonfly80. Dragonfly learned the measured electrochemical stability window of aqueous electrolytes from Otto and used four acquisition functions to adaptively sample based on the performance of each acquisition function in the task through the course of each optimization run. By examining one aqueous electrolyte receipt in one iteration, the cooperative operation of Otto and Dragonfly accomplished the optimization in about 70 cycles.
Application of machine learning in battery research
Batteries are complicated materials systems. Building a better battery requires the solution of multiple scientific and engineering problems from materials discovery and microstructure optimization to the cell and manufacturing process design. After the deployment in a real device, the operation requires the monitoring of battery health and optimization of charge and discharge to maximize the usage value. In the following two sections, we review the success of ML in these individual tasks. We will first review the application of machine learning in battery research in this section and highlight several achievements of machine learning in battery engineering in the section “Machine learning in battery engineering”.
Materials discovery
Among many applications of ML in battery informatics, the exploration of novel battery materials is one of the most active fields. A common approach of ML-guided materials discovery starts with establishing models to accurately predict the performance of a material for a targeted functionality, usually parameterized in one or a few crucial materials properties. The model is then used to inversely predict the functionality for the discovery of candidates with best performance. However, as we discussed in earlier sections, other approaches have been developed to overcome the data scarcity challenge. Below we review the advances of ML-guided materials discovery for important battery materials.
Solid-state electrolyte
Solid electrolyte is a rare class of solids that rival the ionic conductivity typically seen in liquid solutions (10−3–10−2 S cm−1). These materials are of great importance in developing all-solid-state batteries. By replacing the flammable organic electrolyte in current lithium-ion batteries with a solid and lithium-conductive component, all-solid-state battery holds the promise of improved safety, excellent stability, and long cycling life93,120,121,122. An ideal solid-state electrolyte should have several important merits of properties: high ionic conductivity and low electron conductivity, wide window of electrochemical stability, good thermal and chemical stability, suitable mechanical strength, easiness of manufacturing and low materials cost. No single material can meet all of these requirements at this moment, motivating significant interest to the exploration of new solid-state electrolyte with better functionalities.
The challenge to discover new solid-state ionic conductors lies in several aspects. First, the ionic conduction is a complex dynamic process spanning a broad range of time and length scales with the ionic conductivity affected by a number of geometric and chemical factors93. A good model to infer the conductivity thus requires fine feature engineering to capture the underlying physics of conduction. Second, the known ionic conductors are distributed in a wide range of structural and composition space. Solid-state Li-ion conductors, for example, have compositions ranging from oxides, sulfides to nitrides and halides, and a diverse set of crystalline structures including perovskite and antiperovskite123, argyrodite124, garnet125, Li3N126, NASICON127, LGPS128, and Li7P3S11129. The highly diverse sampling challenges the reliability of ML models to explore an unknown space far from any available reference. Finally, the ionic conductivity is sensitive to small compositional variations. Although computational methods such as AIMD can simulate the conductivity in good agreement with experimental measurements, it is still practically challenging to apply the computational demanding method for screening every possible doping in a large and unconstrained configurational space.
Strategies to overcome these challenges have been developed in the past few years. The feature engineering of suitable descriptors can be summarized in three approaches: the domain-knowledge-based chemical features, the physics-based strong descriptors, and the feature abstraction through deep learning. For the domain-knowledge-based chemical features, empirical rules are hand-crafted to vectorize the structural and compositional information of individual compounds. Examples of hand-crafted descriptors include the chemical information of individual elemental constitutes, the local structural features such as bonding coordination and distances, the volume of crystalline cell and packing faction, and the collective statistics of these descriptors. Due to the large pool of hand-crafted features pool typically with low correlation with the targeted property, necessary feature selection is important to avoid overfitting. Sendek crafted 40 empirical features to model the conductivity of Li-containing compounds100. After feature selection, five features were used in the optimal logistic model, including the average number of lithium neighbors for each lithium, the average sublattice bond ionicity, the average anion–anion coordination number in the anion framework, the average shortest lithium–anion distance in angstroms and the average shortest lithium–lithium distance. Nakayama used the histogram statistics of various composition- and/or structure-derived features to construct general vector-form descriptors for Li- and Zn-containing oxides and modeled the Li-migration barrier using Gradient boosting regression119. They found the most critical feature is the radial distribution function of oxygen-oxygen interaction. Jalem et al. constructed a neural network model to simultaneously predict the diffusion barrier and cohesive energy of olivine LiMXO4 compounds130. They found the average bond length of the Li octahedron, distortion index of the Li octahedron and bonding angle Li–O–X are positively correlated with the diffusion barrier, while effective coordination number of lithium, distance between two X tetrahedra near midplane and distortion index of M octahedron are negatively correlated. Using the same neural network architecture, Jalem et al. found six local structure descriptors for the diffusion barrier in tavorite LiMTO4F compounds131. They identified common descriptors to increase the diffusion barrier in olivine and tavorite compounds including bond angle variance of the M octahedron, the average bond length of the Li octahedron while the polyhedral volume of the Li octahedron and effective charge of M cation decreased the barrier.
Some common factors affect the conduction property appeared after consolidating the above studies, including the coordination number of lithium ions, volume of interstitials, local distortion of coordination environment and the charges on non-lithium species. The connection of these factors to conductivity is physically intuitive. For instance, a high coordination number indicates a large energy penalty to break the bond for diffusion. On the other side, for a small cation like lithium high coordination number usually indicates a geometrically frustrated environment, which is beneficial to mitigate the energy difference between favorable and unfavorable bonding environment132,133. The same geometric consideration can be applied to the local distortion of lithium bonding environment. In fact, the majority of lithium-ion conductor has a distorted crystalline structure rather than exposing highly symmetric lattice25,134. The success to identify complex structure-conductivity relation is certainly attributed to the powerful capability of ML to detect buried patterns from data analysis. However, we should be cautious as the results of feature selection may be sensitive to the choice of learning algorithm, selection algorithm and data itself, especially in the circumstance of small availability of training data90.
Physics-based descriptors are constructed from known physics of properties. For example, the ionic conductivity of most solid substances follows an Arrhenius dependence on the temperature σT = Cexp(−Ea/kT). Through this relation, the conductivity at a given temperature can be quickly estimated from the information at other temperatures. Zhu et al. analyzed the mean square displacements (MSDs) obtained from short AIMD simulations at 800 and 1200 K for known superionic conductors. They observed that all known lithium-superionic conductors fall within the regions bounded by MSD800 > 5 Å2 and MSD1200/MSD800 < 7, suggesting the information at high temperature is a strong indicator of diffusion at room temperatures135. In the work of Fujimura et al., the model to predict the conductivity of LISICON compounds used four descriptors, diffusion coefficients at 1600 K, transition temperatures, experimental temperature and average volume of disordered structures, for the prediction of conductivity at 373 K92. Not surprisingly, the diffusivity at high temperatures served as a strong descriptor of low-temperature conductivity and systems having high diffusion coefficients at 1600 K tend to have high condcutivity at 373 K as well.
Deep learning-based feature utilizes the capability of deep learning to learn the feature by itself and thus avoids potentially biased handcrafting. For the exploration of solid-state electrolyte, the representation of the material should appropriately describe the compositional information and the crystalline structure of candidates. Deep learning has achieved significant breakthroughs in representing these two crucial materials features. For the compositional representation, the representations extracted from deep learning models of the formation energy of inorganic compounds abstract the atomic number of each element into patterns correlated to chemical trends41,44,45. It offers the potential to transfer knowledge from learning the formation energies for representing elemental identifies, thus reducing the efforts to craft domain-knowledge-based representation of chemical elements. Meanwhile, the recently introduced crystal graph convolutional neural network (CGCNN) has shown great success for the representation of crystalline structure136,137,138. In the crystal graph convolutional neural network (CGCNN), atoms are treated as nodes in a graph, and the bonds are treated as edges connecting individual nodes. In this way, each individual crystal is represented by a graph with the convolution and pooling layers satisfying the invariance with respect to permutation of atomic indices and choice of unit cell. By introducing global attributes in combination with atom and bond attributes, the CGCNN is generalized to graph network no longer constrained in the family of neural networks9,91. The graph-based deep learning models have shown impressive capability to predict materials properties. Transferring the elemental embedding trained from CGCNN or graph network on a large dataset significantly improved the performance of predicting properties with a limited data availability139. The application of CGCNN to quantify the relation between crystalline structure and ionic conduction remains a promising field for future exploration.
The exploration of new solid-state ionic conductors can be summarized into supervised regression of activation energy barrier or conductivity74,92,117,118,119,130,131,140, supervised classification of superionic or non-superionic materials100,141,142 and unsupervised screening25. In the supervised approach, the model learns the relation between ionic conduction and input features and make a prediction of ionic conductivity accordingly (Fig. 4a). A practical approach to mitigate the data scarcity challenge in this approach is to restrict the modeling in a constrained space of exploration. In battery informatics, this approach is frequently adapted to focus on a specific structural family because many crucial properties of battery materials are highly dependent on the crystalline structure prototypes. Built on the premise that a known family of structure is more likely to yield better functionality of interest, the exploration is therefore converted to the task of optimization in the constrained space of interest. The restriction in the selected structural families efficiently concentrates the data for better pattern extraction, thus reducing the requirement of data availability for a qualitied model. The removal of structure as a variable factoring in the target property also mitigates the technical challenge to represent the crystalline lattice. The typical size of training data used in the past studies ranged from a few hundred for DFT-calculated examples92,130,131 and less than 100 from experimentation74,117,118. The drawback of restrained exploration is that it scarifies the generality of ML model in multiple structural families. To switch different structural families, the training and validation of ML must be re-carried out, usually in a completely independent manner. A possible strategy to overcome this limitation is to transfer the pre-established from one system to the study of a new system because models of conductivity for different crystalline families may share common features of conduction131.

a Supervised regression of the conductivity/activation energy barrier. b Supervised classification of promising and non-promising conductors. c Unsupervised clustering of Li-containing compounds. d Unsupervised clustering of Li-containing compounds based on the anion packing and the discovery of novel inorganic lithium conductors. Reproduced with permission from ref. 25. Copyright Springer Nature 2019.
Another type of supervised exploration is to predict if a candidate has the potential to be promising conductors rather than directly output the ionic conductivity (Fig. 4b). By transforming the regression task into a classification problem, Sendek et al. screened 12000+ Li-containing compounds in unconstrained compositional and structural space using a logistic regression100. Among 317 compounds meeting the requirement of thermodynamic phase stability, low electronic conduction, high electrochemical stability, absence of transition metals, and potentially low materials cost and high earth abundance of the elemental constituents, 21 compounds were predicted to reach the conductivity of >10−4 S cm−1. They further used the output of the logistic regression model to train a new model with only the composition of compounds as input variables141. It extended the screening to compound not included in the database. They predicted that compounds including LiN5P3O, Li3Na4O3, LiPO3, LiMg3K2O4, LiNaMg3O5, Li2K3GaO4, Li5Na2O3, Li4NaGaO4, Li2MgO2, Li5K2O3, and Li5Na2NO2 are promising ionic conductors. The same logistic regression framework predicted LiAuI4 and Ba38Na58Li26N as superionic conductors when Ahmad explored candidates to suppress the growth of Li-dendrites143.
The powerful capability of ML to explore a wide range of unknown space usually yields a list of promising candidates beyond the normal capacity of experimentation for brutal examination. The conductivity of solid electrolyte is especially sensitive to the choice of dopant and defect concentrations, which greatly increases the experimental cost of fine tuning the compositional degree of freedom. To mitigate this challenge, ML-based screening is usually followed by high accurate simulations to further narrow down the choice of candidates. By artificially introducing a lithium vacancy in the supercell, Sendek et al. identified two compounds from the candidates identified through logistic regression, Li5B7S13 and Li2B2S5, with exceptional high conductivities at room temperature142. More rigorously, Li vacancy and excess Li should be introduced through aliovalent doping of immobile species. He et al. proposed the appropriate doping strategy should activate concerted motion of multiple lithium ions by inserting lithium at high-energy sites144,145. They identified aliovalent substitution of LiTaSiO5 and LiAlSiO4 to introduce excess lithium boosted the lithium-ion conductivity at RT145. Confirmed in experiments, Zr-doped Li1.1Ta0.9Zr0.1SiO5 showed a conductivity about two orders of magnitude higher than that of stoichiometric LiTaSiO5146.
Switching the target of ML from accurately predicting the values of conductivity to narrow down the candidates for the examination through expensive simulation or experimentation motivates the screening through unsupervised learning (Fig. 4c). In our work, we used the representation to match the modified periodic anion crystalline lattice of Li-containing compounds into a set of X-ray diffraction intensities at a fixed set of 2θ values25. Through agglomerative hierarchical and spectral clustering, we found most known Li-ion conductors were clustered into two out of a total seven groups with distinctive diffraction fingerprints. It narrowed the screening of initial 2,986 compounds down to the evaluation of ionic conductivity in 82 unique compounds. Through AIMD simulations, we predicted 16 more candidates to have σRT higher than 10–4 S cm−1. Three of these new materials systems, Li8N2Se, Li6KBiO6, and Li5P2N5, have the room temperature conductivity exceeding 10−2 S cm−1 (Fig. 4d). These new predicted candidates comprise new structures, chemistries, and compositions significantly different from known SSLCs, demonstrating the capability of unsupervised learning to discover materials beyond existing chemistries.
Mechanical properties of solid electrolyte
The mechanical property is another important factor to the practical application of solid electrolyte in all-solid-state batteries147. High mechanical strength benefits the suppression of lithium dendrite growth. However, too high mechanical strength may cause the difficulty to wet on lithium anode. Soft electrolyte is more tolerable to compromise the volumetric change of electrodes during cycling. Compared to the conductivity property, the calculation of mechanical property is a more trackable task using first-principles methods. The DFT-calculated elastic properties, including the full elastic tenor, bulk, shear and Young’s moduli and Poisson ratio, of alkali superionic conductors were in good agreement with available experimental data148. The Materials Project database contained the DFT-calculated elastic tensor for more than 13,000 compounds, with the error typically within 15% of the experimental value149. The large availability of calculated data led to the successful prediction of mechanical properties using ML methods9,150,151. To explore candidates of solid-state electrolyte with suitable mechanical properties to suppress the growth of lithium dendrite, Ahmad defined a stability parameter as a function of shear modulus, Poisson’s ratio, and molar volume ratio143. Using the computational database of mechanical modulus from Materials Project, they trained a CGCNN model to predict the stability parameter for 12,950 lithium-containing compounds, among which 3400 were used for training. Twenty dendrite-suppressing interfaces were predicted formed from LiBH4 and LiOH and two polymorphs of Li2WS4.
Solid–electrolyte interface
In addition to the ionic conductivity and mechanical properties, the interface between solid electrolyte and electrode plays a crucial role in determining the performance of all-solid-state battery. The stable operation needs the electrolyte either stable against electrochemical reduction and oxidation or to form stable passivating solid-electrolyte-interface to avoid continuous consumption of active materials. Most known solid electrolytes such as LGPS152, Li1.3Al0.3Ti1.7(PO4)3153, and garnet LLZO154 are reduced once in contact with metallic lithium. On the cathode side, sulfides electrolytes usually exhibit lower stability against oxidation compared to oxides155. Theoretically, the electrochemical stability of solid electrolytes can be evaluated by constructing the grand canonical free energy at varied electrochemical potentials156,157. Utilizing the computational materials database, the interface stability has been evaluated for a large number of lithium and sodium compounds, yielding instructive screening of candidates possessing excellent interface stability and ionic conductivity49,50. To extend the screening beyond the stoichiometric compositional space, Liu et al. incorporated ML to explore the stability of doped garnet LLZO101. They calculated the formation energy of cation doped LLZO and built an automated route to screen all possible reactions between doped materials in contact with metallic lithium. The thermodynamic stability of doped LLZO against the reduction by metallic lithium was found to increase with stronger dopant-oxygen bonding. A binary classification model was then trained to predict whether the Li|LLZO interface is stable or not. They further trained a kernel ridge regression model to predict the reaction energy and found good agreement between the DFT values and KRR predictions. The ML models predicted 18 doped systems stable against Li metal and the predictions were validated in the automated calculations.
Polymer electrolyte
Besides ceramic solid electrolyte, polymer-based electrolyte is an alternative of high processability and appropriate binding properties to the development of all-solid-state batteries158. To balance the requirement of conductivity, mechanical properties, and stability, polymer electrolyte is usually prepared as a composite of a polymer, lithium salts, and other necessary additives. ML provides a powerful tool to optimize the complex receipt for better electrochemical performance. Using a Bayesian neural network, Ibhahim el al. modeled the conductivity in a series of polyethylene oxide (PEO)-lithium salt-solvent-additive systems159,160,161. The neural network was found successful for the prediction of conductivity and impedance of nanocomposite polymer electrolyte system.
ML was used to explore the wide polymer space for potentially novel electrolyte systems. Conventionally, the ionic conduction in PEO-based polymer electrolyte is coupled to the motion of polymer backbone, which higher conductivity is achieved with the cost of lower melting points162. To break this limitation, Hatakeyama-sato et al. constructed a database of Li-ion conductive polymers from published results and used it to train a Gaussian process model of conductivity using the input of chemical structures, composition ratio, and measured temperatures (Fig. 5)163,164. Trained with the data reported up to 2018, the model predicted the conductivities of ~150 representative conductors reported in early 2019 in good agreement with reported values. Applying ML model to explore unknown space led to the discovery that lithium salt in charge-transfer complexes of polyphenylene sulfide (PPS) and dimethyl-substituted PPS (PMPS) and aromatic oxidants such as chloranil and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) could be a promising candidate of electrolytes. They confirmed the prediction in experiments, where the PMPS and PPS electrolytes showed superionic conductivity around 10−3 S·cm−1 at room temperature. More importantly, PPS and PMPS have glass transition temperatures much higher than that of PEO, indicating novel lithium conduction mechanism without involving the movement of polymer chain in these new polymer electrolytes. Considering the vast number of polymer systems and the complexity of polymer electrolytes, great potential exists to apply ML for the exploration, discovery and optimization of new electrolyte candidates for the future development of all-solid-state batteries.

Scheme for predicting properties of the solid polymer electrolytes from consolidating the experimental database to the discovery of new polymer electrolyte. Reproduced with permission from ref. 163. Copyright American Chemical Society 2020.
Electrode materials
In addition to the study of novel electrolyte materials, ML was used for the exploration of novel and better functional electrode materials. By unveiling the complex structure–property relationships underlying the performance of electrode materials, the reported studies include the modeling multiple voltage, structure, and energy landscape of electrode materials. For example, Joshi et al. used DNN, SVR, and KRR to predict the voltage profile diagram of cathode materials. Applying the ML model to screen potential candidates yielded ~5,000 electrode materials for Na- and K-ion batteries with voltages rivaling their Li-ion counterparts165. Wang et al. studied the volume change caused by the delithiation of spinel and layered oxide cathodes166. They found the partial linear square predicted the volumetric change in excellent agreement with DFT-calculated values. Shandiz used a wide range of classification algorithms to predict the crystalline structure in the Li–Si–(Mn, Fe, Co)–O compositional space167. The volume of the unit cell and number of sites showed the highest importance in determining the crystalline lattice, while other factors including formation energies, convex hull energy, and band gap also played an important role. Zhang et al. used machine leaning to model the adsorption energy of lithium polysulfide species on layered sulfides168. By transferring the pre-established model of adsorption on the MoSe2 surface to predict the adsorption on similarly structured WSe2 surface, the ML reduced the computational cost of DFT calculation while maintaining the accuracy in understanding two-dimensional layered compounds as the host materials of lithium-sulfur battery cathode. Table 2 summarizes the data, ML methods, modeled properties, and applications of these studies.
The topotactic lithiation/delithiation of electrode materials usually results in highly disordered lithium and vacancy arrangements after lithium is partially removed from a parent crystalline structure. The classical method to analyze such disordering is footed on the cluster expansion proposed in the seminal work of Sanchez et al.169. The common approach of cluster expansion expresses a lattice model Hamiltonian as a linear combination of orthonormal basis functions of configurational occupancy variables. Recently ML has shown potential as a promising alternative to explore the disordering events. Natarajan and Van der Ven developed a neural network function to relax the constraint of linear Hamiltonian in cluster expansion (Fig. 6a)170. In the case study of spinel LixTiS2, the model using neural network had an error of 36 meV per formula unit compared to the error of 89 meV per formula unit for the linear regression model. Hochins and Visvanathan incorporated a neural network potential to relax the disordered structure determined from grand canonical Monte Carlo simulations of layered oxide cathodes using the cluster expansion Hamiltonian (Fig. 6b)171. After structural relaxation, thermodynamic properties such as lattice parameters, free energy, and entropy were obtained and the predicted voltage profile of LixNiO2 and LixCoO2 were in good agreement with the experimental measurements. Beyond the framework of cluster expansion, Eremin et al. modeled the energy landscape of topotactic delithiation of LiNiO2 and LiNi0.8Co0.15Al0.05O2 cathode through the structure descriptors that encoded the lithium and dopant occupancy information (Fig. 6c)172. They found the energetics was mainly controlled by the topology of Li layers and relative disposition of Li ions and Li and not by the relative dopant positions.

a Schematic of incorporating neural network architecture into cluster expansion and the prediction of the formation energy convex hull in spinel Li3xTi2S4. Reproduced with permission from ref. 169. Copyright Springer Nature 2018. b Machine learning assisted prediction of voltage profile in layered oxides. Reproduced with permission from ref. 170. Copyright American Institute of Physics 2020. c Machine learning explores the configurational space of topotactic delithiation of LiNiO2 and LiNi0.8Co0.15Al0.05O2. Reproduced with permission from ref. 171. Copyright American Chemical Society 2017.
Accelerate the simulation and assist fundamental mechanistic exploration
ML-assisted molecular dynamics
The functionality of battery materials to a large extent originates from the atomistic structure of these materials. The correct understanding of the atomistic structure and reactivity of all materials involved is of paramount important towards the design of better-performed materials. Computational simulation has long become an essential tool in understanding the structure–property relation in complementation to the experimental characterization and analysis techniques. DFT method is now a standard approach with proven accuracy and chemical versality to provide structural, energetic, and electronic insights into the static ground state-of-battery materials. Molecular dynamics simulation, on the other side, provides spatial and temporal knowledge of atomic movements at given conditions. Ab initio molecular dynamics incorporates a molecular dynamics engine to study the dynamic movement of atoms within a simulation cell, where the forces experienced by all atoms are calculated using DFT theory. With the advantage of no prior assumption of potential energy surface, AIMD is becoming a powerful tool to study many dynamic phenomena in battery materials such as ionic transportation and solid-electrolyte interface formation with an excellent accuracy to predict the experimentally measured quantities as well as offering atomistic insight into the physical mechanism173. However, in AIMD simulation each step requires one ionic relaxation of DFT to calculate the force exercised every atom. The high computational cost restricts the simulation cell to a few hundred atoms and the simulation time to at most a few nanoseconds.
An emerging approach to simultaneously maintain the DFT-level accuracy and reduce the cost of AIMD simulation is to create interatomic potentials by ML from quantum-mechanical reference data. More precisely, the ML potential (MLP)-assisted MD simulation learns the potential energy surface from a dataset of accurately computed energies and forces without assuming a specific functional form of the PES. The learned PES is then used in the simulation to avoid the extensive DFT simulation at every MD step (Fig. 7a). Since the introduction about 15 years ago174, ML-assisted MD has been fast developed in the past few years and its application in battery research has led to successfully modeling of a variety of cathode171,175, anode176,177,178,179,180 and solid-state electrolytes12,181,182,183,184,185,186,187,188,189,190,191, as summarized in Table 3. A variety of ML algorithms have been used as the surrogate form of potential energy surface, with the most popular techniques including neural-network potentials171,175,176,178,184,185, gaussian approximation potentials (GAP)177,179,180,192, spectral neighbor analysis potentials186,193, and moment tensor potentials190,194. Leveraging the large amount of data generated during the AIMD simulation, the ML model typically predicts the energy within the error of a few meV per atom and forces with the error of a few hundreds of meV per Å. Due to the low error of the ML model to predict the DFT-calculated energy and forces, the prediction of macroscopic properties through ML-assisted MD can reach the same performance as AIMD simulations. As shown in Fig. 7b, the simulation using the ML on-the-fly (LOTF) potential reached better accuracy to predict the experimental migration energy when benchmarked with AIMD simulations in a range of solid-state electrolytes from very good conductors of β-Li3PS4 and Li7P3S11 to very bad conductors of Li4GeO4181. The diffusivity of lithium in Li7P3S11 was within 14% of that obtained directly from AIMD182, while for LGPS the Li-ion diffusivity at 300 K and the activation energy were predicted to be 12 mS·cm−1 and 226 meV, respectively183, in excellent agreement with the experimental data128.

a Workflow of MLP-assisted molecular dynamics studies. b Diffusivities simulated by AIMD at high temperatures and by LOTF-MD at intermediate temperatures for various solids. Reproduced with permission from ref. 181. Copyright American Institute of Physics 2020. c Supercell of Li372P128O506 for the simulation of amorphous Li3PO4. Reproduced with permission from ref. 184. Copyright American Institute of Physics 2017. d Speed test of DP models on a NVIDIA V100 GPU. Reproduced with permission from ref. 185. Copyright American Institute of Physics 2021. e Schematic of the genetic algorithm sampling approach using the specialized ANN potential. Reproduced with permission from ref. 176. Copyright American Institute of Physics 2018. f Binding energies of sodium on disordered carbon. Reproduced with permission from ref. 180. Copyright Royal Society of Chemistry 2018. g Force prediction correlation plots shown for H in PEO and P atoms in Li4P2O7. Reproduced with permission from ref. 12. Copyright Springer Nature 2019. h MAE of MLMD in the vicinity of phase transition in Li7P3S11 at 500 K. Reproduced with permission from ref. 188. Copyright American Physical Society 2021.
The low computational cost readily extends the time and length scales of MLP-assisted MD simulation compared to conventional AIMD. For example, due to the low conductivity and high migration barrier in Li4GeO4, AIMD had to be performed at temperatures higher than 1200 K, while LOTF-MD simulation was able to extract the conductivity as low as 700 K181. For good conductors, the temperature range reached 300 K while the total simulation was more than 1300 nanoseconds181. For the simulation of amorphous Li3PO4, the expensive cost of AIMD simulations limited the simulation cell to Li46P16O63, while MD simulation using neural network potential extended the cell over 1,000 atoms (Li372P128O506, Fig. 7c)184. Huang et al. examined the speed of MD simulation based on deep potential generator (DP-GEN)185. On one NVIDIA V100 GPUS, the DP-based simulation took around 4 h to simulate a 900-atom LGPS systems for 1 ns and the computational cost scaled linearly with system size up to ~6,000 atoms as shown in Fig. 9d. The high accuracy, ability to simulate low-temperature systems in extended time and length scale make ML-assisted MD simulation a powerful technique for large-scale simulations.
Increasing the size of the simulation cell improves the fidelity of MD results by alleviating size dependence and avoiding fault physics due to artificial interaction across simulation cells. For example, the simulation of Li10SnP2S12 using a supercell of ~200 atoms overestimated the diffusion coefficients by 10 to 100 times especially at low temperatures185. By expanding the simulation cell to 900 and 1600 atoms, the diffusivities converged with a difference of less than 3 × 10−12 m2 s−1. Larger simulation cell used in NN potential MD simulation suppressed the partial crystallization of local structures analogous to those in β-Li3PO4 and γ-Li3PO4 as observed in small cells, suggesting the NN potential simulation better captured the conduction in a real amorphous phase184.
The extended time and lengths scales allows MLP-assisted MD to probe amorphous system, polymer and grain boundaries that conventional AIMD is usually prohibitive due to the large number of atoms necessary to represent the structure and the long simulation time to describe the rare event of melting and structural reconstruction. Arithis et al. incorporated an ANN potential in genetic algorithm and molecular dynamics simulation to generate the phase diagram for lithium intercalation in amorphous silicon anode (Fig. 7e)176. Onat et al. developed an “implanted” neural network that incorporate pre-trained parts to capture the character of different components178. The MD simulation at room temperature predicted the diffusion coefficient of Li in amorphous LixSi in better agreement with experimental measurements than other theoretical results. Fujikake used Gaussian approximation potential to model lithium intercalation in graphite and amorphous carbon structure179. They showed the simulation correctly described the structural and vibration properties of lithium diffusion in carbonaceous frameworks. Deringer and his co-workers used Gaussian approximation potential to model Li- and Na-insertion in disordered carbon anode and obtained lithiation and sodiation behavior in agreement with experimental observations (Fig. 7f)177,180. Mailoa developed a staggered neural network force field structure to predict atomic force vectors through the use of rotation-invariant and -covariant features12. They demonstrated that the simulation can accurately predict the atomic forces accurately for a polyethylene oxide (PEO) run at T = 353 K and amorphous lithium phosphate (Li4P2O7) oxide melted at 3000 K (Fig. 7g). Using electrostatic spectral neighbor analysis potential for the modeling of Li3N, Deng et al. modeled the diffusion on the grain boundary in a simulation box of 5,040 atoms186. They found the diffusivity of Li within the twist grain boundary was about three times the extrapolated value in the bulk phase at 300 K, indicating the important role of grain boundary for conduction in Li3N.
Conventional AIMD is usually carried out at high temperatures to ensure the statistical significance of the sampling on rare events of diffusion and structural reconstruction. By probing the dynamic events directly at low temperatures, MLP-assisted MD has the potential to unveil the physics buried in high-temperature simulations. Miwa and Asahi used the potential constructed by self-Learning and adaptive database (SLAD) approach to study the conduction in Nb-doped LLZO187. The simulation was performed from 400 to 800 K in a supercell containing 1520 atoms. The ML-assisted simulation reproduced the conduction properties in good agreement with experimental results and predicted a negligibly small energy difference between the 24d and 96 h sites, which was likely to benefit fast conduction at room temperatures. Using a sparse Gaussian process potential, Hajibabaei et al. reproduced the melting of Li7P3S11 at 900 K188. As shown in Fig. 7h, they also observed a previously unknown phase transition at temperatures higher than 450 K. By rotating the P2S7 double tetrahedra into a new orientational order, the new polymorph of Li7P3S11 was almost iso-energetic to the initial phase but exhibited Li diffusivity several orders of magnitude smaller. Huang et al. used the DP-GEN models to study the effect of lattice disordering in LGPS phases185. They predict the disordering of Ge4+ and P5+ increased the diffusivity by 2 to 4 times at low temperatures due to the flattening of potential energy surface. Such effect was not seen in high-temperature AIMD simulation, as the benefit diminished in systems with high diffusion coefficients.
ML- analysis of dynamics
Due to the large amount of data generated during molecular dynamics simulation, quantitative analysis to extract relevant dynamic information is a challenge for data analysis. Conventionally, the analysis of molecular dynamics trajectory is carried out through hand-crafted rules in combination with computing the average behavior of atoms. The powerful capability of ML in handling a large amount of data opens new opportunities to post-analyze the MD data to mitigate potential information loss during the analysis137,195,196,197,198. Particularly, an interesting application of ML in analyzing MD data is labeling atoms in distinct coordination environments through unsupervised clustering. The unsupervised labeling analyzes the local configurations and bonding environments of atoms in MD trajectory and uses the clustering to search structurally distinct states107,108,109. Compared to the conventional approach where the system-specific site locations are given a priori, unsupervised learning uses no manually crafted rules and ensures the statistical significance of structural difference. Xie et al. developed a graph dynamical network combined with the Koopman models to map the local configuration of target atoms into a lower-dimensional feature space137. Applying their method to study poly(ethylene oxide) (PEO)/lithium bis-trifluoromethyl sulfonimide (LiTFSI) composite electrolytes, the model identified four coordination states of lithium ion, each of which had distinct solvation environments. Chen et al. developed a method to calculate the nuclear density from the MD trajectories and cluster the data based on the density196. In simulating garnet LLZO, their method yielded 576 available sites in a 2 × 2 × 2 supercell for the conductive cubic phase, and 448 clusters for the less-conductive tetragonal phase. The difference of site availability reflects the conduction characteristics in these two phases. Magdau and Miller developed a machine leaning approach to automate the classification and identification of ion solvation environments in polymer electrolyte based on data from MD simulations197. By concatenating the type-specific Li+ radial distribution functions, they applied two unsupervised algorithms of UMAP to embed the high dimensional feature vectors into a low-dimensional latent space and HDBSCAN to classify the embedded data into specific solvating environments in poly(3,4-propylenedioxythiophene). Understanding the occupancy at different lattice sites is an important first step for subsequent analysis to extract information such as site shape, type and occupancy. In the work of Xie et al.137, the labeling of lithium to different solvation sites identified three relaxation processes. The slowest relaxation is a process to transport a Li-ion into and out of a PEO coordinated environment. The second slowest relaxation corresponds to a movement of the hydroxyl end group. The last relaxation is a Li-ion switching the coordination between PEO and TFSI.
Interpret underlying physics
Another promising application of ML in fundamental mechanistic exploration is to interpret physics underlying measured observables. For some sense all supervised learning models can be regarded as the interpretation of underlying physics because the good model should necessarily discover the relation between property and input features. However, due to the highly complex architecture, ML, especially deep learning-based models, lack the transparent interpretability to understand the physical causality between input and output5. To overcome this limitation, methods such as variable importance measure199, visualizing the hidden layer activations200, attention response map201, physics-leveraging models202,203 have been used for post-hoc interpretation of ML models204,205. In certain ML methods, the interpretability is the strength rather than the weakness of modeling. Symbolic regression, for example, is a ML method that searches the mathematic expression that quantifies fundamental relationships of physical phenomena to each other206. In a number of studies, ML has successfully “rediscovered” important physical equations in both explicit and implicit formats, including the Hamiltonians and Lagrangians for simple harmonic oscillators and double pendulums207, governing equations of dynamic systems208, and partial differential Nave-Stokes equation209. We anticipate symbolic regression could discover a new set of phenomenological equations that leads to the exploration of new physics in future. Another example of ML-based physics interpretation is Bayesian model selection210,211. The Bayesian model selection compares models from different physics and choose that best describes the data from measurement. Thus, the result of Bayesian model selection directly decides the underlying physics of the measured system. In recent work, Park et al. used Bayesian model selection to study the fictitious phase separation in the delithiation of Lix(Ni1/3Mn1/3Co1/3)O2 cathode212. From the operando X-ray diffraction, X-ray microscopy, and electrochemical measurements they found the inter-particle inhomogeneity during delithiation was induced by the limitation of reaction rate. They constructed theoretical models of the reaction- and diffusion-limited delithiation and used Bayesian model selection to decide the correct physics (Fig. 8a). As shown in Fig. 8b, the inter-particle distribution in the fast-delithiation X-ray microscopy data clearly favored a reaction-limited model and rejected the diffusion-limited one. The authors concluded that the anomalous phase separation in layered oxide is caused by electro-autocatalytic reaction instead of originating from diffusion-limited mechanisms.

a Schematic illustration of the reaction-limited and diffusion-limited inhomogeneity evolution. b Bayesian model selection of lithium fraction histogram rejects the diffusion-limited case. Reproduced with permission from ref. 212. Copyright Springer Nature 2021.
Microstructure characterization and design
Microstructure characterization and reconstruction
The electrochemical performance of the complex battery systems heavily depends on not only fundamental materials properties but also the microstructure characteristics and design. Today, advances in experimental methods provide much-needed insights of battery microstructural features using a combination of analysis tools such as X-ray and neutron diffraction, electron microscopy, nuclear magnetic resonance, X-ray spectroscopy and Raman spectroscopy213. ML is becoming a new weapon in the arsenal to provide much desired high-level analysis of the data from these advanced analysis techniques. Leveraging the capability of image analysis beyond manual annotation and object recognition, ML, especially CNN-based method, is well-suited for the in-depth visualization, 3D reconstructing and comprehensive understanding of electrode microstructures214,215,216,217. Jiang et al. trained a Mask R-CNN to perform the segmentation of images taken from the quantitative X-ray phase-contrast nano-tomography of the Ni-rich LiNi0.8Mn0.1Co0.1O2 (NMC) composite cathode (Fig. 9a)214. After training, the ML model automated the segmentation over 650 NMC particles, from which the visualization of the microstructure of the composite electrode and the statistical analysis revealed the mechanism of particle-carbon/binder detachment as well as its correlation to the battery performance. Furat et al. collected the electron backscatter diffraction data for a LiNi0.5Mn0.2Co0.2O2 (NMC532) composite electrode215. A convolutional neural network model of segmentation was trained to identify individual grains in the EBSD images, which allowed the 3D reconstruction and segmentation of grains within NMC particles for further quantification of microstructural features (Fig. 9b). Petrich et al. simulated the morphology evolution during a thermal runaway and trained a classification model to identify particles that are either broken or split by the watershed transformation during the thermal runaway (Fig. 9c)216. The model reached an accuracy of 73% when applied to real-world tomographic images taken from a lab-based X-ray nano-CT. Dixit et al. used synchrotron to track in situ morphology transformation of Li metal electrodes in a Li|LLZO|Li cells during stripping and plating processes217. Segmentation of lithium and pores using a resnet34 based deep convolution neural network quantified microstructural properties such as pore size distribution in lithium metal during cycling experiments.

a Workflow of the machine learning-based segmentation and labeling of NMC cathode using hard X-ray phase contrast nano-tomography. Reproduced with permission from ref. 214. Copyright Springer Nature 2020. b 3D segmentation of NMC cathode using electron backscatter diffraction. Reproduced with permission from ref. 215. Copyright Elsevier 2021. c Broken particle pairs from machine learning reconstruction. Reproduced with permission from ref. 216. Copyright Elsevier 2017. d Unsupervised segmentation of NMC cathode using hyperspectral Raman analysis. Reproduced with permission from ref. 218. Copyright Springer Nature 2019. e Machine learning assisted inverse design of microstructures. Reproduced with permission from ref. 222. Copyright Elsevier 2020.
In the reconstruction of battery microstructures, images are usually taken as a stack, which serves a source of data with good quantity and consistent quality. For example, in the work of Furat et al.215, each stack of EBSD data included 91 individual images for the analysis of convolution neural network. Dixit collected the tomography data with the size greater than 30 GB from each scan217. Their neural network model was trained on 800 images from one electrode in a single electrochemical cycle and tested on another 200 images from the same electrode. Baliyan and Imai used the hyperspectral Raman to characterize the cylinder-type 18650 Li-ion battery cells at different charge states218. Each hyperspectral was composed of 60 × 60 Raman spectra, where the numbers denoted the spatial resolution on the sample analysis. Taking the advantage of the data abundance, They applied principle component analysis to reduce the dimensionality of each Raman spectral and used unsupervised clustering and supervised classification to distinguish the distribution of phases of Li(Ni1−x−yMnxCoy)O2 (NMC) and carbon in the electrode (Fig. 9d). Even for traditional microscopy techniques such as SEM and TEM, the post-processing of images through cropping, flipping and rotation can be utilized to generate more artificial data from a single example, assuming these operations maintain the representative macroscopic property of the original samples219. Furthermore, the established image analysis model can be leveraged to train microstructural image analysis models, reducing potential errors in the model initialization. For example, rather than training the model end-to-end from start, Jiang et al. initialized the weights in neural network from the large-scale ImageNet dataset and optimized the pre-trained weights for the analysis of real image of NMC particles214. Overall, the abundance of image data and the advanced image processing and analysis techniques suggest the great potential of ML for the characterization, reconstruction, and analysis of microstructural characteristics of batteries.
Inverse design of microstructure
In addition to the characterization of microstructural details, ML has been applied to the inverse design of microstructures for the optimized electrochemical performance220. The workflow of inverse design generally includes three essential steps of data generation, training ML models to predict the electrochemical performance directly from the input of microstructural parameters and applying the ML models in the inverse design of microstructures to optimize the electrochemical performance (Fig. 9e)221. Duquesnoy et al. used the experimental data of LiNi1/3Mn1/3Co1/3O2 composite electrode calendaring results to fit mathematical expression of process parameters and microstructure features222. A deep neural network was used to predict the effective properties when the input processing and microstructure parameters changes. The model offered detailed insights into the effect of the calendar pressure, electrode composition and initial porosity on a list of mesoscale electrode properties including the particle network interconnectivity, the electrolyte tortuosity and effective conductivity, the coverage of the current collector by CBD/AM particles and the active surface area. Gao and Lu designed a thick electrode with a bio-inspired electrolyte channel for fast charging/discharging223. To optimize the electrolyte channel design, a DNN model was trained to predict the specific energy, specific capacity and specific power from channel geometric parameters. The obtained DNN model was used for the parameter optimization through gradient descent algorithm. The optimized design showed a 79% increase in specific energy compared to conventional design without electrolyte channels. Takagishi simulated 2100 three-dimensional artificial electrode structures using the stochastic particle packing algorithm224. The artificial neural network was used to predict the reaction resistance, the electrolyte resistance, and the solid diffusion resistance using the input parameters of the volume ratio of the active material, particle size, the pressure in the compaction process and bind/additive volume. Incorporation the ANN prediction in a Bayesian optimization workflow achieved the inverse design of microstructural processing parameters for optimized electrochemical properties of total resistance and high capacity.
Machine learning in battery engineering
In the above sections, we have reviewed the application of ML in a wide range of battery research from materials discovery, materials simulation, and microstructure study. These studies focus on the individual components of a battery, aiming at the improvement of battery performance from enhancing materials functionalities. ML has also achieved significant success going beyond the materials research of battery. In the following section, we briefly overview several applications of ML at the system-level (cell or pack) of battery engineering, highlighting several exciting achievements of ML in battery design, state of health and state of charge estimation and charging protocol optimization Although these problems reside in the different territory with research topics such as materials discovery and mechanistic exploration, the executing of ML follows the same underlying principle to circumvent the complex design and optimization with the surrogate function of observables. Therefore, the exciting achievements of ML in solving battery engineering problems also reflects the promising potential of this data-driven approach towards future better battery technologies.
Optimize battery design
The performance of a battery is strongly determined by the design of individual cell, the packing and stacking of cells and the actual operation conditions. ML is becoming a new tool of optimization for these aspects. To design a better battery by ML, one practice is to parameterize the design and operation conditions, followed by seeking for the correlation of these factors with the battery performance. For example, Li et al. modeled the performance of vanadium flow battery as a function of operating and design parameters225. The parameterized design factors included carbon felt type and thickness, electrode area, cell number, negative electrode/bipolar plate structure, positive electrode/bipolar plate structure, bipolar plate type and area, end plate type, seal type, membrane type and area, flow field type, electrolyte concentration and volume. The operation factors included the compression ratio, cutoff charging voltage and current density. After accumulation the data over more than 100 stacks, they used linear regression to predict the voltage efficiency and energy efficiency, reaching the accuracy of within 1% of mean absolute error. By incorporating the materials cost, the model successfully optimized the best-performed design as well as the low-cost designs of vanadium flow battery stacks.
State of health and State of charge estimation
State of health (SOH) and state of charge (SOC) are two parameters describing the current and future states of battery, defined as the capacity in fully charged state normalized by the capacity of a band new battery, and the capacity in current state normalized by the capacity in fully charged state, respectively. Accurate determination of SOH and SOC is of paramount important in battery management. For instance, SOC allows us to estimate the remaining range of battery usage before the next charge occurs. SOH can be used to predict the reliable remaining useful life of battery, from which appropriate deployment of battery can be developed to increase the remaining value of a battery in other applications.
Traditional means to obtain SOC and SOH relies on the estimation from empirical model and physics-based models226. Equivalent circuit model (ECM), for example, simplifies the battery as a network of electrical components such as resistors and capacitors, and model the battery status with empirical parameters for dynamic diffusion and charge-transfer processes. Because of the computational efficiency, ECM are currently the major choice for online SOC estimations in electric vehicles. However, the accuracy of ECM is restricted by the model parameterization from laboratory test. Physics-based models incorporates internal dynamics of electrochemical process and therefore provides better accuracy of estimation. However, the computational cost of solving the complicated governing partial differential equations in physics-based models makes it less efficient for online estimations.
ML techniques offers new opportunity to develop data-driven models for the estimation of SOC and SOH with the potential to overcome the accuracy-efficiency tradeoff. A variety of ML techniques have been employed to predict SOC and SOH from input variables such as voltages, current, temperature and cycling numbers. On average, the accuracy to predict SOC and SOH had the error percentage of 3–4%, while a few reports reached the accuracy within 99%227. We refer several excellent reviews and perspectives of applying ML for SOH and SOC estimations to readers interested in this topic227,228,229,230.
Optimize charging protocol
The performance of a battery cell highly depends on how the battery is utilized. Good charge and discharge protocol maintaining internal health of battery component are crucial to maximize the usage value to full lifetime expectance. Optimization of a charging protocol on battery performance is thus of great value in battery management. In traditional ways, such optimization would require extensive laboratory experiments to examine a large number of combinations of operation factors. On the other side, the ability to forecast the remaining lifetime of a battery allows us to predict the effect of operation from early cycling data. Hence the combination of lifetime prediction model with a search strategy offers a new avenue towards optimization the operation protocol through data-driven techniques. In recent work, Attia et al. developed a close-loop optimization of fast-charging protocols for commercial high-power lithium iron phosphate/graphite 18650 cylindrical cells231. Their close-loop optimization relied on two ML models. First, an elastic net model was trained to predict the battery lifetime using the early cycling data232. Using the data collected for 124 commercial cells in a temperature-controlled environmental chamber (30 °C) under varied fast charging but identical discharging conditions, the model predicted the cycle life with an error of 9.1%. Next, they coupled the early lifetime prediction model in a Bayesian optimization algorithm to model the effect of charging protocols on the battery lifetimes. The close-loop approach optimized the fast charging protocol from 224 candidates, reducing the time for optimization from over 500 days to 16 days. The successful optimization of fast-charging protocol highlights great potential of ML to find other best charging design space as well as in other aspects of battery optimizations.
Concluding remarks
Batteries are unique compared to other materials systems in terms of their complexities. The observed battery behavior originates from the complex interplays among multiple structural, microstructural, and macrostructural components of batteries. Conventionally, the design and development of a battery starts from the detailed mechanistic understanding of how each individual component works and, in many cases, guided by the domain knowledge, experience and intuition. The employment of ML provides an alternative to circumvent the challenge of understanding the complex mechanism through a surrogate function of observables, thus offering a short cut towards improved battery performance. The recent progress of battery informatics summarized in this review has demonstrated the great success of applying ML to exploit the design space through data interpretation. We should note that the potential of battery informatics is also reflected in making exploration type of findings, such as the discovery of novel inorganic and polymer electrolyte with chemistries significantly differing from existing examples. Yet still in the early stage, the success of ML in solving a variety of challenges in battery domain, ranging from mechanistic understanding and novel materials discovery to the engineering, optimization, and management of battery cells all indicate the promising potential of this data-driven technique for better batteries in future.
A major challenge of battery informatics lies in the lack of available datasets and standards. In our opinion, developing standard battery database with accessibility to the research community is the same importance as advancing algorithms and machine learning pipelines to tackle specific problems in battery research. Although significant advancements have been made for the acquisition of high-quality data in large amounts as well as circumventing the challenge through designing suitable ML pipelines, we believe the situation of data scarcity cannot be fully mitigated without the collaboration of entire community. We note that efforts to foster data sharing in public materials science data repository and the development of modern data infrastructure have been carried out recently233,234. In some journals including npj computational materials, statement of data availability is now a mandatory requirement for publishing. On the other side, public data sharing unavoidably raises the concerns about the intellectual property. Protocols to resolve potential intellectual disputes while promoting data sharing should be considered in our perspective. In addition, the real-world deployment of battery very unlikely conform the constrained lab conditions such as temperature-controlled environmental chamber and standard discharge protocols. The collaboration among battery researchers, developers, and users to share and consolidate the data is urged towards applying ML for more comprehensive and sophisticated design and optimization of batteries.
In a short summary, the ML is becoming a more and more standard tool of battery research to add a new dimension in addition to the conventional materials fabrication, characterization, evaluation, and modeling. We hope this review not only serves as a summary of the research status of battery informatics but sheds light on the exciting opportunities of employing ML for materials-related problems difficult to tackle through traditional means.
References
Historical carbon dioxide emissions from global fossil fuel combustion and industrial processes from 1758 to 2020. https://www.statista.com/statistics/264699/worldwide-co2-emissions/.
Choudhary, A. & Prasad, E. Lithium-ion Battery Market. https://www.alliedmarketresearch.com/lithium-ion-battery-market.
Agrawal, A. & Choudhary, A. Materials informatics and big data: realization of the “fourth paradigm” of science in materials science. APL Mater. 4, 053208 (2016).
Butler, K. T., Davies, D. W., Cartwright, H., Isayev, O. & Walsh, A. Machine learning for molecular and materials science. Nature 559, 547–555 (2018).
Schmidt, J., Marques, M. R. G., Botti, S. & Marques, M. A. Recent advances and applications of machine learning in solid-state materials science. npj Comput. Mater. 5, 83 (2019).
Wang, A. Y.-T. et al. Machine learning for materials scientists: an introductory guide toward best practices. Chem. Mater. 32, 4954–4965 (2020).
Isayev, O. et al. Universal fragment descriptors for predicting properties of inorganic crystals. Nat. Commun. 8, 15679 (2017).
Coley, C. W. et al. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 10, 370–377 (2019).
Chen, C., Ye, W., Zuo, Y., Zheng, C. & Ong, S. P. Graph networks as a universal machine learning framework for molecules and crystals. Chem. Mater. 31, 3564–3572 (2019).
Behler, J. & Parrinello, M. Generalized neural network representation of highdimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Vandermause, J. et al. On-the-fly active learning of interpretable Bayesian force fields for atomistic rare events. npj Comput. Mater. 6, 20 (2020).
Mailoa, J. P. et al. A fast neural network approach for direct covariant forces prediction in complex multi-element extended systems. Nat. Mach. Intell. 1, 471–479 (2019).
Ulissi, Z. W., Medford, A. J., Bligaard, T. & Nørskov, J. K. To address surface reaction network complexity using scaling relations machine learning and DFT calculations. Nat. Commun. 8, 14621 (2016).
Aykol, M. et al. Network analysis of synthesizable materials discovery. Nat. Commun. 10, 2018 (2019).
Granda, J. M., Donina, L., Dragone, V., Long, D.-L. & Cronin, L. Controlling an organic synthesis robot with machine learning to search for new reactivity. Nature 559, 377–381 (2018).
Raccuglia, P. et al. Machine-learning-assisted materials discovery using failed experiments. Nature 553, 73–77 (2016).
Xue, D. et al. Accelerated search for materials with targeted properties by adaptive design. Nat. Commun. 7, 5447 (2016).
Balachandran, P. V., Kowalski, B., Sehirlioglu, A. & Lookman, T. Experimental search for high-temperature ferroelectric perovskites guided by two-step machine learning. Nat. Commun. 9, 1668 (2018).
Carrete, J., Li, W., Mingo, N., Wang, S. & Curtarolo, S. Finding unprecedentedly low-thermal-conductivity half-Heusler semiconductors via high-throughput materials modeling. Phys. Rev. X 4, 011019 (2014).
Kim, C., Pilania, G. & Ramprasad, R. Machine learning assisted predictions of intrinsic dielectric breakdown strength of ABX3 perovskites. J. Phys. Chem. C 120, 14575–14580 (2016).
Seko, A., Hayashi, H., Nakayama, K., Takahashi, A. & Tanaka, I. Representation of compounds for machine-learning prediction of physical properties. Phys. Rev. B 95, 144110 (2017).
Chen, C. et al. A critical review of machine learning of energy materials. Adv. Energy Mater. 10, 1903242 (2020).
Tran, K. & Ulissi, Z. W. Active learning across intermetallics to guide discovery of electrocatalysts for CO2 reduction and H2 evolution. Nat. Catal. 1, 696–703 (2018).
Rickman, J. M. et al. Materials informatics for the screening of multi-principal elements and high-entropy alloys. Nat. Commun. 10, 2618 (2019).
Zhang, Y. et al. Unsupervised discovery of solid-state lithium ion conductors. Nat. Commun. 10, 5260 (2019).
Tshitoyan, V. et al. Unsupervised word embeddings capture latent knowledge from materials science literature. Nature 571, 95–98 (2019).
Liu, Y., Guo, B., Zou, X., Li, Y. & Shi, S. Machine learning assisted materials design and discovery for rechargeable batteries. Energy Storage Mater. 31, 434–450 (2020).
Guo, H., Wang, Q., Stuke, A., Urban, A. & Artrith, N. Accelerated atomistic modeling of solid-state battery materials with machine learning. Front. Energy Res. 9, 695902 (2021).
Liu, H., Ma, S., Wu, J., Wang, J. & Wang, X. Recent advances in screening lithium solid-state electrolytes through machine learning. Front. Energy Res. 9, 639741 (2021).
Deng, L. The MNIST database of handwritten digit images for machine learning research. IEEE Signal Proc. Mag. 29, 141–142 (2012).
Barker, J., Watanabe, S., Vincent, E. & Trmal, J. The fifth ‘CHiME’ Speech Separation and Recognition Challenge: Dataset, task and baselines, In Interspeech 2018 (Hyderabad, India, 2018).
Jain, A. et al. The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Kirklin, S. et al. The Open Quantum Materials Database (OQMD): assessing the accuracy of DFT formation energies. npj Comput. Mater. 1, 15010 (2015).
Saal, J. E., Kirklin, S., Aykol, M., Meredig, B. & Wolverton, C. Materials design and discovery with high-throughput density functional theory: The Open Quantum Materials Database (OQMD). JOM 65, 1501–1509 (2013).
Curtarolo, S. et al. AFLOWLIB.ORG: a distributed materials properties repository from high-throughput ab initio calculations. Comput. Mater. Sci. 58, 227–235 (2012).
Ortiz, C., Eriksson, O. & Klintenberg, M. Data mining and accelerated electronic structure theory as a tool in the search for new functional materials. Comput. Mater. Sci. 44, 1042–1049 (2009).
Landis, D. D. et al. The computational materials repository. Comput. Sci. Eng. 14, 51–57 (2012).
Draxl, C. & Scheffler, M. NOMAD: The FAIR concept for big data-driven materials science. MRS Bull. 43, 676–682 (2018).
Meredig, B. et al. Combinatorial screening for new materials in unconstrained composition space with machine learning. Phys. Rev. B 89, 094104 (2014).
Ye, W., Chen, C., Wang, Z., Chu, I.-H. & Ong, S. P. Deep neural networks for accurate predictions of crystal stability. Nat. Commun. 9, 3800 (2018).
Zeng, S. et al. Atom table convolutional neural networks for an accurate prediction of compounds properties. npj Comput. Mater. 5, 84 (2019).
Bartel, C. J. et al. A critical examination of compound stability predictions from machine-learned formation energies. Npj Comput. Mater. 6, 97 (2020).
Goodall, R. E. A. & Lee, A. A. Predicting materials properties without crystal structure: deep representation learning from stoichiometry. Nat. Commun. 11, 6280 (2020).
Huang, L. & Ling, C. Practicing deep learning in materials science: an evaluation for predicting the formation energies. J. Appl. Phys. 128, 124901 (2020).
Jha, D. et al. ElemNet: deep learning the chemistry of materials from only elemental composition. Sci. Rep. 8, 17593 (2018).
Ceder, G., Hautier, G., Jain, A. & Ong, S. P. Recharging lithium battery research with first-principles methods. MRS Bull. 36, 185–191 (2011).
Jain, A., Hautier, G., Ong, S. P., Dacek, S. & Ceder, G. Relating voltage and thermal safety in Li-ion battery cathodes: a high-throughput computational stu. Phys. Chem. Chem. Physc. 17, 5942–5953 (2015).
Nolan, A., Zhu, Y., he, X., Bai, Q. & Mo, Y. Computation-accelerated design of materials and interfaces for all-solid-state lithium-ion batteries. Joule 2, 2016–2046 (2018).
Aykol, M. et al. High-throughput computational design of cathode coatings for Li-ion batteries. Nat. Commun. 7, 13779 (2016).
Xiao, Y., Miara, L. J., Wang, Y. & Ceder, G. Computational screening of cathode coatings for solid-state batteries. Joule 3, 1252–1275 (2019).
Henkelman, G. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Ceder, G., Ong, S. P. & Wang, Y. Predictive modeling and design rules for solid electrolytes. MRS Bull. 43, 746–751 (2018).
Deng, Z., Zhu, Z., Chu, I.-H. & Ong, S. P. Data-driven first-principles methods for the study and design of alkali superionic conductors. Chem. Mater. 29, 281–288 (2017).
Bergerhoff, G., Hundt, R., Sievers, R. & Brown, I. D. The inorganic crystal structure database. J. CHem. Inf. Comput. Sci. 23, 66–69 (1983).
Gražulis, S. et al. Crystallography Open Database (COD): an open-access collection of crystal structures and platform for world-wide collaboration. Nucleic Acids Res. 40, D420–D427 (2011).
Villars, P., Cenzual, K., Gladyshevskii, R. & Iwata, S. Pauling file—towards a holistic view. Chem. Met. Alloy. 11, 43–76 (2018).
Saha, B. & Goebel, K. A.P.D.R. http://ti.arc.nasa.gov/project/prognostic-data-repository (2007).
Bole, B., Kulkarni, C. & Daigle, M. Adaptation of an electrochemistry-based Li-ion battery model to account for deterioration observed under randomized use, in: F In: Proc. Annual Conference of the Prognostics and Health Management Society, Fort Worth, TX, USA, 29, (2014).
Hogge, E. F., Bole, B. M., Vazquez, S. L. & Celaya, J. Verification of a remaining flying time prediction system for small electric aircraft, In: Proc. Annual Conference of the Prognostics and Health Management Society (2015).
Barkholtz, H. M., Fresquez, A., Chalamala, B. R. & Ferreira, S. R. A database for comparative electrochemical performance of commercial 18650-format lithium-ion cells. J. Electrochem. Soc. 164, A2697 (2017).
Mennen, S. M. et al. The evolution of high-throughput experimentation in pharmaceutical development and perspectives on the future. Org. Process Res. Dev. 23, 1213–1242 (2019).
Shevlin, M. Practical high-throughput experimentation for chemists. ACS Med. Chem. Lett. 8, 601–607 (2017).
Hahn, R. et al. High-throughput battery materials testing based on test cell arrays and dispense/jet printed electrodes. Microsyst. Technol. 25, 1137–1149 (2019).
Liu, P. et al. High throughput materials synthesis methods for lithium ion battery research. J. Materiomics 3, 202–208 (2017).
Mcginn, P. J. Combinatorial electrochemistry—processing and characterization for materials discovery. Mater. Discov. 1, 38–53 (2015).
McGinn, P. J. Thin-film processing routes for combinatorial materials investigations—a review. ACS Comb. Sci. 21, 501–515 (2019).
Yanase, I., Ohtaki, T. & Watanabe, M. Application of combinatorial process to LiCo1−XMnXO2 (0≦X≦0.2) powder synthesis. Solid State Ion. 151, 189–196 (2002).
Fujimoto, K., Takada, K., Sasaki, T. & Watanabe, M. Combinatorial approach for powder preparation of pseudo-ternary system LiO0.5-X-TiO2 (X: FeO1.5, CrO1.5 and NiO). Appl. Surface Sci. 223, 49–53 (2004).
Brown, C. R., McCalla, E., Watson, C. & Dahn, J. R. Combinatorial study of the Li−Ni−Mn−Co oxide pseudoquaternary system for use in Li−ion battery materials research. ACS Comb. Sci. 17, 381–391 (2015).
Carey, G. H. & Dahn, J. R. Combinatorial synthesis of mixed transition metal oxides for lithium-ion batteries. ACS Comb. Sci. 13, 186–189 (2011).
McCalla, E., Rowe, A. W., Camardese, J. & Dahn, J. R. The role of metal site vacancies in promoting Li–Mn–Ni–O layered solid solutions. Chem. Mater. 25, 2716–2721 (2013).
Adhikari, T. et al. Development of high-throughput methods for sodium-ion battery cathodes. ACS Comb. Sci. 22, 311–318 (2020).
Su, L., Ferrandon, M., Kowalski, J. A., Vaughey, J. T. & Brushett, F. R. Electrolyte development for non-aqueous redox flow batteries using a high-throughput screening platform. J. Electrochem. Soc. 161, A1905–A1914 (2014).
Beal, M. S. et al. High throughput methodology for synthesis, screening, and optimization of solid state lithium ion electrolytes. ACS Comb. Sci. 13, 375–381 (2010).
Yada, C. et al. A high-throughput approach developing lithium-niobium-tantalum oxides as electrolyte/cathode interlayers for high-voltage all-solid-state lithium batteries. J. Electrochem. Soc. 162, A722 (2015).
Matsuda, S., Nishioka, K. & Nakanishi, S. High-throughput combinatorial screening of multi-component electrolyte additives to improve the performance of Li metal secondary batteries. Sci. Rep. 9, 6211 (2019).
Matsubara, M., Suzumura, A., Ohba, N. & Asahi, R. Identifying superionic conductors by materials informatics and high-throughput synthesis. Commun. Mater. 1, 5 (2020).
Whitacre, J. F. et al. An autonomous electrochemical test stand for machine learning informed electrolyte optimization. J. Electrochem. Soc. 166, A4181–A4187 (2019).
Dave, A., Gering, K. L., Mitchell, J. M., Whitacre, J. & Viswanathan, V. Benchmarking conductivity predictions of the advanced electrolyte model (AEM) for aqueous systems. J. Electrochem. Soc. 167, 013514 (2019).
Dave, A. et al. Autonomous discovery of battery electrolytes with robotic experimentation and machine learning. Cell Rep. Phys. Sci. 1, 100264 (2020).
Huang, L. & Ling, C. Representing multiword chemical terms through phrase-level preprocessing and word embedding. ACS Omega 4, 18510–18519 (2019).
Olivetti, E. et al. Data-driven materials research enabled by natural language processing. Appl. Phys. Rev. 7, 041317 (2020).
Kononova, O. et al. Opportunities and challenges of text mining in materials research. iScience 24, 102155 (2021).
Kim, E. et al. Machine-learned and codified synthesis parameters of oxide materials. Sci. Data 4, 170127 (2017).
Kim, E. et al. Materials synthesis insights from scientific literature via text extraction and machine learning. Chem. Mater. 29, 9436 (2017).
He, T. et al. Similarity of precursors in solid-state synthesis as text-mined from scientific literature. Chem. Mater. 32, 17399–17404 (2020).
Mahbub, R. et al. Text mining for processing conditions of solid-state battery electrolytes. Electrochem. Commun. 121, 106860 (2020).
Huang, S. & Cole, J. M. A database of battery materials auto-generated using ChemDataExtractor. Sci. Data 7, 260 (2020).
Batra, R., Pilania, G., Uberuaga, B. P. & Ramprasad, R. Multifidelity information fusion with machine learning: a case study of dopant formation energies in Hafnia. ACS Appl. Mater. Interfaces 11, 24906–24918 (2019).
Zhang, Y. & Ling, C. A strategy to apply machine learning to small datasets in materials science. Npj Comput. Mater. 4, 25 (2017).
Chen, C., Zuo, Y., Ye, W., Li, X. & Ong, S. P. Learning properties of ordered and disordered materials from multi-fidelity data. Nat. Comput. Sci. 1, 46–53 (2021).
Fujimura, K. et al. Accelerated materials design of lithium superionic conductors based on first-principles calculations and machine learning algorithms. Adv. Energy Mater. 3, 980 (2013).
Bachman, J. C. et al. Inorganic solid-state electrolytes for lithium batteries: mechanisms and properties governing ion conduction. Chem. Rev. 116, 140–162 (2016).
Thangadurai, V., Narayanan, S. & Pinzaru, D. Garnet-type solid-state fast Li ion conductors for Li batteries: critical review. Chem. Soc. Rev. 43, 4714–4727 (2014).
Kim, E., Huang, K., Kononova, O., Ceder, G. & Olivetti, E. Distilling a materials synthesis ontology. Matter 1, 8–12 (2019).
Raccuglia, P. et al. Machine-learning-assisted materials discovery using failed experiments. Nature 553, 73–77 (2016).
Jia, X. et al. Anthropogenic biases in chemical reaction data hinder exploratory inorganic synthesis. Nature 573, 251–255 (2019).
Artrith, N. et al. Best practices in machine learning for chemistry. Nat. Chem. 13, 505–508 (2021).
Wolpert, D. H. The lack of a priori distinctions between learning algorithms. Neural Comput. 8, 1341–1390 (1996).
Sendek, A. D. et al. Holistic computational structure screening of more than 12,000 candidates for solid lithium-ion conductor materials. Energy Environ. Sci. 10, 306–320 (2017).
Liu, B. et al. Rationalizing the interphase stability of Li|doped-Li7La3Zr2O12 via automated reaction screening and machine learning. J. Mater. Chem. A 7, 19961–19969 (2019).
Balachandran, P. V., Theiler, J., Rondinelli, J. M. & Lookman, T. Materials prediction via classification learning. Sci. Rep. 5, 13285 (2015).
Isayev, O. et al. Materials cartography: representing and mining materials space using structural and electronic fingerprints. Chem. Mater. 27, 735–743 (2015).
Zhou, Q. et al. Learning atoms for materials discovery. Proc. Natl Acad. Sci. 115, E6411–E6417 (2018).
Long, C. J., Hattrick-Simpers, J., Murakami, M., Srivastava, R. C. & Takeuchi, I. Rapid structural mapping of ternary metallic alloy systems using the combinatorial approach and cluster analysis. Rev. Sci. Instrum. 78, 072217 (2007).
Kandasamy, K. et al. Tuning hyperparameters without grad students: scalable and robust bayesian optimisation with dragonfly. J. Mach. Learn. Res. 21, 1–27 (2020).
Auer, P. Using confidence bounds for exploitation-exploration trade-offs. J. Mach. Learn. Res. 3, 397–422 (2002).
Hennig, P. & Schuler, C. J. Entropy search for information-efficient global optimization. J. Mach. Learn. Res. 13, 1809–1837 (2012).
Kushner, H. J. A new method of locating the maximum point of an arbitrary multipeak curve in the presence of noise. J. Fluid Eng. 86, 97–106 (1964).
Jones, D. R., Schonlau, M. & Welch, W. J. Efficient global optimization of expensive black-box functions. J. Glob. Optim. 13, 455–492 (1998).
Qin, C., Klabjan, D. & Russo, D. Improving the Expected Improvement Algorithm, In: Conference on Neural Information Processing Systems (2017).
Frazier, P., Powell, W. & Dayanik, S. The knowledge-gradient policy for correlated normal beliefs. Inf. J. Comput. 21, 599 (2009).
Thompson, W. R. On the likelihood that one unknown probability exceeds another in view of the evidence of two samples. Biometrika 25, 285–294 (1933).
Sonek, J. et al. Scalable Bayesian optimization using deep neural networks. In: International Conference on Machine Learning https://arxiv.org/abs/1502.05700 (2015).
Hutter, F., Hoos, H. H. & Leyton-Brown, K. Sequential Model-Based Optimization for General Algorithm Configuration. In: International Conference on Learning and Intelligent Optimization (2011).
Rasmussen, C. E. & Williams, C. K. Gaussian Processes for Machine Learning (University Press Group Limited, 2006).
Homma, K. et al. Optimization of a heterogeneous ternary Li3PO4–Li3BO3–Li2SO4 mixture for Li-ion conductivity by machine learning. J. Phys. Chem. C. 124, 12865–12870 (2020).
Harada, M. et al. Bayesian-optimization-guided experimental search of NASICON-type solid electrolytes for all-solidstate Li-ion batteries. J. Mater. Chem. A 8, 15103–15109 (2020).
Nakayama, M. et al. Data-driven materials exploration for Li-ion conductive ceramics by exhaustive and informatics-aided computations. Chem. Rec. 18, 1–9 (2018).
Zhang, Z. et al. New horizons for inorganic solid state ion conductors. Energy Environ. Sci. 11, 1945–1976 (2018).
Manthiram, A., Yu, X. & Wang, S. Lithium battery chemistries enabled by solid-state electrolytes. Nat. Rev. Mater. 2, 16103 (2017).
Famprikis, T., Canepa, P., Dawson, J. A., Islam, M. S. & Masquelier, C. Fundamentals of inorganic solid-state electrolytes for batteries. Nat. Mater. 18, 1278–129 (2019).
Ibarra, J. et al. Influence of composition on the structure and conductivity of the fast ionic conductors La2/3−xLi3xTiO3 (0.03 ≤ x ≤ 0.167). Solid State Ion. 134, 219–228 (2000).
Boulineau, S., Courty, M., Tarascon, J.-M. & Viallet, V. Mechanochemical synthesis of Li-argyrodite Li6PS5X (X = Cl, Br, I) as sulfur-based solid electrolytes for all solid state batteries application. Solid State Ion. 221, 1–5 (2012).
Kumazaki, S. et al. High lithium ion conductive Li7La3Zr2O12 by inclusion of both Al and Si. Electrochem. Commun. 13, 509–512 (2011).
Li, W. et al. Li+ ion conductivity and diffusion mechanism in α-Li3N and β-Li3N. Energy Environ. Sci. 3, 1524–1530 (2010).
Aono, H., Sugimoto, E., Sadaoka, Y., Imanaka, N. & Adachi, G.-Y. Ionic conductivity and sinterability of lithium titanium phosphate system. Solid State Ion. 40/41, 38–42 (1990).
Kamaya, N. et al. A lithium superionic conductor. Nat. Mater. 10, 682–686 (2011).
Mizuno, F., Hayashi, A., Tadanaga, K. & Tatsumisago, M. New, highly ion-conductive crystals precipitated from Li2S-P2S5 glasses. Adv. Mater. 17, 918–921 (2005).
Jalem, R., Nakayama, M. & Kasuga, T. An efficient rule-based screening approach for discovering fast lithium ion conductors using density functional theory and artificial neural network. J. Mater. Chem. A 2, 720–734 (2014).
Jalem, R., Kimura, M., Nakayama, M. & Kasuga, T. Informatics-aided density functional theory study on the Li ion transport of tavorite-type LiMTO4F (M3+–T5+, M2+–T6+). J. Chem. Inf. Model. 6, 1158–1168 (2015).
Düvel, A. et al. Is Geometric frustration-induced disorder a recipe for high ionic conductivity? J. Am. Chem. Soc. 139, 5842–5848 (2017).
Stefano, D. D. et al. Superionic diffusion through frustrated energy landscape. Chem 5, 2450–2460 (2019).
Wang, Y. et al. Design principles for solid-state lithium superionic conductors. Nat. Mater. 14, 1026–1031 (2015).
Zhu, Z., Chu, I.-H. & Ong, S. P. Li3Y(PS4)2 and Li5PS4Cl2: new lithium superionic conductors predicted from silver thiophosphates using efficiently tiered ab initio molecular dynamics simulations. Chem. Mater. 29, 2474–2484 (2017).
Xie, T. & Grossmann, J. Crystal graph convolutional neural networks for an accurate and interpretable prediction of material properties. Phys. Rev. Lett. 120, 145301 (2018).
Xie, T., France-Lanord, A., Wang, Y., Shao-Horn, Y. & Grossman, J. C. Graph dynamical networks for unsupervised learning of atomic scale dynamics in materials. Nat. Commun. 10, 2667 (2019).
Xie, T. & Grossman, J. C. Hierarchical visualization of materials space with graph convolutional neural networks. J. Chem. Phys. 149, 174111 (2018).
Lee, J. & Asahi, R. Transfer learning for materials informatics using crystal graph convolutional neural network. Comput. Mater. Sci. 190, 110314 (2021).
Jalem, R. et al. Bayesian-driven first-principles calculations for accelerating exploration of fast ion conductors for rechargeable battery application. Sci. Rep. 8, 5845 (2018).
Cubuk, E. D., Sendek, A. D. & Reed, E. J. Screening billions of candidates for solid lithium-ion conductors: A transfer learning approach for small data. J. Chem. Phys. 150, 214701 (2019).
Sendek, A. D. et al. Machine learning-assisted discovery of solid li-ion conducting materials. Chem. Mater. 31, 342–352 (2019).
Ahmad, Z., Xie, T., Maheshwari, C., Grossman, J. C. & Viswanathan, V. Machine learning enabled computational screening of inorganic solid electrolytes for suppression of dendrite formation in lithium metal anodes. ACS Cent. Sci. 4, 996–1006 (2018).
He, X. et al. Crystal structural framework of lithium super‐ionic conductors. Adv. Energy Mater. 9, 1902078 (2019).
He, X., Zhu, Y. & Mo, Y. Origin of fast ion diffusion in super-ionic conductors. Nat. Commun. 8, 15893 (2017).
Xiong, S. et al. Computation-guided design of LiTaSiO5, a new lithium ionic conductor with sphene structure. Adv. Energy Mater. 9, 1803821 (2019).
Ke, X., Wang, Y., Ren, G. & Yuan, C. Towards rational mechanical design of inorganic solid electrolytes for all-solid-state lithium ion batteries. Energy Storage Mater. 26, 313–324 (2020).
Deng, Z., Wang, Z., Chu, I.-H., Luo, J. & Ong, S. P. Elastic properties of alkali superionic conductor electrolytes from first principles calculations. J. Electrochem. Soc. 163, A67–A74 (2016).
de Jong, M. et al. Charting the complete elastic properties of inorganic crystalline compounds. Sci. Data 2, 150009 (2015).
de Jong, M. et al. A statistical learning framework for materials science: application to elastic moduli of k-nary inorganic polycrystalline compounds. Sci. Rep. 6, 34256 (2016).
Zhu, Y. et al. Accelerating materials discovery with Bayesian optimization and graph deep learning. Mater. Today 51, 126–135 (2021).
Wenzel, S. et al. Direct observation of the interfacial instability of the fast ionic conductor Li10GeP2S12 at the lithium metal anode. Chem. Mater. 28, 2400–2407 (2016).
Liu, Y. et al. Stabilizing the interface of NASICON solid electrolyte against Li metal with atomic layer deposition. ACS Appl. Mater. Interfaces 10, 31240–31248 (2018).
Ma, C. et al. Interfacial stability of Li metal–solid electrolyte elucidated via in situ electron microscopy. Nano Lett. 16, 7030–7036 (2016).
Richards, W. D., Miara, L. J., Wang, Y., Kim, J. C. & Ceder, G. Interface stability in solid-state batteries. Chem. Mater. 28, 266–273 (2016).
Xiao, Y. et al. Understanding interface stability in solid-state batteries. Nat. Rev. Mater. 5, 105–126 (2020).
Zhu, Y., He, X. & Mo, Y. Origin of outstanding stability in the lithium solid electrolyte materials: insights from thermodynamic analyses based on first-principles calculations. ACS Appl. Mater. Interfaces 7, 23685–23693 (2015).
Long, L., Wang, S., Xiao, M. & Meng, Y. Polymer electrolytes for lithium polymer batteries. J. Mater. Chem. A 4, 10038–10069 (2016).
Johan, M. R. & Ibrahim, S. Neural networks for Nyquist plots prediction in a nanocomposite polymer electrolyte (PEO–LiPF6–EC–CNT). Ionics 17, 683 (2011).
Ibrahim, S. & Johan, M. R. Conductivity, thermal and neural network model nanocomposite solid polymer electrolyte (PEO–LiPF6–EC–CNT). Int. J. Electrochem. Sci. 6, 5565–5587 (2011).
Johan, M. R., Yasin, S. M. M. & Ibrahim, S. Bayesian neural networks model for ionic conductivity of nanocomposite solid polymer electrolyte system (PEO–LiCF3SO3–DBP–ZrO2). Int. J. Electrochem. Sci. 7, 222–233 (2011).
Xue, Z., He, D. & Xie, X. Poly(ethylene oxide)-based electrolytes for lithium-ion batteries. J. Mater. Chem. A 3, 19218–19253 (2015).
Hatakeyama-Sato, K., Tezuka, T., Umeki, M. & Oyaizu, K. AI-assisted exploration of superionic glass-type Li+ conductors with aromatic structures. J. Am. Chem. Soc. 142, 3301–3305 (2020).
Hatakeyama-Sato, K., Tezuka, T., Nishikitani, Y., Nishide, H. & Oyaizu, K. Synthesis of lithium-ion conducting polymers designed by machine learning-based prediction and screening. Chem. Lett. 48, 130–132 (2019).
Joshi, R. P. et al. Machine learning the voltage of electrode materials in metal-ion batteries. ACS Appl. Mater. Interfaces 11, 18494–18503 (2019).
Wang, X., Xiao, R., Li, H. & Chen, L. Quantitative structure-property relationship study of cathode volume changes in lithium ion batteries using ab-initio and partial least squares analysis. J. Materiom. 3, 178–183 (2017).
Shandiz, M. A. & Gauvin, R. Application of machine learning methods for the prediction of crystal system of cathode materials in lithium-ion batteries. Comput. Mater. Sci. 117, 270–278 (2016).
Zhang, H., Wang, Z., Ren, J., Liu, J. & Li, J. Ultra-fast and accurate binding energy prediction of shuttle effect-suppressive sulfur hosts for lithium-sulfur batteries using machine learning. Energy Storage Mater. 35, 88–98 (2021).
Sanchez, J. M., Ducastelle, F. & Gratias, D. Generalized cluster description of multicomponent systems. Physica A 128, 334–350 (1984).
Natarajan, A. R. & Van der Ven, A. Machine-learning the configurational energy of multicomponent crystalline solids. Npj Comput. Mater. 4, 56 (2018).
Houchins, G. & Viswanathan, V. An accurate machine-learning calculator for optimization of Li-ion battery cathodes. J. Chem. Phys. 153, 054124 (2020).
Eremin, R. A., Zolotarev, P. N., Ivanshina, O. Y. & Bobrikov, I. A. Li(Ni,Co,Al)O2 cathode delithiation: a combination of topological analysis, density functional theory, neutron diffraction, and machine learning techniques. J. Phys. Chem. C 121, 28293–28305 (2017).
Leung, K. & Budzien, J. L. Ab initio molecular dynamics simulations of the initial stages of solid–electrolyte interphase formation on lithium ion battery graphitic anodes. Phys. Chem. Chem. Phys. 12, 6583–6658 (2009).
Behler, J. & Parrinello, M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 98, 146401 (2007).
Eckhoff, M. et al. Closing the gap between theory and experiment for lithium manganese oxide spinels using a high-dimensional neural network potential. Phys. Rev. B 102, 174102 (2020).
Artrith, N., Urban, A. & Ceder, G. Constructing first-principles phase diagrams of amorphous LixSi using machine-learning-assisted sampling with an evolutionary algorithm. J. Chem. Phys. 148, 241711 (2018).
Deringer, V. L. et al. Towards an atomistic understanding of disordered carbon electrode materials. Chem. Commun. 54, 5988–5991 (2018).
Onat, B., Cubuk, E. D., Malone, B. D. & Kaxiras, E. Implanted neural network potentials: application to Li-Si alloys. Phys. Rev. B 97, 094106 (2018).
Fujikake, S. et al. Gaussian approximation potential modeling of lithium intercalation in carbon nanostructures. J. Chem. Phys. 148, 241714 (2018).
Huang, J.-X., Csányi, G., Zhao, J.-B., Cheng, J. & Deringer, V. L. First-principles study of alkali-metal intercalation in disordered carbon anode materials. J. Mater. Chem. A 7, 19070–19080 (2019).
Wang, C., Aoyagi, K., Wisesa, P. & Mueller, T. Lithium ion conduction in cathode coating materials from on-the-fly machine learning. Chem. Mater. 32, 3741–3752 (2020).
Park, C. W. et al. Accurate and scalable graph neural network force field and molecular dynamics with direct force architecture. Npj Comput. Mater. 7, 73 (2021).
Miwa, K. & Asahi, R. Molecular dynamics simulations of lithium superionic conductor Li10GeP2S12 using a machine learning potential. Solid State Ion. 361, 115567 (2021).
Li, W., Ando, Y., Minamitani, E. & Watanabe, S. Study of Li atom diffusion in amorphous Li3PO4 with neural network potential. J. Chem. Phys. 147, 214106 (2017).
Huang, J. et al. Deep Potential generation scheme and simulation protocol for the Li10GeP2S12-type superionic conductors. J. Chem. Phys. 154, 094703 (2021).
Deng, Z., Chen, C., Li, X.-G. & Ong, S. P. An electrostatic spectral neighbor analysis potential for lithium nitride. npj Comput. Mater. 5, 75 (2019).
Miwa, K. & Asahi, R. Molecular dynamics simulations with machine learning potential for Nb-doped lithium garnet-type oxide Li7−xLa3(Zr2−xNbx)O12. Phys. Rev. Mater. 2, 105404 (2018).
Hajibabaei, A., Myung, C. W. & Kim, K. S. Sparse Gaussian process potentials: application to lithium diffusivity in superionic conducting solid electrolytes. Phys. Rev. B 103, 214102 (2021).
Marcolongo, A., Binninger, T., Zipoli, F. & Laino, T. Simulating diffusion properties of solid-state electrolytes via a neural network potential: performance and training scheme. ChemSystemsChem 2, e1900031 (2019).
Qi, J. et al. Bridging the gap between simulated and experimental ionic conductivities in lithium superionic conductors. Mater. Today Phys. 1, 110463 (2021).
Miwa, K. & Ohno, H. Interatomic potential construction with self-learning and adaptive database. Phys. Rev. Mater. 1, 053801 (2017).
Bartók, A. P., Payne, M. C., Kondor, R. & Csányi, G. Gaussian approximation potentials: the accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 104, 136403 (2010).
Thompson, A. P., Swiler, L. P., Trott, C. R., Foiles, S. M. & Tucker, G. J. Spectral neighbor analysis method for automated generation of quantum-accurate interatomic potentials. J. Comput. Phys. 285, 316–330 (2015).
Shapeev, A. V. Moment tensor potentials: a class of systematically improvable interatomic potentials. Multiscale Model. Simul. 14, 1153–1173 (2016).
Molinari, N. et al. Spectral denoising for unsupervised analysis of correlated ionic transport. Phys. Rev. Lett. 127, 025901 (2021).
Chen, C., Lu, Z. & Ciucci, F. Data mining of molecular dynamics data reveals Li diffusion characteristics in garnet Li7La3Zr2O12. Sci. Rep. 7, 40769 (2017).
Magdău, I.-B. & Miller, T. F. III Machine learning solvation environments in conductive polymers: application to ProDOT-2Hex with solvent swelling. Macromolecules 54, 3377–3387 (2021).
Kahle, L., Musaelian, A., Marzari, N., Molinari, N. & Kozinsky, B. Unsupervised landmark analysis for jump detection in molecular dynamics simulations. Phys. Rev. Mater. 3, 055404 (2019).
Bartel, C. J. et al. New tolerance factor to predict the stability of perovskite oxides and halides. Sci. Adv. 5, eaav069 (2019).
Stenev, V. et al. Machine learning modeling of superconducting critical temperature. npj Comput. Mater. 4, 29 (2018).
Ziletti, A., Kumar, D., Scheffler, M. & Ghiringhelli, L. M. Insightful classification of crystal structures using deep learning. Nat. Commun. 9, 2775 (2018).
Wang, S., Pillai, H. S. & Xin, H. Bayesian learning of chemisorption for bridging the complexity of electronic descriptors. Nat. Commun. 11, 6132 (2020).
Huang, L. & Ling, C. Leveraging transfer learning and chemical principles towards interpretable materials properties. J. Chem. Inform. Model. 61, 4200 (2021).
Sun, N., Yi, J., Zhang, P., Shen, H. & Zhai, H. Deep learning topological invariants of band insulators. Phys. Rev. B 98, 085402 (2018).
Zhang, P., Shen, H. & Zhai, H. Machine learning topological invariants with neural networks. Phys. Rev. Lett. 120, 066401 (2018).
Wang, Y., Wagner, N. & Rondinelli, J. M. Symbolic regression in materials science. MRS Commun. 9, 793–805 (2019).
Schmidt, M. & Lipson, H. Distilling free-form natural laws from experimental data. Science 324, 81–85 (2009).
Brunton, S. L., Proctor, J. L. & Kutz, J. N. Discovering governing equations from data by sparse identification of nonlinear dynamical systems. Proc. Natl Acad. Sci. 113, 3932–3937 (2016).
Rudy, S. H., Brunton, S. L., Proctor, L. L. & Kutz, J. N. Data-driven discovery of partial differential equations. Sci. Adv. 3, e1602614 (2017).
Mark, C. et al. Bayesian model selection for complex dynamic systems. Nat. Commun. 9, 1803 (2018).
Wasserman, L. Bayesian model selection and model averaging. J. Math. Psychol. 44, 92–107 (2000).
Park, J. et al. Fictitious phase separation in Li layered oxides driven by electro-autocatalysis. Nat. Mater. 20, 991 (2021).
Gao, Y. et al. Classical and emerging characterization techniques for investigation of ion transport mechanisms in crystalline fast ionic conductors. Chem. Rev. 120, 5954–6008 (2020).
Jiang, Z. et al. Machine-learning-revealed statistics of the particle-carbon/binder detachment in lithium-ion battery cathodes. Nat. Commun. 11, 2310 (2020).
Furat, O. et al. Mapping the architecture of single lithium ion electrode particles in 3D, using electron backscatter diffraction and machine learning segmentation. J. Power Sources 483, 229148 (2021).
Petrich, L. et al. Crack detection in lithium-ion cells using machine learning. Comput. Mater. Sci. 136, 297–305 (2017).
Dixit, M. B. et al. Synchrotron imaging of Li metal anodes in solid state batteries aided by machine learning. ACS Appl. Energy Mater. 3, 9534–9542 (2020).
Baliyan, A. & Imai, H. Machine learning based analytical framework for automatic hyperspectral Raman analysis of lithium-ion battery electrodes. Sci. Rep. 9, 18241 (2019).
Kondo, R., Yamakawa, S., Masuoka, Y., Tajima, S. & Asahi, R. Microstructure recognition using convolutional neural networks for prediction of ionic conductivity in ceramics. Acta Mater. 141, 29–38 (2017).
Gao, X. et al. Designed high-performance lithium-ion battery electrodes using a novel hybrid model-data driven approach. Energy Storage Mater. 36, 435–458 (2021).
Xu, H. et al. Guiding the design of heterogeneous electrode microstructures for Li-ion batteries: microscopic imaging, predictive modeling, and machine learning. Adv. Energy Mater. 11, 2003908 (2021).
Duquesnoy, M., Lombardo, T., Chouchane, M., Primo, E. N. & Franco, A. A. Data-driven assessment of electrode calendering process by combining experimental results, in silico mesostructures generation and machine learning. J. Power Sources 480, 229103 (2020).
Gao, T. & Lu, W. Physical model and machine learning enabled electrolyte channel design for fast charging. J. Electrochem. Soc. 167, 110519 (2020).
Takagishi, Y., Yamanaka, T. & Yamaue, T. Machine learning approaches for designing mesoscale structure of Li-ion battery electrodes. Batteries 5, 54 (2019).
Li, T. et al. Cost, performance prediction and optimization of a vanadium flow battery by machine-learning. Energy Environ. Sci. 13, 4353–4361 (2020).
Farmann, A., Waag, W., Morongiu, A. & Sauer, D. U. Critical review of on-board capacity estimation techniques for lithium-ion batteries in electric and hybrid electric vehicles. J. Power Sources 281, 114–130 (2015).
Ng, M.-F., Zhao, J., Yan, Q., Conduit, G. & Seh, Z. W. Predicting the state of charge and health of batteries using data-driven machine learning. Nat. Mach. Intell. 2, 161–170 (2020).
Roman, D., Saxuena, S., Robu, V., Pecht, M. & Flynn, D. Machine learning pipeline for battery state-of-health estimation. Nat. Mach. Intell. 3, 447–456 (2021).
Aykol, M. et al. Combining physics and machine learning to predict battery lifetime. J. Electrochem. Soc. 168, 030525 (2021).
Vidal, C., Malysz, P., Kollmeyer, P. & Emadi, A. Machine learning applied to electrified vehicle battery state of charge and state of health estimation: state-of-the-art. IEEE Access 8, 52796–52814 (2020).
Attia, P. M. et al. Closed-loop optimization of fast-charging protocols for batteries with machine learning. Nature 578, 397–402 (2020).
Severson, K. A. et al. Data-driven prediction of battery cycle life before capacity degradation. Nat. Energy 4, 383–391 (2019).
Soedarmadji, E., Stein, H. S., Suram, S. K., Guevarra, D. & Gregoire, J. M. Tracking materials science data lineage to manage millions of materials experiments and analyses. Npj Comput. Mater. 5, 79 (2019).
Liu, S. et al. An infrastructure with user-centered presentation data model for integrated management of materials data and services. Npj Comput. Mater. 7, 88 (2021).
Acknowledgements
I wish to appreciate the support from my colleagues, Debasish Banerjee and Ryuta Sugiura, for their continuous support on the research work.
Author information
Authors and Affiliations
Contributions
The author is responsible for conceiving the idea, composing the review and writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ling, C. A review of the recent progress in battery informatics. npj Comput Mater 8, 33 (2022). https://doi.org/10.1038/s41524-022-00713-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-022-00713-x
This article is cited by
-
A document-level information extraction pipeline for layered cathode materials for sodium-ion batteries
Scientific Data (2024)
-
Recent advances in functional materials and devices for Zn-Ion hybrid supercapacitors
NPG Asia Materials (2024)
-
The value of negative results in data-driven catalysis research
Nature Catalysis (2023)
-
Fog robotics-based intelligence transportation system using line-of-sight intelligent transportation
Multimedia Tools and Applications (2023)
-
Growth of TiS2 nanoflakes using CVD approach for sodium-ion battery application
Journal of Nanoparticle Research (2023)
