
Abstract
Thermal conductivity is one of the most fundamental properties of materials with the value being determined by nearly all-scale structural features and multiple physical processes. Rapidly judging material’s thermal conductivity is extremely important but challenging for the applications. The material genome paradigm offers a revolutionary way to efficiently screen and discover materials with designed properties by using accessible indicators. But such a performance indicator for thermal conductivity is quite difficult to propose due to the existence of multiple mechanisms and processes, especially for the materials with complex structures such as chalcogenides. In this study, the number mismatch between cations and anions is proposed as a practical performance indicator for lattice thermal conductivity in complex copper and silver chalcogenides, which can be used to explain the observed experimental data and find new low thermal conductivity materials. Such a number mismatch brings about rich phenomena to affect thermal conductivity including the complication of the unit cell and the creation of chemical hierarchy, point defects, rattling modes and lone-pair electrons. It is expected that this rich-connotation performance indicator can be also extended to other complex materials to discover designed thermal conductivities.
Similar content being viewed by others
Introduction
Thermal conductivity (κ) is a material’s basic property that is used to characterize its heat conducting capability. High-κ materials are widely used in heat dissipating applications while low-κ ones are usually used in thermal insulation. In particular, thermal conductivity is one of the key parameters in thermoelectric materials. It also plays an important role in other energy conversion and storage materials such as solar cells and battery materials. Understanding thermal conductivity is thus of great significance in various disciplines. For semiconductors, heat conduction comes mainly from the vibration of lattice (phonons), i.e., the lattice thermal conductivity (κL)1. It is determined by heat capacity CV, sound speed v and phonon relaxation time τ through the simplified relationship2,3 κL ∝ CVv2τ. The structures on atomic and/or unit cell levels basically decide the phonon dispersion, elastic properties, and the intensity of phonon–phonon normal and Umklapp scattering etc.4,5, while the microstructures in all scales contribute additional boundaries, defects, and interfaces as phonon scattering centers6. The physical mechanisms on different levels have been developed to explain material’s thermal conductivity observed in experiment. Particularly, a few conceptual directions have been proposed towards low κL, such as large cell with heavy atoms1,7, small elastic parameters8, and the existence of lone-pair electrons9,10, soft bonds8, rattling atoms11, and liquid-like ions12.
For the fields of thermoelectrics, design and discovery of new materials with low thermal conductivity is a key task. Materials genome initiative (MGI)13 offers a revolutionary scenario to efficiently find new materials with desired properties14, which is particularly suitable for thermal conductivity. MGI requires simple yet straightforward performance indicators for initial but fast high-throughput screening to pick up possible candidates. For lattice thermal conductivity, intuitive parameters such as average atomic mass (M) and primitive unit cell volume (VPUC) are available in the database, but they are far below the requirements for materials with complex crystal structures and lattice dynamics. For example, in chalcogenides, chemical bonding inequality, and hierarchy are diverse, leading to various factors affecting thermal transport such as abundant intrinsic defects, loose bonding, lone-pair, and rattling atoms. They are likely to coexist and interact with each other. Thus, it is quite difficult to find simple performance indicators to quickly guide the thermal conductivity.
In this work, we report that the number mismatch (δ) between cations (Ncation) and anions (Nanion), δ = (Ncation − Nanion)/Nanion, can be regarded as the simple performance indicator to quickly and directly explain and discover materials with low lattice thermal conductivity (κL) in complex chalcogenides. Furthermore, new low-κL materials such as Cu2Sn4S9 are screened by the proposed large number mismatch between cations and anions, and confirmed by experiment (Fig. 1).

The red line is a guide to the eyes. The green square is Cu4Sn7S16, which is used for the case study. Cu2Sn4S9 and Cu4SnS4 (red circle) are the screened low κL materials based on proposed number mismatch between cations and anions. The raw data and the source for each material can be found in Supplementary Table 1.
Results and discussion
We go back to check the room-temperature lattice thermal conductivity of 73 complex ternary copper and silver chalcogenides (Fig. 1 and Supplementary Table 1). It ranges from 0.18 W m−1 K−1 in Ag8GeTe6 to 6.8 W m−1 K−1 in CuInTe2, a quite large range. For each material, the δ value is calculated, and the room temperature κL varying with δ is shown in Fig. 1. Despite the diversity in composition and crystal structure, a clear decreasing trend of κL is observed when δ departs from 0, no matter whether the compounds are cation or anion rich. Particularly, all the materials with |δ| ≥ 0.5 have the κL values smaller than 0.5 W m−1 K−1. As shown in Supplementary Fig. 1, the significant, negative correlation between κL and |δ| is quantified by the Spearman correlation coefficient ρs = −0.79, which is closer to −1 (totally negative relation) than either κL-M or κL-VPUC. Furthermore, for each specific family of Cu-based materials, the decrease of normalized κL with |δ| is more straightforward (see Supplementary Fig. 2). These results strongly indicate that δ can be an effective performance indicator for low lattice thermal conductivity in Cu- and Ag-based complex chalcogenides. The detailed physical mechanisms for the low κL can be included in the simple parameter δ that can be immediately obtained from the chemical formula.
Cu4Sn7S16: a case study
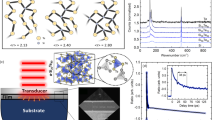
Here we pick up Cu4Sn7S16 as a case study to show how the simple parameter δ can be a performance indicator for the low κL. Complex semiconductor Cu4Sn7S16 is a largely distorted derivative of diamond-like compound with δ = −0.312515. As shown in Fig. 2a, Cu4Sn7S16 exhibits a low κL < 1 W m−1 K−1 above room temperature, and the dependence of κL on T is quite weak, which is comparable with multiple-filled skutterudites16 and Type-I clathrates17.

a κL as a function of temperature for various Cu-based chalcogenides. Data are taken from the followings: CuFeS2 (ref.22), Cu2SnS3 (this work), skutterudites (ref.16), clathrates (ref.17), Cu12Sb4S13 (ref.23), Cu2Se (ref.12), Cu3SbSe3 (ref.24), and Cu4Sn7S16 (this work, Supplementary Fig. 4). b Cp/T versus T2 for Cu4Sn7S16. The black squares are the experimental data. The red solid line represents the fitted curve by taking account the electronic, one Debye and two Einstein modes, which are shown by the dashed lines. Fitting parameters are given in Supplementary Table 2. c Phonon scattering mechanisms in Cu4Sn7S16. U, B, PD, and RES denote the Umklapp process, grain-boundary scattering, point-defect scattering and resonant scattering, respectively. Fitting parameters are given in Supplementary Table 3. d Crystal structure of Cu4Sn7S16 and local coordination of three types of Cu atoms.
There are a few reasons for such an observed low κL. Firstly and apparently, Cu4Sn7S16 possesses a complex crystal structure. In this structure with the R\(\overline {\mathrm{3}}\)m (Z = 3) symmetry, sulfur atoms S1 (18 h), S2 (18 h), S3 (6c), and S4 (6c) constitute triangular, tetrahedral, and octahedral frameworks, accommodating Cu and Sn atoms15 (Fig. 2d). This complex structure with diverse local structure units leads to a very large-cell volume (1695 Å3) with 81 atoms per unit cell15,18. The primitive unit cell volume VPUC is 565 Å3 for Cu4Sn7S16, which is considerably larger than typical diamond-like materials (VPUC is 150–250 Å3). Intuitively, a large and complex cell corresponds to numerous optical phonon modes (see Fig. 3d). These optical modes store a majority of heat (~78% at 20 K as shown in Fig. 2b) but exhibit a low group velocity while the acoustic modes contribute only ~20%. In addition, the optical modes may also suppress and even interact with the acoustic modes, thus suppressing the thermal transport.

a–c Molecular dynamics trajectories for Cu4Sn7S16; d Phonon dispersion and projected phonon density of states.
Secondly, Cu1 atoms randomly occupy half of the 6c sites15. Therefore, high-density intrinsic vacancies are present and strongly scatter high-frequency phonons, reducing the κL by one order of magnitude (from 11.1 to 1.68 W m−1 K−1) at room temperature (Fig. 2c).
Thirdly, our ab initio molecular dynamics (AIMD) simulations (Fig. 3a–c) and crystallographic data19,20 show that Cu2 and Cu3 atoms exhibit a noticeably large vibration amplitude along c-axis and within ab-plane like rattling modes, respectively. These rattling-like behaviors lead to low-lying optical modes which start from as low as 2 meV and are intensely overlapped with the acoustic modes as indicated in Fig. 3d. In addition to altering the acoustic phonon dispersion near the boundary of Brillouin zone (particularly along Γ-A/L direction), the low-lying optical modes also introduce resonant scattering on acoustic phonons21, further suppressing κL to 0.78 W m−1 K−1 at 300 K as shown in Fig. 2c. The reduction of κL is more directly demonstrated by the spectra data shown in Supplementary Fig. 3, where areas II and III are the contributions by point-defect scattering and resonant scattering, respectively.
As shown above, the large |δ| value in Cu4Sn7S16 compound is unambiguously related to the low κL through the detailed physical mechanisms of large cell, rattling modes, and point defects. In fact, such a relationship can be easily extended to other materials. For example, Cu3SbSe3 has a large δ value of 0.33 and a low κL of 0.5–0.6 W m−1 K−1 at 300 K with the mechanisms of Cu rattling11,25 and/or lone-pair electrons9. Similarly, tetrahedrite Cu12Sb4S13 has a δ of 0.23 and a low κL about 0.65 W m−1 K−1 at room temperature26,27. All these detailed mechanisms of suppressing κL can be included in the simple performance indicator δ, which is schematically depicted in Fig. 4a, b and will be expanded in the next part.

a, b Schematic depiction of the effects of number mismatch between cations and anions on thermal transport in Cu-based chalcogenides. Ionic number mismatch varying with c primitive cell volume for Cu- and Ag-based compounds and d local bond distortion for Cu-based compounds.
Effects of δ on thermal transport
δ and large-cell effect
In chalcogenides, the relatively simple crystal structure cannot persist when |δ| is large enough. Thus, a larger cell with more atoms and lower symmetry tends to be adopted in order to accommodate the diverse bonding. In Fig. 4c, when |δ| is large, the primitive cell volume is large too. Although the data are scattered, the positive correlation between |δ| and VPUC is quite clear. For large cells, the lattice complexity shrinks the first Brillouin zone and induces more optical modes. As a consequence, a majority of heat is damped in slowly propagating optical waves, and the acoustic branches are smothered or even interacted by the optical modes4. All these factors lead to the great reduction of κL.
δ and vacancies
Large δ means that certain crystallographic positions are not fully occupied. As an extreme case of point defects, vacancies have zero mass and free space to introduce large lattice fluctuation to scatter phonons. In fact, phonon scattering by point defects is a well-known strategy to reduce κL, which have been widely employed for a few dozens of years.
δ and local bonding distortion, rattling and lone pair
Large δ also brings about various local bonding distortions including the change in bond lengths and angles. Particularly, some atoms can be loosely bonded along certain directions or within certain planes, showing rattling-like vibrations to dramatically alter heat storage and transport. This effect has been demonstrated in the filled skutterudites16,28, Cu3SbSe311, Cu12Sb4S1326, and Ag-based materials8,29,30. An extreme case of bonding inequality is the lone-pair electrons, which can further enhance the lattice anharmonicity9,10. Here we defined the microscopic distortion index Δ to qualitatively describe the degree of local chemical bond distortion, which is simply assumed as the sum of distortions in bond length Δl and bond angle Δθ. For most Cu-based ternary chalcogenides, the cations are tetrahedrally or octahedrally bonded with anions in an ideal or distorted way. For the ith cation coordination, it is defined
where lmax and lmin are the largest and smallest bond lengths, respectively, and θmax and θmin are the largest and smallest bond angles, respectively. Here the data of ideally coordinated bonds are taken as the standard values. The detailed calculation method is described in Supplementary Fig. 5. For example, θideal are 109.5° and 90° for tetrahedral and octahedral bonding, respectively. The total distortion degree of a material is the sum of the ones for each cation divided by the cation number:
For the case of lone pairs, it is assumed that the distance between the center cation and the lone pair is half of the normal bond length.
We calculated the Δ values based on the crystal structure of 19 Cu-based materials. Only 16 materials have the value of κL (Supplementary Table 4). As shown in Supplementary Fig. 6a, the lattice thermal conductivity exhibits a clear decreasing trend with Δ. The correlation between Δ and δ is relatively weak and scattered at the first sight (Supplementary Fig. 6b). Nonetheless, we found that this poor relativity is mainly caused by the treatment of vacancy-containing materials, in which the bonding between the anions and the virtue void is not included. After removing the five vacancy-containing materials (Cu4Sn7S16, Cu2SnSe4, Cu2GeS4, Cu4SnS6, and CuSn3.75S8), the positive correction is clearly seen in Fig. 4d between the microscopic chemical bonding distortion parameter and the macroscopic ionic number mismatch. Thus, when δ deviates from 0, the degree to local chemical bond distortion is increased to greatly suppress κL.
κ L-δ diagram in Cu–Sn–S family
Parameter δ is an effective indicator for κL in the materials with structural complexity and local bonding distortions. Large absolute value of δ means low κL. Thus the δ criterion can be used to discover low κL materials. Here we still take Cu–Sn–S family as the “material base” from the reported phase diagram31. It includes a variety of low-cost and environmentally friendly compounds and part of them can be potential candidates for various applications like thermoelectric, solar cells, and electrode materials. As shown in Fig. 5, the available experimental data (green symbols) of this family well follow the “δ criterion”, further confirming the validity of this simple performance indicator. For the compounds31 (the short vertical lines at the bottom of Fig. 5) without κL values being reported in the previous studies, the “δ criterion” suggests that Cu2Sn2S9, CuSn3.75S8, Cu2Sn4S9, Cu9Sn2S9, and Cu5SnS4 are likely to exhibit low lattice thermal conductivities because of their large |δ| values. In experiment, we selected the nominally valence-state-balanced compound Cu2Sn4S9 (δ = −0.33) to check its κL. A low value of 1.0 W m−1 K−1 is observed at room temperature, well consistent with our prediction. In addition, the compound Cu4SnS4 has a large δ of 0.25, but there are some discrepancies on the reported κL32,33. Particularly, Goto et al. reported a high value of 4 W m−1 K−133, which is in contradictive with the “δ criterion”. We rechecked this compound and found its κL at room temperature is 0.36 W m−1 K−1, an extremely low value in chalcogenides. Furthermore, the κL data of the two compounds show weak dependences on temperature (Supplementary Fig. 14), which are in close analogy to Cu4Sn7S16 with similarly large δ while in sharp contrast to Cu2SnS3. All the data have substantiated the effectiveness of the concise δ criterion in screening low-κL materials.

The squares are the experimental data. The red line is fitted to guide the eyes. The short vertical line donates the mismatch of various Cu–Sn–S compounds from the reported phase diagram31 whose lattice thermal conductivity has not been reported. Materials with large |δ| values are expected to exhibit low κL.
Limitations of δ
There are certainly some limitations for this simple indicator. Fundamentally, the case of δ = 0 is pre-assumed to be the tetrahedrally bonded diamond-like semiconductors. The real case is more complicated. A typical example for this deviation is the Sb/Bi-containing compounds (e.g., CuBiS2 and AgSbS2), where the lone-pair electrons of Sb/Bi make the structure largely different from the chalcopyrite despite the same 1–1–2 composition. This is the main reason why the κL data are largely scattered for δ = 0 (Fig. 1). In addition, the mismatch criterion does not consider the difference in atomic mass. Therefore, this indicator is more effective and reliable for homologs, i.e., a family of compounds with the same elements but different compositions. A good demonstration has been shown above for the Cu–Sn–S materials.
In real applications, the total thermal conductivity is of greater interest. We found that most Cu/Ag-based ternary chalcogenides have extremely low κe (<5% of κtotal) at room temperature. Also, there seems no clear correlation between κe and |δ|. However, things can be more complicated when κe is nonnegligible in other systems, and the possible relationship between κe and |δ| needs further investigation.
In summary, this work proposed that the mismatch (δ) between the number of cations and anions is a practical performance indicator for low κL in ternary Cu- and Ag-based chalcogenides. When the mismatch exists, multiple phenomena and effects such as large cell, point defects, chemical bonding distortion and inequality, rattling atoms and lone pair may be induced to lower κL. This criterion and the underlying mechanisms have been illustrated in the Cu4Sn7S16 compound and well extended to other ternary Cu- and Ag-based chalcogenides. Guided by the κL-δ relationship, low-κL Cu2Sn4S9 compound was predicted and confirmed. Such an intuitive indicator provides a unique perspective for understanding thermal transports and is powerful for efficiently screening and designing low κL materials for energy conversion and thermal management applications.
Methods
Synthesis
Cu4Sn7S16, Cu2SnS3, Cu4SnS4, and Cu2Sn4S9 polycrystalline samples were synthesized by different methods with the details described in Supplementary Information.
Characterization
The phase purity and crystal structure were detected by X-ray diffraction with Cu Kα X-ray sources (XRD, Rigaku D/max 2550 V). The elemental distribution was characterized by scanning electron microscopy (ZEISS supra 55) and energy dispersive X-ray spectroscopy (Oxford) with accelerate voltage set as 20 kV. Samples’ phase purity and homogeneity are shown in Supplementary Figs. 7–13. High-temperature thermal conductivity was derived from the formula κ = ρCpλ, where ρ is the density, CP is the heat capacity calculated by Dulong–Petit law, and λ is thermal diffusivity measured between 300 and 700 K by using the laser flash method (LFA 457, NETZSCH). Low-temperature CP and κL were measured by Physical Property Measurement System (PPMS Quantum Design). The uncertainty for κL is within 10%.
Computation
The AIMD was performed for Cu4Sn7S16 at 300 K. A large supercell, 3 × 3 × 1 of the 81-atom unit cell (729 atoms in total), was constructed for the AIMD. The simulation time was over 18 ps with a step of 2 fs. The AIMD was carried out by the Vienna ab initio simulation package34, with the projector augmented wave method35 and Perdew–Burke–Ernzerhof generalized gradient approximation functional36. The temperature dependent effective potential method37,38 was adopted to extract the harmonic interatomic force constants and phonon dispersions from the AIMD trajectories and forces.
Data availability
The data supporting the findings of this study are available within the main text and its Supplementary Information files and are available from the corresponding authors upon reasonable request.
References
Berman, R. Thermal Conduction in Solids. (Clarendon Press, 1976).
Toberer, E. S., Baranowski, L. L. & Dames, C. Advances in thermal conductivity. Annu. Rev. Mater. Res. 42, 179–209 (2012).
Yang, J. in Thermal Conductivity: Theory, Properties and Applications (ed Terry, M. Tritt) Ch. 1.1, 1–20 (Plenum, 2004).
Toberer, E. S., Zevalkink, A. & Snyder, G. J. Phonon engineering through crystal chemistry. J. Mater. Chem. 21, 15843–15852 (2011).
Chen, Z., Zhang, X. & Pei, Y. Manipulation of phonon transport in thermoelectrics. Adv. Mater. 30, 1705617 (2018).
Biswas, K. et al. High-performance bulk thermoelectrics with all-scale hierarchical architectures. Nature 489, 414–418 (2012).
Slack, G. A. The thermal conductivity of nonmetallic crystals. Solid State Phys. 34, 1–71 (1979).
Lin, S. et al. High thermoelectric performance of Ag9GaSe6 enabled by low cutoff frequency of acoustic phonons. Joule 1, 1–15 (2017).
Skoug, E. J. & Morelli, D. T. Role of lone-pair electrons in producing minimum thermal conductivity in nitrogen-group chalcogenide compounds. Phys. Rev. Lett. 107, 235901 (2011).
Nielsen, M. D., Ozolins, V. & Heremans, J. P. Lone pair electrons minimize lattice thermal conductivity. Energy Environ. Sci. 6, 570–578 (2013).
Qiu, W. et al. Part-crystalline part-liquid state and rattling-like thermal damping in materials with chemical-bond hierarchy. Proc. Natl Acad. Sci. USA. 111, 15031–15035 (2014).
Liu, H. et al. Copper ion liquid-like thermoelectrics. Nat. Mater. 11, 422–425 (2012).
de Pablo, J. J. et al. New frontiers for the materials genome initiative. npj Comput. Mater. 5, 41 (2019).
Xi, L. et al. Discovery of high-performance thermoelectric chalcogenides through reliable high-throughput material screening. J. Am. Chem. Soc. 140, 10785–10793 (2018).
Chen, X.-a, Wada, H., Sato, A. & Mieno, M. Synthesis, electrical conductivity and crystal structure of Cu4Sn7S16 and Cu12Sb4S13. J. Solid State Chem. 139, 144–151 (1998).
Shi, X. et al. Multiple-filled skutterudites: high thermoelectric figure of merit through separately optimizing electrical and thermal transports. J. Am. Chem. Soc. 133, 7837–7846 (2011).
Shi, X. et al. On the design of high-efficiency thermoelectric clathrates through a systematic cross-substitution of framework elements. Adv. Funct. Mater. 20, 755–763 (2010).
Deng, T. et al. Thermoelectric properties of n-type Cu4Sn7S16-based compounds. RSC Adv. 9, 7826–7832 (2019).
Lemoine, P. et al. High temperature neutron powder diffraction study of the Cu12Sb4S13 and Cu4Sn7S16 phases. J. Solid State Chem. 247, 83–89 (2017).
Jemetio, J. P. F., Zhou, P. & Kleinke, H. Crystal structure, electronic structure and thermoelectric properties of Cu4Sn7S16. J. Alloy. Compd. 417, 55–59 (2006).
Pohl, R. O. Thermal conductivity and phonon resonance scattering. Phys. Rev. Lett. 8, 481–483 (1962).
Li, Y. et al. Thermoelectric transport properties of diamond-like Cu1−xFe1+xS2 tetrahedral compounds. J. Appl. Phys. 116, 203705 (2014).
Kosaka, Y. et al. Effects of Ge and Sn substitution on the metal–semiconductor transition and thermoelectric properties of Cu12Sb4S13 tetrahedrite. Phys. Chem. Chem. Phys. 19, 8874–8879 (2017).
Wei, T.-R., Wu, C.-F., Sun, W., Pan, Y. & Li, J.-F. Is Cu3SbSe3 a promising thermoelectric material? RSC Adv. 5, 42848–42854 (2015).
Qiu, W., Wu, L., Ke, X., Yang, J. & Zhang, W. Diverse lattice dynamics in ternary Cu-Sb-Se compounds. Sci. Rep. 5, 13643 (2015).
Lai, W., Wang, Y., Morelli, D. T. & Lu, X. From bonding asymmetry to anharmonic rattling in Cu12Sb4S13 tetrahedrites: when lone-pair electrons are not so lonely. Adv. Funct. Mater. 25, 3648–3657 (2015).
Suekuni, K. et al. Retreat from stress: rattling in a planar coordination. Adv. Mater. 30, e1706230 (2018).
Sales, B., Mandrus, D. & Williams, R. K. Filled skutterudite antimonides: a new class of thermoelectric materials. Science 272, 1325–1328 (1996).
Jiang, B. et al. An argyrodite-type Ag9GaSe6 liquid-like material with ultralow thermal conductivity and high thermoelectric performance. Chem. Commun. 53, 11658–11661 (2017).
Tan, G., Hao, S., Zhao, J., Wolverton, C. & Kanatzidis, M. G. High thermoelectric performance in electron-doped AgBi3S5 with ultralow thermal conductivity. J. Am. Chem. Soc. 139, 6467–6473 (2017).
Fiechter, S., Martinez, M., Schmidt, G., Henrion, W. & Tomm, Y. Phase relations and optical properties of semiconducting ternary sulfides in the system Cu–Sn–S. J. Phys. Chem. Solids 64, 1859–1862 (2003).
Choudhury, A. et al. New insights into the structure, chemistry, and properties of Cu4SnS4. J. Solid State Chem. 253, 192–201 (2017).
Goto, Y., Kamihara, Y. & Matoba, M. Effect of indium substitution on the thermoelectric properties of orthorhombic Cu4SnS4. J. Electron. Mater. 43, 2202–2205 (2014).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Hellman, O., Abrikosov, I. & Simak, S. Lattice dynamics of anharmonic solids from first principles. Phys. Rev. B 84, 180301 (2011).
Hellman, O. & Abrikosov, I. A. Temperature-dependent effective third-order interatomic force constants from first principles. Phys. Rev. B 88, 144301 (2013).
Acknowledgements
This work is supported by the National Key Research and Development Program of China (2018YFB0703600), the National Natural Science Foundation of China (51625205, 91963208, and 51802333), the Key Research Program of Chinese Academy of Sciences (KFZD-SW-421) and the Youth Innovation Promotion Association of CAS (2016232).
Author information
Authors and Affiliations
Contributions
T.D. and T.-R.W. contributed equally to this work. T.-R.W., J.Y., and X.S. proposed the idea and designed the experiments. T.D. performed material syntheses, sample measurements, and data collection. T.D. and T.-R.W. analyzed the data. J.Y. and H.H. performed the ab initio molecular dynamics (AIMD) calculations. T.D. and T.-R.W. wrote the papers with the guidance of J.Y., X.S., and L.C. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Deng, T., Wei, TR., Huang, H. et al. Number mismatch between cations and anions as an indicator for low lattice thermal conductivity in chalcogenides. npj Comput Mater 6, 81 (2020). https://doi.org/10.1038/s41524-020-00355-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-020-00355-x
