
Abstract
We present Parameter Quantification Network (PQ-Net), a regression deep convolutional neural network providing quantitative analysis of powder X-ray diffraction patterns from multi-phase systems. The network is tested against simulated and experimental datasets of increasing complexity with the last one being an X-ray diffraction computed tomography dataset of a multi-phase Ni-Pd/CeO2-ZrO2/Al2O3 catalytic material system consisting of ca. 20,000 diffraction patterns. It is shown that the network predicts accurate scale factor, lattice parameter and crystallite size maps for all phases, which are comparable to those obtained through full profile analysis using the Rietveld method, also providing a reliable uncertainty measure on the results. The main advantage of PQ-Net is its ability to yield these results orders of magnitude faster showing its potential as a tool for real-time diffraction data analysis during in situ/operando experiments.
Similar content being viewed by others


Introduction
Over the past decade, advancements in X-ray sources, optics and detector technologies have led to a dramatic increase in the volume and data quality of experimental powder diffraction patterns1,2,3,4,5,6,7. These technical advances are beginning to make high-throughput powder diffraction measurements a reality not just at synchrotron facilities but also at the laboratory8,9. It is now not uncommon to acquire diffraction patterns with sufficient signal-to-noise ratio in matters of minutes with laboratory diffractometers and in milliseconds at X-ray diffraction (XRD) dedicated beamlines at synchrotron facilities. In situ and operando ultra-fast and/or spatially-resolved multi-dimensional XRD experiments of functional materials and devices previously considered technically infeasible have already been demonstrated10,11. For example, we have previously demonstrated the first 5D operando tomographic diffraction imaging experiment (three spatial, one scattering and one dimension to denote time/imposed state) to study a multi-component catalytic reactor for the partial oxidation of methane12. However, these advances come at a cost and this is related to the handling of the big data collected during these experiments.
It is now possible to acquire many Terabytes (TBs) of XRD data per dynamic experiment and this is expected to increase significantly with the advent of the fourth generation synchrotron facilities all around the world, such as the Extremely Brilliant Source (EBS) of the European Synchrotron Radiation Facilities (ESRF) and MAX IV (Sweden) and the scheduled upgrades for Diamond-II (United Kingdom), Petra IV (Germany) and Advanced Photon Source-Upgrade (USA)13. It is currently well-accepted that it is the data analysis that is emerging as the bottleneck for measurement science and not the data acquisition and/or the experiment itself. Conventional data analysis methods, such as least-squares minimisation approaches, are not able to keep up with the data collections rates and there is a need for alternative methods which can provide both fast and accurate results14. As an example, the Rietveld method, often employed in XRD data when performing full profile analysis, can yield invaluable physicochemical information regarding the material system under investigation (e.g., lattice parameters, crystallite sizes/strain, atom/site occupancies and weight percentages for mixed phase systems) but does not scale well with big data. Equally important is the data crunching/visualization capabilities which are not developed in tandem with the data acquisition capabilities and as a result dynamic beamtime experiments are largely driven ‘blind’.
In recent years, deep learning methods involving convolutional neural networks (CNNs) have gained popularity and attracted a lot of attention due to the aforementioned properties and also due to their unique scalability (i.e., ability to handle big data)15,16,17. Although primarily explored for applications in medical imaging and tomography18,19, their potential for materials science and especially for spectroscopic/scattering techniques is gaining momentum20,21,22,23,24,25,26,27,28. Regarding XRD, previous studies have mainly focused on developing CNNs for crystal structure prediction, space group classification and phase identification19,29,30,31,32,33,34. For example, Park et al. demonstrated a classification CNN model that can extract crystal-system and space-group information from an XRD pattern29. Lee et al. proposed a CNN structure used for XRD pattern identification, which also has potential to predict phase fractions19. Wang et al. demonstrated a phase identification CNN which was trained with simulated patterns with noise extracted from experimental data34. Tatlier used an Artificial Neural Network (ANN) to predict the structures of crystalline materials and zeolites from XRD patterns and it was claimed that the ANN performed better than manual regression35. There are also some algorithms that can achieve phase identification without neural networks, but they are generally less efficient and/or less accurate than neural network models36,37,38,39,40,41. It should be pointed out though that most studies focus on developing classification models for phase identification; predicting whether a phase is present or not in an XRD pattern.
In this work, we demonstrate a neural network for parameter quantification (PQ) that we term PQ-Net; a regression CNN model applied in 1D powder diffraction data for extracting physico-chemical information. Specifically, PQ-Net is trained with simulated 1D XRD patterns and it is shown that it can yield accurate predictions for scale factors, crystallite sizes and lattice parameters for both simulated and experimental XRD data. For the latter, a very challenging X-ray diffraction computed tomography (XRD-CT) dataset was chosen in order to test the performance of the network and its feasibility for deployment for real world applications. We focus on the scale factors, lattice parameters and crystallite sizes, because they are most often sufficient to describe and understand materials and process performance under operating conditions12,42,43,44,45,46,47,48. This exemplar dataset was acquired using a multi-component Ni-Pd/CeO2-ZrO2/Al2O3 catalyst and consists of ca. 20,000 diffraction patterns. The results from the analysis of this five-phase system are also compared with results obtained using the Rietveld method with the TOPAS software49.
Results
Single-phase PQ-Net architecture and simulated XRD-CT dataset
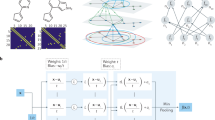
The architecture of the PQ-Net for a single-phase of cubic symmetry is presented in Fig. 1. The PQ-Net architecture can be divided into three main parts. The first part (pattern-block), which decreases the size of the XRD pattern and extracts local features, involves three convolutional layers (128 filters, kernel size of 35 and stride equal to one) followed by a max-pooling layer (stride equal to two). The second part, which is replicated for each crystalline phase used in the model, includes a sequence of five convolutional and max-pooling layers ending with a flatten layer; this extracts features from the XRD pattern related to the specific phase (phase-block). The third and final part (parameter-block) contains two fully connected (dense) layers ending with one single output per phase parameter. A glossary explaining the various layers in more depth is available in the Supporting Information (see also Supplementary Fig. 1 and Supplementary Tables 1–2).

CONV represents 1-D convolutional layers, Pool represents max-pooling layers, FC represents fully connected layers, and Dropout represents dropout layers with 10% dropout rate. The network consists of eight convolutional layers, six max-pooling layers, one flatten layer, nine fully connected layers and six dropout layers in total. There are three routes connected to the flatten layer which give predictions for scale factor, crystallite size and lattice parameter a, respectively. Each route has three fully connected layers whose scales are shown in the figure. The number of filters, kernel sizes and the stride of convolutional layers are also given in the figure. All max-pooling layers have stride equal to 2.
The parameter-block part of the architecture contained the majority of the trained weights so our initial work focused on minimizing both the depth and width of the dense layers. It was found that the width of the second dense layer has to be doubled for the crystallite size and lattice parameters as these are more challenging parameters to quantify compared to scale factors. A dropout layer has been used after each fully connected layer (apart from the final layers) to prevent overfitting. Regarding the training process, it was seen that using mean absolute error (MAE) as the loss function instead of mean squared error (MSE) yielded a more accurate model; this is not surprising as MAE is known to better handle outliers present in the training data. This feature is crucial as there are diffraction patterns in the training data that contain low intensity peaks and the network has to adapt and learn to ignore them. It should be noted that the various parameters were normalised prior to the training of the network since their magnitude varied significantly (i.e., between scale factors, lattice parameters and crystallite sizes). Adam was used as the optimisation algorithm and the learning rate, after initial tuning, was set to 0.0005 for all case studies presented in this work50. In material science aspect
As a proof-of-concept study, the PQ-Net was first trained with noiseless and zero-background XRD patterns of a Ni fcc structure (ICSD: 64989) simulated with the TOPAS software v749. These diffraction patterns were used as the training data with a 10 % validation split. The impact of the diffraction library size (i.e. size of the training data) was investigated and the results are summarised in Supplementary Table 3; there was no apparent change in performance with libraries containing more than 100 K patterns. There is also a sudden drop in MAE above 10 K patterns which implies that accurate results could be obtained with libraries as small as 20 K. It should also be pointed out that the MAE values provide an indication of accuracy regarding the predictions of the network but a PQ-Net model trained with relatively high MAE can potentially still yield accurate predictions.
To evaluate the performance of the PQ-Net we simulated a phantom XRD-CT dataset using a scale factor map derived from the Rietveld analysis of an experimental XRD-CT dataset from a catalyst sample51. One advantage of this dataset is the realistic nature of this intensity map compared to idealised ones, such as the Shepp-Logan phantom. More important though is that one can easily segment the various particles, assign different properties to them and generate multiple XRD-CT datasets with varying chemistry in an efficient manner. More details regarding the phantom simulation can be found in our previous work52. These 120 × 120 (14,400) diffraction patterns were used as test data for the single-phase PQ-Net. The results from a single-phase PQ-Net trained with 100 K patterns applied to this test set are presented in panel in Fig. 2. The ground truth (GT) maps for the various parameters are presented in the first row, the results from the PQ-Net in the second row and the difference between the two maps in the third row. The scale factor is the easiest parameter to train the PQ-Net for, so it is not surprising that it yields very accurate maps with a relative error less than 5 % (Supplementary Fig. 2). Impressively though it can be seen that the error for the crystallite size is below 1 nm for all catalyst particles and similarly the error for the lattice parameter is below 10−3 Å. The Rwp also is below 5% for the majority of particles (Supplementary Fig. 2). To illustrate this further, the mean diffraction pattern from three catalyst particles exhibiting different properties were extracted and analysed with the PQ-Net. The results are presented in panel b of Fig. 2 and it can be clearly seen that the network is able to accurately predict the three patterns (Rwp of 3.144, 1.191 and 1.835% respectively). The corresponding Ni parameters are summarised in Supplementary Table 4.

a Ni scale factors, crystallite sizes (nm) and lattice parameter (Å) ground truth maps, results obtained with the PQ-Net and their absolute difference. b Comparison between the average diffraction patterns extracted from three particles of interest and the TOPAS generated patterns using the parameters predicted by the PQ-Net.
Multi-phase deep ensemble PQ-Net and simulated XRD-CT dataset
The next logical step was to test the PQ-Net against simulated multi-phase XRD patterns which is a closer approximation to most experimental data and closer to real world applications. Here, we chose a five-phase system that corresponds to the chemistry encountered in the experimental XRD-CT dataset presented in the next section. The choice of this material system was based on the complexity and its challenging nature; it is a multi-component system with the various phases exhibiting different symmetries and their physico-chemical properties vary spatially as it will be shown in the next section.
The multi-phase PQ-Net architecture used here is presented in Supplementary Fig. 3 and it can be seen that it is an extension of the one presented in Fig. 1. The five crystalline phases modelled here are NiO (ICSD: 9866), PdO (ICSD: 24692), CeO2 (ICSD: 72155), ZrO2 (ICSD: 66781) and theta-Al2O353. The parameters refined here are the scale factors, lattice parameters and crystallite size for each phase. Poisson noise and a varying linear background (2nd degree Chebyshev polynomial) were also introduced in the diffraction libraries used to train the multi-phase PQ-Net (Supplementary Table 5). To improve the performance and robustness of our model, necessary for complex tasks such as this, we implemented and evaluated deep ensembles of the PQ-Net54,55,56. Deep ensembles are based on the assumption that different initial weights of a CNN will lead to different local minima which may not necessarily be a minimum on the validation data, but these local minima are evenly distributed in parameter space and therefore sample different possible solutions efficiently57. Therefore, simply retraining a CNN model several times and taking the average of the outputs can usually have better results than using only one model.
Supplementary Fig. 4 shows the influence of deep ensemble acting on the results’ mean absolute error (MAE). It can be seen that the more individual models in a deep ensemble model, the more accurate results it can generate (Supplementary Table 6). Another benefit of deep ensembles is that the method can give a referable uncertainty of the results. By taking the standard deviation among all individual models’ predictions on a parameter, one can easily estimate the precision of a deep ensemble result. However, because the time taken by training and merging individual models can rise proportionally as the number of models in deep ensemble increases and the curve tends to flatten out, using a deep ensemble of size between five to ten is the most realistic option for real world applications. It is worth mentioning that the MAEs for the validation dataset have a lower limit which can not be surpassed by increasing the size of the training dataset or the deep ensemble, because the crystallite sizes and lattice parameters are very hard to predict when scale factors are approaching zero.
The performance of the trained multi-phase PQ-Net was evaluated with a simulated XRD-CT dataset using the same intensity image as for the single-phase dataset. Here, the same strategy was followed creating five single-phase XRD-CT datasets, one for each component which were then added together to form the multi-phase simulated XRD-CT dataset consisting of 14,400 diffraction patterns (test data). This dataset was then passed to the PQ-Net for analysis and quantification of the various parameters. The scale factor results from the deep ensemble PQ-Net consisting of 10 models and trained with 100 K patterns (training data) are presented in Fig. 3. It can be clearly seen that the PQ-Net is able to create accurate phase distribution maps for all components and retain the local features and relative intensities. Importantly, it is able to also create accurate crystallite size and lattice parameter maps for all phases (Supplementary Figs. 5–9) while the Rwp remains <10 % for all particles (Supplementary Fig. 10).

Scale factor ground truth maps, results obtained with the PQ-Net and their absolute difference for the simulated multi-phase NiO-PdO-CeO2-ZrO2-Al2O3 system.
To further illustrate the ability of the PQ-Net to handle multi-phase systems, we also extracted three diffraction patterns from the XRD-CT which correspond to the mean diffraction patterns from three particles. The results from the analysis of these three patterns are presented in Fig. 4 where it is clearly shown that the PQ-Net is able to model them accurately (Rwp of 5.024, 5.419 and 4.798 % respectively; see also Supplementary Tables 7–11).

Comparison between the average diffraction patterns extracted from three particles of interest and the TOPAS generated patterns using the parameters predicted by the PQ-Net. The main reflections for NiO (blue ticks), PdO (green ticks), CeO2 (red ticks) and ZrO2 (cyan ticks) are also presented.
Experimental XRD-CT dataset
The final and most challenging dataset is an experimental XRD-CT dataset acquired using the Ni-Pd/CeO2-ZrO2/Al2O3 catalyst. Details regarding its preparation and phase identification are provided in our previous work12. Here we use the previous deep ensemble PQ-Net consisting of 10 models and trained with 100 K patterns (training data) to analyse the 22,801 XRD patterns (test data) present in the XRD-CT dataset (XRD-CT images of 151 × 151 pixels). We also perform conventional Rietveld analysis which serves as an approximation to the ground truth and the benchmark to assess the performance of the PQ-Net.
The results for the scale factors for all phases using the Rietveld method and the PQ-Net are presented in Fig. 5. It can be seen that the PQ-Net is able to accurately predict the scale factors of both the main phases, such as Al2O3, NiO and ZrO2, and also minor components such as the PdO. It should be noted here that the PdO should not have been detectable at this resolution due to its low content (<1 wt.%) and it is only possible due to areas of high concentration near the surface of some particles. Importantly, the PQ-Net results are consistent with the ones obtained with the Rietveld method showing for example that the ZrO2 (or Zr-rich CexZr1-xO2 phase, where x ≤ 1) is located only at the periphery of the catalyst particles while the CeO2 (or Ce-rich CexZr1-xO2 phase) is present also at the interior of the catalyst particles. The Al2O3 distribution is correctly shown to be homogeneous in all catalyst particles while NiO, the primary catalyst active component, is also present in all particles. The uncertainty maps shown in the last row of Fig. 5 correspond to the standard deviation of the calculated values from the 10 models of the deep ensemble PQ-Net (Supplementary Fig. 11).

Scale factor maps obtained with the Rietveld method, results obtained with the PQ-Net, their absolute difference for the experimental multi-phase NiO-PdO-CeO2-ZrO2-Al2O3 system and the uncertainty maps of the deep ensemble PQ-Net (last row).
Importantly, apart from the scale factors, the PQ-Net is able to capture the chemical gradients present in this challenging XRD-CT dataset. As shown in Fig. 6, it is able to resolve the heterogeneities in the crystallite size and lattice parameter of the CeO2-ZrO2 phases. As we have previously reported, the CeO2 crystallite size and lattice parameter follow an egg-shell distribution in this catalyst with lower values for lattice parameter and crystallite size at the shell compared to the core of the particles. As expected, this result is reproducible with the Rietveld method but it can also be seen that the PQ-Net yields the same results. Moreover, the difference between the CeO2-ZrO2 lattice parameter maps obtained with the Rietveld method and the PQ-Net is below 2 × 10−2 Å for the majority of the particles while for the crystallite size maps the difference is in the order of 1–2 nm. The uncertainty maps presented in the last row of Fig. 6 indicate that the error for the crystallite size for both phases is in the order of 1 nm.

Crystallite size (colorbar axis corresponding to nm) and lattice parameter a (colorbar axis corresponding to Å) maps for CeO2 and ZrO2 obtained with the Rietveld method, results obtained with the PQ-Net, their absolute difference for the experimental multi-phase NiO-PdO-CeO2-ZrO2-Al2O3 system and the uncertainty maps of the deep ensemble PQ-Net.
Diffraction patterns from two representative regions-of-interest were extracted from the XRD-CT for further analysis. As shown in Fig. 7, one pattern is derived from the periphery of three particles (light blue region) while the second is derived from the whole area of two other particles (magenta region). The first region corresponds to an area where the ZrO2 phase is present while the second where it is absent. Both patterns are the mean XRD patterns from the respective regions in the sample. These two patterns were then analysed with the PQ-Net and the results are presented in Fig. 7. It can be clearly seen that the PQ-Net is able to model accurately the data (Rwp of 8.353 and 7.749 % respectively).

Comparison between the average diffraction patterns extracted from two regions of interest and the TOPAS generated patterns using the parameters predicted by the PQ-Net. The main reflections for NiO (blue ticks), PdO (green ticks), CeO2 (red ticks) and ZrO2 (cyan ticks) are also presented.
Discussion
The results presented in this work demonstrate that the PQ-Net model is able to extract accurate physico-chemical information from XRD patterns. Its performance was evaluated with different datasets of increasing complexity, varying from simulated noiseless single-phase to experimental five-phase systems. In all cases, PQ-Net was able to quantify the scale factors, lattice parameters and crystallite sizes of the various phases providing predictions with errors within acceptable ranges (i.e. compared to ground truth and Rietveld results for the simulated and experimental data respectively). The work presented here therefore serves also as an exemplar study demonstrating PQ-Net’s flexibility and scalability as the method applied to datasets of different complexity and it can be scaled up or down depending on the data requirements. The robustness of the PQ-Net was further improved through the implementation of deep ensembles that allow for uncertainty quantification.
The deep ensemble PQ-Net model was able to provide results with less than 2% difference in Rwp compared to the one obtained from the analysis of the same data using the Rietveld method. These results were obtained (i.e. analysis of ca. 20 K XRD patterns) in ca. 10 s while the state-of-the-art Rietveld for 9 K XRD patterns required ca. 4.4 h. A comparison table and corresponding figures demonstrating how the performance of PQ-Net and Rietveld scale with increasing data size are provided in the Supporting Information (Supplementary Tables 12–14 and Supplementary Figs. 12–14). It should be noted though that the PQ-Net was not designed to replace the Rietveld method but to assist and provide very fast and accurate predictions for the various physico-chemical parameters; conventional least-square minimisation techniques, such as the Rietveld method, can be used for fine parameter tuning using the PQ-Net results as the starting point. The PQ-Net in its current form, similar to the Rietveld method, requires some a priori knowledge of the sample and its chemistry. However, in case there is evolving/unpredictable chemistry or unknown phases form/are present in the system, it can be easily spotted through the Rwp, the difference between the experimental and predicted patterns and the uncertainty maps provided by the deep ensemble.
The key advantage of the PQ-Net is that the diffraction libraries can be generated prior to an experiment and used to pretrain the model. This approach can allow for real-time assessment of diffraction data acquired during a dynamic experiment and enable the user to better guide the experiment (e.g., through the applied operating conditions) and intervene when necessary. In future, we plan to increase the complexity of the PQ-Net and explore its ability to model other parameters such as complex backgrounds, to quantify amorphous/crystalline components and to perform simultaneous calculation of weight percentages and strain analysis. It should be pointed out that the PQ-Net could in principle also be used to benefit X-ray Free Electron Laser (XFELs) facilities, like the European XFEL (Germany), for fast analysis of powder diffraction data. We will also try to apply the PQ-Net on other analytical scattering/spectroscopic techniques such as X-ray absorption fine structure spectroscopy (XAFS) data which is a technique suitable for large volume data analysis (e.g., XAFS-CT). Last but not least, we expect that the PQ-Net has the potential to become an essential tool for diffraction applications beyond synchrotron experiments such as real-time quality inspection in manufacturing/synthesis.
Methods
XRD-CT measurements
A Ni-Pd/CeO2-ZrO2/Al2O3 catalyst was measured with XRD-CT at beamline station ID31 of the ESRF using a 70 keV monochromatic X-ray beam focused to have a spot size of 20 × 20 μm. Information regarding the sample preparation can be found in our previous work12. The Pilatus3 X CdTe 2 M hybrid photon counting area detector was used for the acquisition of the 2D powder diffraction patterns. The total acquisition time per point was 20 ms. 225 translation steps (translation step size of 20 μm) covering 0–180° angular range, in steps of 1.125° (i.e., 160 line scans) were used for the tomographic measurements. A CeO2 NIST standard was used for the detector calibration. Every 2D powder diffraction pattern was transformed to a 1D powder diffraction pattern after applying a 1% trimmed mean filter to remove outliers; this was done using in-house developed MATLAB scripts58. The tomographic images were reconstructed using the filtered back projection algorithm and were cropped to a 151 × 151 × 2048 reconstructed data volume (22801 diffraction patterns).
Rietveld analysis
Full profile analysis of the spatially-resolved diffraction patterns present in the XRD-CT data was performed using the Rietveld method. Inhouse developed MATLAB scripts were used in combination with the TOPAS software v7 to perform the analysis in a sequential manner, processing one line at a time (i.e., corresponding to 225 diffraction patterns). A mask was applied so that only the pixels in the images corresponding to sample regions would be processed (decreasing significantly the number of patterns to be analysed in each line) decreasing the number of patterns to 9027. Full profile analysis was initially performed using the mean diffraction pattern of the XRD-CT dataset before performing the batch fitting (i.e., in order to have a good starting model for the Rietveld analysis). The background was fairly linear and a 2nd degree Chebyshev polynomial was used to model it. For each phase, the scale factor, lattice parameters and crystallite size were refined in the model.
Diffraction libraries
All diffraction libraries were created using inhouse developed MATLAB scripts and the TOPAS software v7 using CPU. The neural networks were trained using GPU and a CPU/GPU comparison Table is provided in the Supporting Information (Supplementary Table 1). A 10% validation split was applied to the training data for each diffraction library used in this work to train the PQ-Net. The scale factors, lattice parameters and crystallite size values for each pattern were created using random sampling from a fixed range. Details for each parameter are presented in the Supporting Information. Each parameter corresponds to a 1D array with length equal to the library size. For each parameter, the minimum value was subtracted from the corresponding array and then it was normalised. This step proved very important for the stability of the network as it allowed all parameters to have values at the same range (0–100). The minimum and maximum values are recorded in a text file, then the models’ predictions on the experimental dataset can be easily converted to the real values before normalisation. A 3XS Data Science Workstation C264X2 with 2x Intel Xeon Silver 4216, 350 GB RAM and 2x Quadro RTX 8000 was used for the development and training of the neural networks used in this work and the Rietveld analysis of the diffraction patterns.
Data availability
The training and test datasets used in this work are publicly available through https://doi.org/10.5281/zenodo.4664597. All other data are available from the corresponding authors on reasonable request.
Code availability
The codes developed in this study are available from the authors upon reasonable request.
References
Vaughan, G. B. M. et al. X-ray transfocators: focusing devices based on compound refractive lenses. J. Synchrotron Radiat. 18, 125–133 (2011).
Schroer, C. G. & Lengeler, B. X-Ray Optics. In Springer Handbook of Lasers and Optics (ed. Träger, F.) 1461–1474 (Springer Berlin Heidelberg, 2012).
Ebensperger, T., Stahlhut, P., Nachtrab, F., Zabler, S. & Hanke, R. Comparison of different sources for laboratory X-ray microscopy. J. Instrum. 7, C10008–C10008 (2012).
Morgan, A. J. et al. High numerical aperture multilayer Laue lenses. Sci. Rep. 5, 9892 (2015).
Murray, K. T. et al. Multilayer Laue lenses at high X-ray energies: performance and applications. Opt. Express 27, 7120–7138 (2019).
Hornberger, B., Kasahara, J., Gifford, M., Ruth, R. & Loewen, R. A compact light source providing high-flux, quasi-monochromatic, tunable X-rays in the laboratory. Proc. SPIE 11110, Advances in Laboratory-based X-Ray Sources, Optics, and Applications VII, 111100 (2019).
Ponchut, C., Tartoni, N. & Pennicard, D. X-ray imaging at synchrotron research facilities. Radiat. Meas. 140, 106459 (2021).
Xia, M. et al. Lab-scale in situ x-ray diffraction technique for different battery systems: designs, applications, and perspectives. Small Methods 3, 1900119 (2019).
Gjørup, F. H., Ahlburg, J. V. & Christensen, M. Laboratory setup for rapid in situ powder X-ray diffraction elucidating Ni particle formation in supercritical methanol. Rev. Sci. Instrum. 90, 073902 (2019).
Beale, A. M., Jacques, S. D. M., Gibson, E. K. & Di Michiel, M. Progress towards five dimensional diffraction imaging of functional materials under process conditions. Chem. Coord. Rev. 277–278, 208–223 (2014).
Poulsen, H. F. & Vaughan, G. B. M. Multigrain crystallography and three-dimensional grain mapping. In International Tables for Crystallography: Powder Diffraction 601–616 (International Union of Crystallography, 2019).
Vamvakeros, A. et al. 5D operando tomographic diffraction imaging of a catalyst bed. Nat. Commun. 9, 1–11 (2018).
Pacchioni, G. An upgrade to a bright future. Nat. Rev. Phys. 1, 100–101 (2019).
Wang, C., Steiner, U. & Sepe, A. Synchrotron big data science. Small 14, 1802291 (2018).
Guo, Y. et al. Deep learning for visual understanding: a review. Neurocomputing 187, 27–48 (2016).
Wang, B., Yager, K., Yu, D. & Hoai, M. X-ray scattering image classification using deep learning. in 2017 IEEE Winter Conference on Applications of Computer Vision (WACV) 697–704 (IEEE, 2017).
Pelt, D. M. & Sethian, J. A. A mixed-scale dense convolutional neural network for image analysis. Proc. Natl Acad. Sci. 115, 254–259 (2018).
Liu, X. et al. A comparison of deep learning performance against health-care professionals in detecting diseases from medical imaging: a systematic review and meta-analysis. Lancet Digit. Health 1, e271–e297 (2019).
Lee, J.-W., Park, W. B., Lee, J. H., Singh, S. P. & Sohn, K.-S. A deep-learning technique for phase identification in multiphase inorganic compounds using synthetic XRD powder patterns. Nat. Commun. 11, 1–11 (2020).
Ke, T. W. et al. A convolutional neural network-based screening tool for X-ray serial crystallography. J. Synchrotron Radiat. 25, 655–670 (2018).
Schleder, G. R., Padilha, A. C., Acosta, C. M., Costa, M. & Fazzio, A. From DFT to machine learning: recent approaches to materials science–a review. J. Phys. Mater. 2, 032001 (2019).
Sullivan, B. et al. BraggNet: integrating Bragg peaks using neural networks. J. Appl. Crystallogr. 52, 854–863 (2019).
Ito, S., Ueno, G. & Yamamoto, M. DeepCentering: fully automated crystal centering using deep learning for macromolecular crystallography. J. Synchrotron Radiat. 26, 1361–1366 (2019).
Sullivan, B. et al. Volumetric Segmentation via Neural Networks Improves Neutron Crystallography Data Analysis. In 2019 19th IEEE/ACM International Symposium on Cluster, Cloud and Grid Computing (CCGRID) 549–555 (2019).
Aguiar, J. A., Gong, M. L., Unocic, R. R., Tasdizen, T. & Miller, B. D. Decoding crystallography from high-resolution electron imaging and diffraction datasets with deep learning. Sci. Adv. 5 (2019).
Muto, S. & Shiga, M. Application of machine learning techniques to electron microscopic/spectroscopic image data analysis. Microscopy 69, 110–122 (2020).
Aguiar, J. A., Gong, M. L. & Tasdizen, T. Crystallographic prediction from diffraction and chemistry data for higher throughput classification using machine learning. Comput. Mater. Sci. 173, 109409 (2020).
Ponce, A., Aguilar, J. A., Tate, J. & Yacamán, M. J. Advances in the electron diffraction characterization of atomic clusters and nanoparticles. Nanoscale Adv. 3, 311–325 (2021).
Park, W. B. et al. Classification of crystal structure using a convolutional neural network. IUCrJ 4, 486–494 (2017).
Ryan, K., Lengyel, J. & Shatruk, M. Crystal structure prediction via deep learning. J. Am. Chem. Soc. 140, 10158–10168 (2018).
Ziletti, A., Kumar, D., Scheffler, M. & Ghiringhelli, L. M. Insightful classification of crystal structures using deep learning. Nat. Commun. 9, 2775 (2018).
Oviedo, F. et al. Fast and interpretable classification of small X-ray diffraction datasets using data augmentation and deep neural networks. Npj Comput. Mater. 5, 60 (2019).
Liu, C.-H., Tao, Y., Hsu, D., Du, Q. & Billinge, S. J. L. Using a machine learning approach to determine the space group of a structure from the atomic pair distribution function. Acta Crystallogr. Sect. A 75, 633–643 (2019).
Wang, H. et al. Rapid identification of x-ray diffraction patterns based on very limited data by interpretable convolutional neural networks. J. Chem. Inf. Model. 60, 2004–2011 (2020).
Tatlier, M. Artificial neural network methods for the prediction of framework crystal structures of zeolites from XRD data. Neural Comput. Appl. 20, 365–371 (2011).
Bunn, J. K., Hu, J. & Hattrick-Simpers, J. R. Semi-Supervised approach to phase identification from combinatorial sample diffraction patterns. Jom 68, 2116–2125 (2016).
Bunn, J. K. et al. Generalized machine learning technique for automatic phase attribution in time variant high-throughput experimental studies. J. Mater. Res. 30, 879 (2015).
Barr, G., Dong, W. & Gilmore, C. J. High-throughput powder diffraction. II. Applications of clustering methods and multivariate data analysis. J. Appl. Crystallogr. 37, 243–252 (2004).
Obeidat, S. M., Al-Momani, I. & Haddad, A. & Bani Yasein, M. Combination of ICP-OES, XRF and XRD techniques for analysis of several dental ceramics and their identification using chemometrics. Spectroscopy 26, 141–149 (2011).
Chen, Z. P. et al. Enhancing the signal-to-noise ratio of X-ray diffraction profiles by smoothed principal component analysis. Anal. Chem. 77, 6563–6570 (2005).
Lee, D., Lee, H., Jun, C.-H. & Chang, C. H. A variable selection procedure for X-ray diffraction phase analysis. Appl. Spectrosc. 61, 1398–1403 (2007).
Martens, I. et al. X-ray transparent proton-exchange membrane fuel cell design for in situ wide and small angle scattering tomography. J. Power Sources 437, 226906 (2019).
Li, T. et al. Design of next-generation ceramic fuel cells and real-time characterization with synchrotron X-ray diffraction computed tomography. Nat. Commun. 10, 1497 (2019).
Martens, I. et al. Holistic multi-scale imaging of oxygen reduction reaction catalyst degradation in operational fuel cells. ArXiv Prepr. ArXiv200804770 (2020).
Matras, D. et al. In situ X-ray diffraction computed tomography studies examining the thermal and chemical stabilities of working Ba0.5Sr0.5Co0.8Fe0.2O3−δ membranes during oxidative coupling of methane. Phys. Chem. Chem. Phys. 22, 18964–18975 (2020).
Finegan, D. P. et al. Spatial quantification of dynamic inter and intra particle crystallographic heterogeneities within lithium ion electrodes. Nat. Commun. 11, 631 (2020).
Matras, D. et al. Effect of thermal treatment on the stability of Na–Mn–W/SiO2 catalyst for the oxidative coupling of methane. Faraday Discuss. https://doi.org/10.1039/C9FD00142E (2021).
Vamvakeros, A. et al. Real-time tomographic diffraction imaging of catalytic membrane reactors for the oxidative coupling of methane. Catal. Today 364, 242–255 (2021).
Coelho, A. TOPAS and TOPAS-Academic: an optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Crystallogr. 51, 210–218 (2018).
Kingma, D. P. & Ba, J. Adam: A Method for Stochastic Optimization (2017).
Vamvakeros, A. et al. Real-time multi-length scale chemical tomography of fixed bed reactors during the oxidative coupling of methane reaction. J. Catal. 386, 39–52 (2020).
Vamvakeros, A. et al. DLSR: a solution to the parallax artefact in X-ray diffraction computed tomography data. J. Appl. Crystallogr. 53, 1531–1541 (2020).
Kovarik, L. et al. Unraveling the Origin of Structural Disorder in High Temperature Transition Al2O3: Structure of θ-Al2O3. Chem. Mater. 27, 7042–7049 (2015).
Lakshminarayanan, B., Pritzel, A. & Blundell, C. Simple and Scalable Predictive Uncertainty Estimation using Deep Ensembles. (2017).
Suk, H.-I., Lee, S.-W. & Shen, D. Deep ensemble learning of sparse regression models for brain disease diagnosis. Med. Image Anal. 37, 101–113 (2017).
Kozlovskaia, N. & Zaytsev, A. Deep ensembles for imbalanced classification. In 2017 16th IEEE International Conference on Machine Learning and Applications (ICMLA) 908–913 (IEEE, 2017).
Fort, S., Hu, H. & Lakshminarayanan, B. Deep ensembles: a loss landscape perspective. ArXiv Prepr. ArXiv191202757 (2019).
Vamvakeros, A. et al. Removing multiple outliers and single-crystal artefacts from X-ray diffraction computed tomography data. J. Appl. Crystallogr. 48, 1943–1955 (2015).
Acknowledgements
We would like to thank Marco di Michiel (ID15A, ESRF) and Jakub Drnec (ID31, ESRF) for preparing beamline instrumentation and setup and for their help with the experimental XRD-CT data acquisition. We acknowledge ESRF for beamtime. Finden acknowledges funding through the Innovate UK Analysis for Innovators (A4i) program (Project No: 106107). A.M.B. acknowledges EPSRC (grants EP/R026815/1 and EP/S016481/1).
Author information
Authors and Affiliations
Contributions
H.D. and A.V. designed and implemented the PQ-Net. H.D., A.V. and S.D.M.J. created the MATLAB, Python and TOPAS scripts for the creation of the diffraction libraries. S.W.T.P. and Y.O. were responsible for aspects of testing. H.D., A.V. and D.M. performed the Rietveld analysis of the XRD-CT data. K.B. advised on the deep ensembles while R.K. and A.T. advised on the CNN architectures. A.V., S.D.M.J., D.M., V.M. and A.M.B. designed the XRD-CT experiments and acquired the tomographic data. H.D. developed and tested the deep ensembles. H.D. and A.V. are responsible for writing the manuscript with feedback given by all authors. S.D.M.J., A.M.B. and A.V. directed the research.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dong, H., Butler, K.T., Matras, D. et al. A deep convolutional neural network for real-time full profile analysis of big powder diffraction data. npj Comput Mater 7, 74 (2021). https://doi.org/10.1038/s41524-021-00542-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-021-00542-4
This article is cited by
-
Automated classification of big X-ray diffraction data using deep learning models
npj Computational Materials (2023)
-
Validating neural networks for spectroscopic classification on a universal synthetic dataset
npj Computational Materials (2023)
-
Recent advances and applications of deep learning methods in materials science
npj Computational Materials (2022)
-
MaterialsAtlas.org: a materials informatics web app platform for materials discovery and survey of state-of-the-art
npj Computational Materials (2022)
-
Virtual diffraction simulations using the quasi-coarse-grained dynamics method to understand and interpret plasticity contributions during in situ shock experiments
Journal of Materials Science (2022)
