
Abstract
Future lithium (Li) energy storage technologies, in particular solid-state configurations with a Li metal anode, opens up the possibility of using cathode materials that do not necessarily contain Li in its as-made state. To accelerate the discovery and design of such materials, we develop a general, chemically, and structurally agnostic methodology for identifying the optimal Li sites in any crystalline material. For a given crystal structure, we attempt multiple Li insertions at symmetrically in-equivalent positions by analyzing the electronic charge density obtained from first-principles density functional theory. In this report, we demonstrate the effectiveness of this procedure in successfully identifying the positions of the Li ion in well-known cathode materials using only the empty host (charged) material as guidance. Furthermore, applying the algorithm to over 2000 candidate cathode empty host materials we obtain statistics of Li site preferences to guide future developments of novel Li-ion cathode materials, particularly for solid-state applications.
Similar content being viewed by others


Introduction
The need for rechargeable energy storage with higher energy density and longer operating life-time is a technical challenge for a future, carbon-neutral society. Although many battery chemistries have been commercialized in different usage scenarios, Li-ion batteries have proven to be the most successful, showing tremendous growth since its first implementation in 19911. While current Li-ion technology is ubiquitous in portable electronics, there are numerous barriers that limit the wide-spread adoption in other sectors, such as transportation and the electric grid. A critical concern in current Li-ion technology is safety, due to the use of a highly oxidized cathode together with a combustible organic electrolyte2. Another issue is cost and element criticality—cobalt-containing cathodes already account for a significant portion of the current cobalt market, and it is highly uncertain whether projected market growth can be accommodated3,4.
All-solid-state batteries replace the flammable organic electrolyte with an inorganic solid electrolyte, and are predicted to improve on both safety as well as chemical diversity5. In particular, the solid-state electrolyte enables the use of a metal anode, which eliminates the need for a cathode as the source of the working ion and hence opens up the playing field to a broader chemical and structural space, also considering different application specifics, beyond those traditionally tailored for portable electronics. To accelerate the discovery and design of novel cathode materials, particularly focusing on materials where the working ion is not present in the as-made material, we here develop a software and data analysis infrastructure to identify possible intercalating structures within the framework of the Materials Project6.
As of Feburary 2020, the Materials Project includes 125,134 material entries, 21,584 of which contain at least one transition-metal redox element in addition to either oxygen or sulfur but none of the following possible working ions—Li+, Ca2+, Mg2+, and Na+. These candidate intercalation host materials cover 6862 distinct combinations of atomic species, which we call chemical systems. Computational investigation of any material for cathode applications require first-principles calculations of the structure at different states of charge, e.g., different working ion compositions. A thorough study of the full set of these chemical systems has not been possible until now due to several challenges, the primary one being: automated identification the preferred positions of the working ion in the host cathode structure. Naively, one may hypothesize that the best position to place a working ion is the most "empty” site in the structure, but this concept is not well-defined. In this work, we present an algorithmic framework that suggests possible insertion sites in any crystal structure using the charge density of the host material. While the method is inherently agnostic to the working ion, e.g., can be applied to Na+, Mg2+, Ca2+, etc., we will hereon focus on Li-ion insertion as the most immediately applicable technology. We validate the approach by predicting viable Li sites based on the charge density of the delithiated phases of well-known cathode materials and compare the results to the original discharged compound. Going further, we then attempt Li insertions into 2271 candidate cathode materials available within the Materials Project, which allows us to systematically investigate the effect of inserting Li into different chemical systems as well as the local coordination preference of Li, across a large diverse set of chemistries and structures. Statistics of Li insertion site preferences and chemical environments are presented.
Results
Validation of insertion algorithm
A charge-density-based cation insertion algorithm is described in the “Methods” section. To demonstrate the insertion algorithm, we apply it to two well-known cathode materials: lithium manganese oxide (LMO) (LixMnO2) in the λ phase and lithium iron phosphate (LixFePO4, or LFP) in the olivine phase, which provides two very different testing materials. The Wyckoff symbols for each site are obtained from a structure with all candidate (e.g., Li) sites occupied using pymatgen’s interface to spglib library7. LMO presents a 3D percolating network of tetrahedral (8b) and octahedral (16d) Li sites, where typically only the 8b sites are occupied in the as-made material. In contrast, the olivine structure exhibits 1D channels of Li sites at the 4a Wyckoff position. Since both LMO and LFP are synthesized in their lithiated state, we first remove the Li ions to obtain the corresponding “empty” host structures, on which we demonstrate our insertion algorithm. The resulting relaxed atomic structures of spinel MnO2 and olivine FePO4 are shown in Fig. 1 with green/blue/yellow spheres indicating each local minima in the charge density. The positions of their local minima (in crystal coordinates), their average charge density, and the symmetry-equivalent group each local minima belongs to are shown in Supplementary Tables 1 and 2.

Atomic structure and low-charge-density iso-surfaces of a spinel MnO2 and b olivine FePO4. The purple isosurfaces indicate regions of low charge density, the A(green) B(yellow) and C(blue) sites are groups of symmetry-equivalent local minima in the charge density.
For the 12-atom unit cell of spinel MnO2, we observe 8 local minima in the charge density, providing candidate sites for Li insertion. These sites form the three symmetry-equivalent groups as shown in Fig. 1a. The site labels correspond to Wyckoff symbols in the unit cell A ↦ 4d, B ↦ 2b and C ↦ 2a. Within the conventional (48 atom) cubic cell used in literature8, the Wyckoff symbols are: A ↦ 16d, B ↦ 8b and C ↦ 8a. The two lowest charge density sites are the A (4d, octahedral) site and the B (2b, tetrahedral) site. Within a sphere of radius r0 = 0.4 Å, each A-site exhibits an average charge density (\(\bar{\rho }\)) of 8.6 me/Å3 and each B-site has an \(\bar{\rho }=12.9\) me/Å3, respectively. There is a third group of symmetrically equivalent local minima, C sites, which are enclosed by MnO6 octahedra, and exhibit a much higher average charge density of \(\bar{\rho }=161.1\) me/Å3.
The candidate Li insertion sites of olivine FePO4 can also be organized into three groups with their corresponding Wyckoff symbols A ↦ 4a, B ↦ 4b and C ↦ 4c. The A and B-sites fall within two copies of the same low charge density regions in FePO4 unit cell as shown in Fig. 1b, and exhibits average charge densities of 6.8 me/Å3 and 7.8 me/Å3 respectively. Similar to the previous case, the C-type local minima in FePO4 exhibits much higher charge density—58.2 me/Å3.
For each identified, symmetry-unique Li site, we perform a single structure optimization with a Li ion inserted into that site. The resulting energies are then used to determine the most stable insertion site in LMO and LFP. It should be acknowledged that since we are often inserting into the primitive cells of the delithiated materials, the unit cells tend to be smaller (the smallest lattice vector is 5.83 Å for MnO2 and 4.89 Å for FePO4) which may incur finite-size effects. Also, we emphasize that any effects of Li arrangement and orderings in larger simulation cells are not considered explicitly.
To analyze the energy differences for the different candidate sites, using LMO as an example, we define the relative stability of the inserted structures, indexed by α, by the binding energy:
where the notation (Li:α)MnO2 denotes the relaxed structure after Li was inserted at site α ∈ {A, B, C} and E[Limetal] is the energy per atom of bulk Li metal. The relative binding energy ΔEB of a particular structure is defined as
which is the difference in energy between a given inserted structure and the most stable inserted structure for that material. This relative binding energy ΔEB is used henceforth as a measure of the Li site preference Li in a given host structure.
The relaxed atomic configurations and binding energies of inserting Li onto different sites in LMO and LFO are shown in Fig. 2. For the A and B sites in each material, the insertion of Li:
is defined as a topotactic reaction if the host crystal structure is unchanged after a single Li atom was inserted and the new structure has been optimized. The definition of "unchanged” inherently depends on a predefined metric. Here, we utilize the structure matching capabilities in pymatgen9 with the default tolerances parameters for the fractional length (ltol = 0.2), site position (stol = 0.3) and relative angles (angletol = 5∘).

Relaxed atomic structures after inserting Li into MnO2 (a–c) and FePO4 (d–f) are shown. Three Li insertions were attempted for each material at the A, B, and C site as defined in Fig. 1, corresponding to the subplot columns. The binding energy of inserting a Li atom into the unit cell is also reported in each case. In the case of C-site insertion in FePO4, the final relaxed structure is identical to the relaxed structure of the A-site insertion.
Using this definition, in MnO2, only the A-site and B-site insertions were classified as topotactic reactions, in agreement with the known insertion sites in the LMO spinel. On the other hand, the C-site insertion occurs at a site with much higher charge density, indicating severe repulsive interactions with the surrounding ions in LMO. As a result, when the structure with Li at the C-site is relaxed, the Li atom disrupts the MnO2 lattice, and displaces the surrounding atoms to create space as shown in Fig. 2. The resulting configuration is more than 4 eV/atom higher in energy than the A and B-site inserted structures.
In FePO4, all three insertions were found to be topotactic. The Li atoms inserted into the A and B positions did not move significantly or cause large distortions in the lattice. The C-site insertion resulted in an atomic configuration that was symmetrically equivalent to the A-site insertion, indicating that the initial C-site position was too far from a local minimum in the potential energy surface. The A-site in FePO4 was found to be the most stable, in agreement with known Li insertion into the olivine phase of LiFePO410, while the B-site is a meta-stable 4b site that connects the 4a sites within the 1D percolating channels11,12.
From the binding energies reported in Fig. 2, we observe that the charge-density provides a useful descriptor for site finding and correctly generates new structures with the stable and meta-stable Li sites for MnO2 and FePO4. Moreover, the stable/metastable Li sites all result from Li insertion at positions with low average charge density, hence providing a good descriptor for Li insertion sites at dilute Li concentrations. Incidentally, we note that, for both of these materials, migration between symmetry-equivalent A and B-sites creates a percolating network between neighboring sites of similar low charge densities, indicating a possibility of favorable migration.
While the site finder algorithm performed well for the MnO2 spinel and FePO4 olivine cathode materials, a more systematic way to gauge its performance across different chemistries and structures is needed to confidently apply it to a broader compound space such as e.g., the Materials Project. To further test the accuracy of the algorithm, we selected eight cathode material from the Materials Project with experimentally verified structures, ionic positions and ICSD entries representing a variety of chemical and structural motifs. To further test the limitations of our insertion process, and to highlight its effectiveness in close-packed structures, we also included two Li-ion conductor materials with a varying packing efficiency. For the loose-packed example, we use a new super-ionic conductor material recently realized by Di Stefano et al.13 LiTi2(PS4)3 (LTPS), which presents TiS6 octahedra connected by edge-sharing thiophosphate (PS4) groups resulting in a porous network of open cavities, where the Li ions are delocalized at room temperature. For the close-packed example, we include a hypothetical structural variant on the same chemistry, LiTi2(PS4)3, in the NASICON structure where the low-energy Li sites are known. These materials are listed in Table 1, and for each material, we removed the Li from the structure and performed a structural relaxation on the host lattice, obtaining the charge density of the "empty” host material. We insert a Li atom into each symmetry-unique site using the insertion algorithm described above. For each material in Table 1, we perform step-wise Li insertion: (i) relax the atomic structure of the host material, (ii) analyze the charge density of the host material to identify a list of symmetry-unique sites, (iii) create new structures by inserting one Li atom into each symmetry-unique site, and (iv) relax each new structure again, finally (v) we utilize the new structure with the lowest energy and repeat from step (ii) until we reach the Li content of the original material.
For each material we examine the charge density minima (\(\{{\tilde{{\bf{s}}}}_{i}\}\)) of the relaxed delithiated structure to carefully evaluate the performance of the insertion algorithm. The Wyckoff symbols of the first three candidate sites with the lowest average charge densities are given in Table 1. For seven out of the ten materials, the local minima with the lowest average charge density (labeled \({\tilde{{\bf{s}}}}_{1}\)) coincided with a Li site of the original structure. In all cases, except the novel LTPS from ref. 13, repeated Li insertions into the most stable candidate site was enough to recover the original, fully lithiated, structure. In the case of LTPS, Li does not exhibit typical crystallographic sites (e.g, octahedral or tetrahedral)—instead showing delocalized occupancy over broader "pocket” regions, which provides a highly challenging test case.
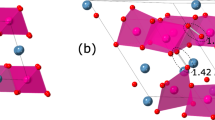
For the spinel phases LiTi2O4 and LiMn\({}_{\frac{3}{2}}\)Ni\({}_{\frac{1}{2}}\)O4, the positions of \({\tilde{{\bf{s}}}}_{1}\) are symmetrically distinct from the Li atoms in the original crystal. The atomic structures of these two materials are shown Fig. 3 along with the three groups of candidate sites with the lowest \(\bar{\rho }\). For LiTi2O4, the host structure and fractional coordinates of the candidate sites are identical to those of λ-MnO2. Again, the two lowest charge density sites exhibit Wyckoff symbols 4d and 2b, respectively. In both materials, the ΔEB for these sites were very similar, with the 4d site being favored in MnO2 and the 2b site being favored in TiO2. For LiMn3/2Ni1/2O8, the lithium positions coincide with the eight \({\tilde{{\bf{s}}}}_{3}\)(8c) local minima in the charge density. We note that in such cases, where the energy difference between sites are small, it is likely that Li–Li interactions, which are not taken into account here, affect the final site preference.

The relaxed delithiated structure of Ti2O4 (a), Mn3NiO8 (b), and Ti2(PS4)3 (c). The insertions sites with the three lowest average charge densities are shown in green, yellow, and blue sequentially. The original sites of the removed working ions are indicated with a transparent blue spheres.
The novel ionic conductor (LTPS) reported by ref. 13 exhibits two candidate site regions with the lowest average charge densities. These regions correspond to the centers of the large (2d) and small (4c) cavities of the structure. As expected in such large empty cavities, during relaxation, the Li ion moves towards the wall of the cavity, mimicking absorption. The third lowest charge density site is located in a crevice of the pore linkers. Interestingly, when relaxed, the large cavity (2d) site \({\tilde{s}}_{1}\) ends up the least stable (2.1 eV higher in the energy), whereas the \({\tilde{s}}_{2}\) and \({\tilde{s}}_{3}\) sites are close in energy, 78 meV apart, with the \({\tilde{s}}_{3}\) site corresponding to one of the regions of high Li concentration observed in the ref. 13. We speculate that, given the high degree of Li ion delocalization observed in this structure, there is a collection of sites exhibiting a flat energy landscape which enables fast ion migration. Among our three candidate sites, our framework was able to successfully identify two relevant regions of Li occupancy, however, we emphasize that further exploration and validation may be necessary for broad applications to porous structural families.
To demonstrate that the method can be applied to other complex, covalent network structures, we also attempted Li insertion on a hypothetical NASICON14 configuration of Ti2(PS4)3, e.g., chemically identical to LTPS. For this hypothetical material, the typical NASICON working-ion sites coincide with the local minima in the charge density of the relaxed Ti2(PS4)3 structure with 6e and 2b Wyckoff symbols, which exhibit the two lowest charge densities.
Since the effect of explicit Li insertion in most cases is accompanied with lattice relaxation and changes in the local environment, we do not expect the charge density be a reliable indicator of the ranking of stable Li sites. However, for each material in Table 1, the original Li positions were always among the three lowest-density sites identified by the charge density method. Additionally, the insertion site with the lowest binding energy (the underlined entries in Table 1) always coincided with a Li position in the original structure. This lends the confidence to our site-finding algorithm and indicates that the optimal site can be found by systematic insertion of the active cation into a handful of candidate individual sites, while recording the resulting energy. Structures with higher lithium content can be obtained by repeated insertions on the lowest energy structure, until one of the following criteria are met: (i) the new structure is no longer topotactically related to the original one, (ii) the new structure is too far above the convex-hull or (iii) the minimum redox state has been achieved. For the materials in Table 1 this type of repeated insertion was sufficient to produced the original, known lithiated structure. Although these results are highly promising, it is important to note that finite-size effects such as Li–Li interactions can influence the insertion sequence, especially for candidate sites with very similar energies.
Statistical analysis of topotactic insertion
Using our benchmarked algorithm to explore possible Li intercalation into any structure, we are poised to investigate the success rate of topotactic insertions into the broad range of structurally distinct transition metal oxides and sulfides available in the Materials Project. However, 21,584 materials present a formidable number of possible Li insertions, and hence we limit the number of possible compounds that exhibit a computed energy above hull Ehull < 0.1 eV15 and limited cell size (less or equal to 20 atoms per unit cell). This selection process results in a candidate set of reasonably stable, computationally tractable 15,484 transition-metal compounds. Attempting Li insertion into all of the symmetry-unique sites in all these structures is still intractable given current computational resources; however, we can elucidate the site preference of Li across diverse transition metal systems by attempting insertions on a smaller, randomly selected subset of these structures. Hence, we applied Li insertion framework developed above on this set (which we will call \({{\mathcal{S}}}_{{\rm{inserted}}}\)) of 2271 materials.
To understand the chemical diversity of the materials in \({{\mathcal{S}}}_{{\rm{inserted}}}\), we plot the occurrences of different transition metals in Fig. 4. Interestingly, while the Materials Project does not enforce uniform representation of different chemical systems, similar numbers of entries are found on the diagonal of Fig. 4, representing materials that only contain one type of TM. That is, the random selection fortuitously resulted in similar numbers of single TM-containing compounds. Materials that contain more than one kind of TM atom manifest as off-diagonal cells in Fig. 4, and they are less common in \({{\mathcal{S}}}_{{\rm{inserted}}}\).

Frequency of TM occurrence in the randomly sampled collection of TM oxides and sulfides. Each cell represents the number of materials in our database that contain the transition metal atoms given in the row and column.
For each material in \({{\mathcal{S}}}_{{\rm{inserted}}}\), we attempt a single Li insertion into each candidate site identified by the charge density. A material is deemed to exhibit a successful topotactic insertion if any one of the attempted Li insertions was topotactic. For each combination of TM atoms, the percentage of materials which exhibit successful topotactic insertions is given in Fig. 5. Even though multiple TM-containing compounds are less common, they still represent a significant portion of the materials in our dataset. Although there is some variation in the topotactic Li insertion success rate across different combinations of TMs, the overall success rate for the 2271 TM containing host materials is 88% (or 2059 materials).

The fraction of materials in each TM chemistry that resulted in successful topotactic insertion for at least one of the attempted cation insertions sites.
In addition to the chemical diversity, the set of materials \({{\mathcal{S}}}_{{\rm{inserted}}}\) also exhibit significant diversity in their crystal structure, which were analysed using the Robocrystallographer software16. Of the 2271 structures in \({{\mathcal{S}}}_{{\rm{inserted}}}\), less than half (971) matched any well-known prototype mineral structure. For the ones that did, 50 distinct mineral types can be identified within the set of 971 materials. The mineral types with more than ten matching structures in \({{\mathcal{S}}}_{{\rm{inserted}}}\) are tabulated in Table 2 along with the number of matching structures. For each mineral type, the percentage of structures with successful topotactic Li insertions is also shown. From Table 2, we can see that well-known cathode structures like 3-D spinel structure and the 2-D molybdenite structure have high rates of successful topotacitic insertion. However, more tightly packed structures like the cubic and orthorhombic perovskite structures exhibit much lower rates of successful insertion.
As a first observation, regardless of structure or chemical composition, we find that the thermodynamic prerequisites for topotatic insertion of Li is a common phenomenon. We emphasize that in order for Li to actually enter and leave the structure, good kinetics is also required. However, sites and structure-tolerance to intercalation is a non-negotiable to cathode performance. Our results presents a promising, as well as daunting, prospect as almost all of the transition metal chemical space is open for a broader search of new Li-ion cathode materials.
Local environment preference of Li
The coordination preference of Li in compounds of various structure and chemistry has been a long-standing question in cathode research17,18. The Li site insertion methodology presented here can address this question and investigate the coordination preference of Li in a systematic way. For all structures with a single topotactically inserted Li atom, we compute the coordination number of that Li atom using the ChemEnv19,20 local environment descriptor. For any given structure and target ion, ChemEnv provides a continuous descriptor which attributes a fraction of commonly observed local-environment motifs to the chemical environment of the target ion, where the sum of all the fractions are enforce to equal unity. The assignment of local-environment fractions is sigmoidal such that small deviations from the ideal geometry still produce similar results. As an example, a perfectly octahedral environment would exhibit a fraction of 1.0 for octahedral environment and zero for all others. If a Jahn–Tellar distortion is applied where the z-direction bonds have been lengthened by 10% and the xy-plane bonds have been reduced by 10% the local environment still have an octahedral fraction of >0.99 with a small addition of square planar. However, if a distortion of 20% is applied, the local environment will be only 0.17 octahedral and 0.83 square planar.
We first examine the dataset comprised of all compounds that exhibited a successful topotactic insertion, per the procedure described above. We emphasize that this set may include several varieties of the same compound, at the same Li composition, as metastable (but relaxed) sites would be included. The distribution of the coordination number for this set of all topotactically inserted Li is shown by the blue bar histogram in Fig. 6. Not surprisingly, from Fig. 6, we find that topotactitically inserted Li has a general preference for coordination numbers 4 (CN4) and 6 (CN6). Final relaxed positions with CN4 accounts for 36% of all the topotactically inserted Li atoms, while sites with CN6 accounts for 33%. Together, these two coordination environments account for a majority of topotactic Li insertions in our dataset.

The distribution of coordination numbers for all topotactically inserted Li atoms are shown in blue, the distribution for the only topotacitic site with ΔEB = 0 is shown in yellow.
One may question whether the coordination preference changes if we consider the dataset comprising only the most stable sites (lowest ΔEB) as a function of Li concentration, for each material. This analysis is provided as a comparative yellow bar histogram in Fig. 6. Interestingly, we find that the site preference distribution of the stable sites is quite similar to the full set of all topotactic sites, indicating that the obtained Li coordination preference is robust, even when metastable (but relaxed) sites are considered as well. It further supports that the insertion site algorithm together with subsequent relaxation is able to locate suitable sites, as no coordination preference is included in this process.
Within the subsets of Li positions with CN4 and CN6, we compute the ChemEnv coordination parameters for further analysis. For a given coordination number, there are a number of well-known coordination environments recommended by the IUPAC21. For CN4 the possible motifs are tetrahedral, square co-planar, and see-saw; for CN6 the motifs are octahedral, pentagonal pyramidal, and trigonal prism. A given site is identified as having a particular local environment if the fraction of a given motif is above the cutoff criterion of 0.85. For the 10,956 Li insertion positions among the 2271 materials, 8535 exhibits topotactic Li sites of which 4549 are identifiable with one of the CN4 or CN6 motifs, according to the cutoff criterion. If we restrict ourselves to topotactic sites that represent the stable (lowest energy) Li insertion in a given material, 1120 out of the 2010 stable topotactic sites were identifiable. The distributions of these motifs for the CN4 and CN6 sites are shown in Fig. 7.

Occurrences of identifiable local environment motifs for the CN4 and CN6 topotactic Li insertion sites for the entire set of topotactic insertions (blue) and for the most stable topotactic insertion (yellow).
The results presented in Fig. 7 indicate that the octahedral local environment is the most common motif, but tetrahedral topotactic insertions are also common. If we restrict ourselves to materials with stable topotactic insertion sites for Li, we find that 80% of these sites are four or six coordinated, with tetrahedral and octahedral sites accounting for the majority of identifiable structure motifs. While these structural motifs are clearly preferred, out of the 2010 stable sites only about half (1120) were identifiable with any CN4 or CN6 motif, while 76% where identifiable with any motif at all using a ChemEnv fraction cutoff of >0.85. Since the set of stable topotactic insertion site show such great diversity in their local environment, the precise site preference should always be explicitly investigated using our insertion algorithm.
Discussion
In summary we have developed a generic atom insertion algorithm for suggesting atomic insertion sites by analysing the electronic charge density and demonstrated how this algorithm performs for Li insertions in candidate cathode, transition metal-containing material systems. We showed that the local minima of the charge density provide good initial guesses for the final lithium positions, such that the lowest energy sites can be easily recovered using a small number of calculations on these candidate sites. In fact, the most stable site is often found among the local minima with the lowest surrounding charge density. By iteratively inserting Li atoms and optimizing the new structures to identify the most stable new Li position, the fully intercalated structures of known Li cathodes can be recovered from the completely delithiated structures. Using this method, we attempted Li insertions on a set of 2271 randomly selected transition metal compounds and showed that Li can be inserted topotactically on an overwhelming majority of these structures. Finally, we demonstrated that the long-held belief that Li has a strong preference for 4 and 6 coordinated sites is valid across a large set of transition metal-containing compounds, with the octahedral coordination being the most common local environment for topotactically inserted Li atoms.
Methods
The total energy of each atomic arrangement was calculated from first principles using density functional theory with the generalized-gradient approximation (GGA) functional of Perdew–Burke–Ernzerhof (PBE), as implemented in the Vienna Ab initio Simulation Package (VASP). To obtain accurate and directly comparable energies across chemical systems, we use GGA when appropriate, GGA+U otherwise, and mix energies from the two calculation methodologies using the procedure described by Jain et al.22. Structural relaxation was performed until the maximum force on each atom was less than 0.05 eV/Å.
Our insertion algorithm relies on analysis of the charge density—ρ(r) of the original host compound, without a priori information of the preferred Li-ion intercalation sites. For a given host structure, ρ can either be obtained from the Materials Project or computed and stored in a database using the atomate23 framework. A set of local minima si of ρ(r), as well as the average charge density in the local neighborhood of radius r0 around each local minima, are identified. For the remaining discussions, we assume that r0 = 0.4 Å whenever the radius is not specified (i.e., \(\bar{\rho }({\bf{s}})={\bar{\rho }}_{0.4}({\bf{s}})\)). We note that the charge density of the host structure will often exhibit high symmetry, which will be reflected by the charge density. Hence, many of the local minima are symmetrically equivalent, and the set {si} can be reduced to a (often much) smaller subset of symmetrically unique sites \(\{{\tilde{{\bf{s}}}}_{{\mathrm{i}}}\}\). Starting with a host structure, we perform an independent structure relaxation calculation with a single Li inserted at each \({\tilde{{\bf{s}}}}_{i}\) site, in order to obtain and identify new, suitable lithiated structures.
Data availability
The data that support the findings of this study are available on the Materials Project website (materialsproject.org). The material ids of the 2271 materials in the set \({\mathcal{S}}_{\rm{inserted}}\) of materials with attempted Li insertions are listed in the Supplementary Table 3.
Code availability
Code for workflow generation and supporting analysis is available in the atomate code repository (atomate.org) and in the pymatgen code repository (pymatgen.org) respectively.
References
Nagaura, T. & Tozawa, K. Progress in batteries and solar cells. JEC Press 9, 209 (1990).
Abada, S. et al. Safety focused modeling of lithium-ion batteries: a review. J. Power Sources 306, 178–192 (2016).
Olivetti, E. A., Ceder, G., Gaustad, G. G. & Fu, X. Lithium-ion battery supply chain considerations: analysis of potential bottlenecks in critical metals. Joule 1, 229–243 (2017).
Lee, J. et al. Reversible Mn2+/Mn4+ double redox in lithium-excess cathode materials. Nature 556, 185–190 (2018).
Zheng, F., Kotobuki, M., Song, S., Lai, M. O. & Lu, L. Review on solid electrolytes for all-solid-state lithium-ion batteries. J. Power Sources 389, 198–213 (2018).
Jain, A. et al. The materials project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Togo, A. & Tanaka, I. Spglib: a software library for crystal symmetry search. arXiv. https://arxiv.org/abs/1808.01590 (2018).
Ning, F. et al. Ab initio investigation of Jahn–Teller-distortion-tuned Li-ion migration in λ-MnO2. J. Mater. Chem. A 5, 9618–9626 (2017).
Ong, S. P. et al. Python Materials Genomics (pymatgen): a robust, open-source python library for materials analysis. Comput. Mater. Sci. 68, 314–319 (2013).
Padhi, A. K., Nanjundaswamy, K. S. & Goodenough, J. B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144, 1188–1194 (1997).
Dathar, G. K. P., Sheppard, D., Stevenson, K. J. & Henkelman, G. Calculations of Li-ion diffusion in olivine phosphates. Chem. Mater. 23, 4032–4037 (2011).
Morgan, D., Van der Ven, A. & Ceder, G. Li conductivity in Li x MPO4 (M = Mn, Fe, Co, Ni) olivine materials. Electrochem. Solid State Lett. 7, A30 (2003).
Di Stefano, D. et al. Superionic diffusion through frustrated energy landscape. Chem 5, 2450–2460 (2019).
Kohler, H., Schulz, H. & Melnikov, O. Structural investigations of NASICON (Na1+xZr2SixP3-xO12; x = 3) with x-ray diffraction at 298 K and 403 K. Mater. Res. Bull. 18, 589–592 (1983).
Sun, W. et al. The thermodynamic scale of inorganic crystalline metastability. Sci. Adv. 2, e1600225 (2016).
Ganose, A. M. & Jain, A. Robocrystallographer: automated crystal structure text descriptions and analysis. MRS Commun. 9, 874–881 (2019).
Goodenough, J. B., Manthiram, A. & Wnetrzewski, B. Electrodes for lithium batteries. J. Power Sources 43, 269–275 (1993).
Bhattacharya, J. & Wolverton, C. Prediction of quaternary spinel oxides as Li-battery cathodes: cation site preference, metal mixing, voltage and phase stability. J. Electrochem. Soc. 161, A1440–A1446 (2014).
Waroquiers, D. et al. Statistical analysis of coordination environments in oxides. Chem. Mater. 29, 8346–8360 (2017).
Waroquiers, D. et al. ChemEnv: a fast and robust coordination environment identification tool. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 76, 683–695, https://doi.org/10.1107/S2052520620007994 (2020).
Hartshorn, R. M., Hey-Hawkins, E., Kalio, R. & Leigh, G. J. Representation of configuration in coordination polyhedra and the extension of current methodology to coordination numbers greater than six (IUPAC Technical Report). Pure Appl. Chem. 79, 1779–1799 (2007).
Jain, A. et al. Formation enthalpies by mixing GGA and GGA + U calculations. Phys. Rev. B 84, 045115 (2011).
Mathew, K. et al. Atomate: a high-level interface to generate, execute, and analyze computational materials science workflows. Comput. Mater. Sci. 139, 140–152 (2017).
Johnston, W. D., Heikes, R. R. & Sestrich, D. The preparation, crystallography, and magnetic properties of the LixCo(1-x)O system. J. Phys. Chem. Solids 7, 1–13 (1958).
Antaya, M., Cearns, K., Preston, J. S., Reimers, J. N. & Dahn, J. R. In situ growth of layered, spinel, and rock-salt LiCoO2 by laser ablation depostion. J. Appl. Phys. 76, 2799–2806 (1994).
Kataoka, K. & Akimoto, J. Single-crystal growth, crystal structure analysis and physical properties of lithium overstoichiometric Li1 + xCoO2. Solid State Ionics 262, 106–109 (2014).
Barré, M. & Catti, M. Neutron diffraction study of the βxn–svg and γ phases of LiFeO2. J. Solid State Chem. Fr. 182, 2549–2554 (2009).
Hirano, A. et al. Relationship between non-stoichiometry and physical properties in LiNiO2. Solid State Ionics 78, 123–131 (1995).
Laubach, S. et al. Changes in the crystal and electronic structure of LiCoO2 and LiNiO2 upon Li intercalation and de-intercalation. PCCP 11, 3278–3289 (2009).
Cava, R. J., Murphy, D. W., Zahurak, S., Santoro, A. & Roth, R. S. The crystal structures of the lithium-inserted metal oxides Li0.5TiO2 anatase, LiTi2O4 spinel, and Li2Ti2O4. J. Solid State Chem. 53, 64–75 (1984).
Akimoto, J., Gotoh, Y., Kawaguchi, K. & Oosawa, Y. Preparation of LiTi2O4 single crystals with the spinel structure. J. Solid State Chem. 96, 446–450 (1992).
Takahashi, Y., Gotoh, Y. & Akimoto, J. Structure and electron density analysis of oxide spinel LiTi2O4. J. Phys. Chem. Solids 63, 987–990 (2002).
Berger, R. & Tergenius, L.-E. Room temperature synthesis and structural characterization of monoclinic LiCuO2 by X-ray and neutron diffraction. J. Alloys Compd. 203, 203–207 (1994).
Lii, K. H., Li, C. H., Cheng, C. Y. & Wang, S. L. Hydrothermal synthesis, structure, and magnetic properties of a new polymorph of lithium vanadyl(IV) orthophosphate: β-LiVOPO4. J. Solid State Chem. 95, 352–359 (1991).
Wang, S.-L., Wu, C.-h & Liu, S.-N. Synthesis and structural characterization of lithium iron diarsenate, LiFeAs2O7. J. Solid State Chem. 113, 37–40 (1994).
Rousse, G., Rodríguez-Carvajal, J., Wurm, C. & Masquelier, C. Spiral magnetic structure in the iron diarsenate LiFeAs2O7: a neutron diffraction study. Phys. Rev. B 88, 214433 (2013).
Lee, E.-S. & Manthiram, A. Influence of doping on the cation ordering and charge–discharge behavior of LiMn 1.5 Ni 0.5-x M x O 4 (M = Cr, Fe, Co, and Ga) spinels between 5.0 and 2.0 V. J. Mater. Chem. A 1, 3118–3126 (2013).
Acknowledgements
J.X.S. acknowledges the primary support of BASF through the California Research Alliance (CARA) and the many useful conversations with our colleagues at BASF. Integration with the Materials Project infrastructure was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division under Contract No. DE-AC02-05-CH11231 (Materials Project program KC23MP). This research used resources of the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility operated under Contract No. DE-AC02-05CH11231.
Author information
Authors and Affiliations
Contributions
J.X.S. performed the calculations and developed the charge-density-based insertion algorithm. M.K. provided expertise on the site analysis algorithms. K.A.P. supervised the project at all stages. All three authors contributed significantly in the preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, JX., Horton, M. & Persson, K.A. A charge-density-based general cation insertion algorithm for generating new Li-ion cathode materials. npj Comput Mater 6, 161 (2020). https://doi.org/10.1038/s41524-020-00422-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-020-00422-3
This article is cited by
-
Topological graph-based analysis of solid-state ion migration
npj Computational Materials (2023)
-
Automated classification of big X-ray diffraction data using deep learning models
npj Computational Materials (2023)
-
Large scale dataset of real space electronic charge density of cubic inorganic materials from density functional theory (DFT) calculations
Scientific Data (2022)
-
A representation-independent electronic charge density database for crystalline materials
Scientific Data (2022)
-
Equivariant graph neural networks for fast electron density estimation of molecules, liquids, and solids
npj Computational Materials (2022)
