
Abstract
Inverse design is an outstanding challenge in disordered systems with multiple length scales such as polymers, particularly when designing polymers with desired phase behavior. Here we demonstrate high-accuracy tuning of poly(2-oxazoline) cloud point via machine learning. With a design space of four repeating units and a range of molecular masses, we achieve an accuracy of 4 °C root mean squared error (RMSE) in a temperature range of 24–90 °C, employing gradient boosting with decision trees. The RMSE is >3x better than linear and polynomial regression. We perform inverse design via particle-swarm optimization, predicting and synthesizing 17 polymers with constrained design at 4 target cloud points from 37 to 80 °C. Our approach challenges the status quo in polymer design with a machine learning algorithm, that is capable of fast and systematic discovery of new polymers.
Similar content being viewed by others

Introduction
Polymers are ubiquitous in both structural and functional systems, owing to their highly tunable physical, chemical, and electrical properties.1,2,3,4 The development of polymers has historically been based on an Edisonian approach. Herein, we develop a machine-learning framework to predict polymer structure (topology, composition, functionality, and size), on the basis of target-phase properties, specifically the cloud point. This framework accommodates the complex disorder across multiple length scales that distinguishes polymers from small molecules,5,6,7 inorganic crystals,8 and systems-structure optimization.9,10,11
Phase properties, which describe the order of a polymer across multiple length scales, are determined by interactions of polymers with other polymers, the solution, and themselves. One such phase property is the cloud point, the temperature at which polymers are no longer miscible in solution.12 Numerous studies tabulate simple relationships between cloud point and one or two experimental variables (e.g., structure13 and temperature14,15), or offer polynomial fits to the data.16 Ramprasad et al. applied machine learning to density-functional theory (DFT) calculations to predict optoelectronic17,18 and physical19 bulk polymer properties.4,19 However, this approach is computationally expensive,7,20 particularly for polymer systems,21 and does not enable scalable inverse design over a wide range of conditions with high accuracy.22,23
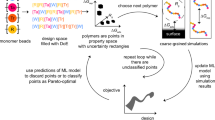
In this study, we combine machine learning, domain expertise, and experiment to solve the inverse-design problem for polymers. Our framework (Fig. 1) has three parts: (1) data curation (defining material descriptors) that relates poly(2-oxazolines) cloud point, size, and relative ratios of four different monomer units; (2) machine-learning algorithm selection and hyperparameter tuning to enable fast forward prediction of cloud point based on the structure with the evaluation of algorithmic robustness over systematic error and differing data quality; and (3) use of said algorithm for inverse design using particle-swarm optimization (PSO) with design selection using an ensemble of neural networks. We demonstrate the accuracy of our inverse-design paradigm by predicting the compositions of, and synthesizing, 17 polymers, not previously reported in the literature, with cloud points between 37 and 80 °C, using a modular combination of four repeating monomer units. We achieve ~4 °C error, nearly within experimental error (1–3 °C).

Study framework. First, we train a machine-learning model to predict cloud point on the basis of the poly(2-oxazoline) structure, with varying ratios of four monomer units (building blocks) and molecular weights. Second, we demonstrate inverse design using the trained algorithm and particle-swarm optimization, predicting 17 polymer structures from user-defined cloud points. The model accommodates the inherent complexity of polymers over multiple length scales. The scatter plots for model training and inverse design correspond to Figs 3e and 4d, respectively
Results and discussion
We combine and curate literature and experimental data to create the input into our machine-learning framework. Historical cloud-point data for poly(2-oxazoline)s16,24,25,26,27,28,29 were curated into a set of input variables ((1) molecular weight of the polymers; (2) polydispersity index; (3) polymer type (homo, statistical, or block); (4) the total number of each monomer unit in the final polymer (A: EtOx, B: nPropOx, C: cPropOx, D: iPropOx, E: esterOx)) and output variables (cloud point in °C) (Table S1). We synthesized 87 poly(2-oxazoline)s by similar methods to augment this data (Table S2). Cloud point was evaluated by dynamic-light scattering (DLS) in accordance with best practices,30 particularly since DLS affords greater weightage to the modal mass as a correction for the asymmetric molecular weight distributions (MWD) of our synthesized polymers (details in Supplementary Materials under the heading “Curation and synthesis of polymer library”). Due to data scarcity, esterOx was neither synthesized nor considered in inverse design. The relationships of individual input variables to the output cloud point are plotted in Fig. 2.

Data summary. The dependencies cloud points to the mole fraction of (a) EtOx; (b) nPropOx; (c) cPropOx; (d) iPropOx; and (e) molecular mass (M), where all zero values were filtered from the graphs, and, (f) the number distribution of cloud point, where zero represents polymers without a CP
We test whether machine-learning methods have superior predictive accuracies to simple regression methods in this multi-variable parameter space.31,32,33 We compare the root-mean-squared errors (RMSE) of simple linear and quadratic regressions against more robust machine-learning methods, including support vector regressions (SVR), (ensembles of) neural networks (NN), and gradient boosting regression with decision trees (GBR) (Fig. 3; S3). The accuracies of the various models are determined by splitting the input data set into training, validation, and test sets, with training and validation performed from the historical data, while testing is performed with the experimental data. The RMSE and inference times are reported in Table S3.

Machine-learning performance. (Top row) Comparison of three regression methods (a–c: linear, polynomial (order-2), and gradient boosting (with decision trees) regressions). The literature data are split into 68 training data points and 7 validation data points. Test data points are 42 experimental data points produced in the lab. The results were compared using the root-mean-squared error. We observe that GBR achieves the best generalization. (Bottom row) Final GBR model performance on three different random train-test splits of the combined data set
Linear and polynomial regressions, while significantly faster than the others, performed poorly when compared with SVR, NN, and GBR. Of the latter three, GBR was the more accurate out-of-the-box without extensive hyperparameter tuning. Moreover, it possesses fast inference speed, which is essential for efficient exploration of the parameter space in inverse design. We chose GBR as our primary forward model to balance fast inference speed and good test RMSE. The predictive accuracy was further improved by tuning via a cross-validation grid search on hyperparameters. We used both historic and experimental data, with a test set of 10%, to validate our choice of hyper-parameters with the test error on three randomly split training and test sets (Fig. 3). We now observe improved performance with an increased data set and thorough tuning.
This algorithm is shown to generalize well across the variation in polymer data set of varying polydispersity. The historical data sets had narrow polydispersity indices with the assumption of symmetrical MWDs, while the synthesized polymers had broad and unsymmetrical MWD. Nevertheless, the model trained on the historical data still performed adequately on data from our synthesized polymers. The robustness of this algorithm in handling variations in the data renders this far more powerful than less sophisticated algorithms, which may require highest quality of data. With a sufficiently accurate model, we finally retrain (using the tuned hyper-parameters) on the entire data set to produce a finalized forward model that we use for subsequent inverse design.
The feature importance ranking based on Gini importance or gain (roughly the mean improvement in objective due to splits in the chosen feature, see the Ref. 34 for more details) (Fig. S4) indicates that “units of A” and “molecular mass” are the two most important features defining cloud point. We note that these insights are not trivially derived from Fig. 2, which indicates similarly strong dependences of variables a–c on cloud point. Also, the molecular mass correlating most strongly with cloud point is the mode, not the median or mean (Fig. S2), which we speculate could indicate a critical threshold, e.g., of polymers with molecular mass above a certain concentration necessary to induce globule formation. However, we note that this statistical relationship depends on the model and fitting algorithm employed, and certainly does not imply the presence of causal relationships, for which more rigorous theoretical and experimental studies must be conducted.
While a forward predictive models in machine-learning approaches for materials science are fairly common, inverse design is far more challenging. This is because the descriptors, which are usually high dimensional, are difficult to predict from outputs which are low dimensional. In the case of our polymer data set, the output of cloud point is a single number, attributed to the five numbers representing molecular mass and composition of the polymer.
Inverse design would provide the ability to design polymers based on a desired final property, and accelerate the synthesis process of target polymers based on design constraints to meet desired cloud points. To further realize new material discovery, we propose to extrapolate from our training data set by designing terpolymers, which are nonexistent in our training set, and limiting EtOx composition, which is common.
Typically, inverse optimization on piecewise constant functions provides a large number of different predicted designs. These may achieve our optimization and constraint target according to the fitted GBR model. However, the quality of these designs vary, particularly in the case of extrapolation. By extrapolation, we mean designs that are different in class from the training data set (e.g., binary vs. ternary systems), or in a more precise sense, those that lie outside of the convex hull of the training data points, which is the smallest convex set containing all the points. Validating all of produced designs experimentally would be inefficient and so a filtering method with an ensemble of M three-layer fully connected neural networks (NN) was employed to select the most promising design candidates for experimental validation. Each NN’s trainable parameters are initialized with distinct, random values, resulting in different fitted predictors \(\{ \hat f_1, \ldots ,\hat f_M\}\), due to the non-convex nature of the objective function and random initialization. Note that this is even the case when a deterministic training algorithm is used (e.g., full-batch gradient descent), hence this heterogeneity is inherent in our model choice. For each design x, we then compared the ensemble of NN-predicted cloud points \(\{ \hat f_1(x), \ldots ,\hat f_M(x)\}\) with the GBR prediction \(\hat f(x)\) and only experimentally validated designs where \(\hat f\left( x \right) \approx \frac{1}{M}\mathop {\sum }\nolimits_{i = 1}^M \hat f_i(x)\)(NN predictions agree with GBR) and \(Var\{ \hat f_1(x), \ldots ,\hat f_M(x)\}\) was small. This ensures that x is predicted with high confidence and not an ad-hoc extrapolation. As far as we are aware, there is no concrete theory analyzing the relationship between generalization properties of neural networks with the variance of the ensemble predictions, in which each network is trained with random initial conditions. However, we found experimentally that this is an effective filtering strategy. Figure 4 illustrates the principle of this approach. Although the NNs are also good approximators for the cloud point, they were not used as the forward model for producing inverse-design candidates because the feed-forward step of the NN ensemble is still too slow compared with GBR, which consists of simple summing of piecewise constant functions.

Inverse Design. (a) Framework of the selection criteria, where the data set is used to train GBR and NN ensemble, PSO predicts polymer design (x*) a desired CP (y*), and the design is verified for accuracy by the NN ensemble where CP agreement is a downselection criteria. (b) Illustration of the validity of the filtering procedure. We observe that given limited training data, not all extrapolated points are valid. However, when an ensemble of neural networks trained with distinct initializations agree on a certain input, then we have a much greater confidence in the validation of their predictions. (c) Final PSO-based inverse-design performance, with experimental values (orange triangles) showing an RMSE of 3.9 °C. (d) Forward model (NN ensemble) performance of the polymers synthesized from design (orange triangles)
Using this technique, we downselected 17 polymers over our four desired cloud points (37, 45, 60, 80 °C) designing polymers with more than two components—unseen in the training data. Several design constraints were imposed in order to narrow the search space, based on a weightage to minimize EtOx and also to limit the polymer design within the bounds of what could be made with our laboratory resources. From this series of design and downselection, we observe that a significant proportion of the target and obtained designs (~35%) lie strictly outside the convex hull of the training data (see Table S4). Hence, some of these designs are also extrapolations in a precise mathematical sense.
These polymers were synthesized, although an average of three iterations were required to achieve the target mass and composition of the designs, owing to the difficulties with terpolymer synthesis, where the Mayo–Lewis equation does not apply in calculating required feed ratio of monomer for desired final copolymer composition. The mass and composition of the synthesized polymers are reported in Table S4, showing minimal deviation from algorithmic design, along with their cloud points (an average of three measurements). The RMSE of the obtained cloud points was 3.9 °C, however, when the polymer structure of the new polymers is fed back into the NN ensemble, a larger RMSE is observed (6.1 °C) (Fig. 4). Deviation from the target cloud points was within test RMSE between 37 and 60 °C, but above it at 80 °C, and can be attributed to sparseness of the data set at higher temperatures (Fig. 2f)—an in-depth analysis is provided in Supplementary Materials under the heading “Machine-Learning Validation”. These results show that our combination of slow and fast algorithms are able to design polymers with unique compositions with control over the desired physical property and structural design.
Overall, a significant conceptual advance in polymer design has been achieved via judicious application of machine-learning methods. This was done in three important steps. First, we curated and categorized historical and new data. Second, we selected and fine-tuned a machine-learning model based on gradient boosting regression with decision trees, resulting in a cloud point predictive accuracy of 3.9 °C (RMSE). The model was able to generalize well with both well-defined historic data sets as well as newly synthesized polymers of unsymmetrical MWDs. Third, polymer inverse design by particle-swarm optimization which predicted the design of new polymers based on desired cloud points spread over the range of the cloud points of the training data (37, 45, 60, 80 °C). We discuss how our inverse-design methodology is scalable to more than one objective function. We also demonstrated how we could extrapolate beyond the training set via an ensemble of neural networks as a cross-validation technique to downselect 17 polymers with the lowest variance across predictions. The RMSE of predicted polymers were similar to those of the forward model. This methodology offers unprecedented control of polymer design, which may significantly accelerate polymer design for one or more objective properties well beyond cloud points.
Methods
Materials
2-n-propyl-2-oxazoline (nPropOx),1 2-cyclopropyl-2-oxazoline (cPropOx),2 and 2-isopropyl-2-oxazoline (iPropOx)3 were synthesized as described in the literature, and distilled over calcium hydride and stored with molecular sieves (size 5 Å) in a glovebox. In all, 2-ethyl-2-oxazoline (EtOx, Sigma-Aldrich) was distilled over calcium hydride and stored with molecular sieves (size 5 Å) in glovebox. All other reagents were used as supplied unless otherwise stated.
Analytical methods
Nuclear magnetic resonance (NMR)
The compositions of the polymers were determined using 1H NMR spectroscopy. 1H NMR spectra were on JEOL 500 -MHz NMR system (JMN-ECA500IIFT) in CDCl3. The residual protonated solvent signals were used as reference.
Size exclusion chromatography (SEC)
Gel permeation chromatography (GPC) measurements were performed in THF (flowrate: 1 mL/min) on a Viscotek GPC Max module equipped with Phenogel columns (10−3 and 10−5 Å) (size: 300 × 7.80 mm) in series heated to 40 °C. The average molecular weights and polydispersities were determined with a Viscotek TDA 305 detector calibrated with poly(methyl methacrylate) standards.
Dynamic-light scattering (DLS)
Measurements at various temperatures were conducted using a Malvern Instruments Zetasizer Nano ZS instrument equipped with a 4 mV He–Ne laser operating at l = 633 nm, an avalanche photodiode detector with high quantum efficiency, and an ALV/LSE-5003 multiple tau digital correlator electronics system. on Malvern Nano ZS. Solutions of polymers (5 mg/mL) were prepared by dissolving polymer in deionized water at room temperature. The solutions were then heated to 100 °C and cooled down to remove thermal memory, before measurements were taken.
Experimental methods
For all polymerizations, the polymerization mixture was prepared in vials that were dried in 100 °C oven overnight before use, and crimped air-tight in a glovebox. The mixture contained the monomers (EtOx, nPropOx, cPropOx, iPropOx) of desired ratios, with a total monomer concentration of 4 M, anhydrous acetonitrile (ACN) and methyl tosylate (MeOTs) as initiator. The amount of methyl tosylate added was determined by the various [M]/[I] ratios. Temperature controlled polymerizations were performed in sealed vials in a microwave reactor equipped with IR temperature sensor at 140 °C for different length of time. The mixture was then cooled to ambient temperature and quenched by addition of tetramethylammonium hydroxide (2.5 wt% in methanol, 2 equivalence relative to initiator). The solutions were concentrated by removing some of the solvent under reduced pressure, then precipitated in cold diethyl ether. The product was collected and dried under reduced pressure overnight. All polymers were redissolved in THF for SEC, CDCl3 for 1H NMR and deionized water for DLS. 1H NMR of P((EtOx)w(nPropOx)x(cPropOx)y(iPropOx)z) (500 MHz, CDCl3, δ, ppm): 0.8 (d, 66.5 Hz, 4 yH, CHCH2 CH2), 0.96 (s, 3x H, CH2CH2CH3), 1.11 (s, 6z H, CHCH3CH3), 1.12 (s, 3w H, CH2CH3), 1.64 (s, 2x H, CH2CH2CH3) 2.30 (d, 56.5 Hz, 2x H, NCOCH2CH2CH3), 2.38 (s, 2w H, NCOCH2CH3), 2.70 (d, 61.0 Hz, y H, CHCH2CH2), 2.80 (d, 123.5 Hz, z H, CHCH3CH3), 3.49 (s, 2(w+x+y+z) H, CH2 backbone). Whereby w, x, y, and z are the mole ratio of EtOx, nPropOx, cPropOx, and iPropOx, respectively.
Data availability
The data generated and analyzed during the current study can be found in the Supplementary Materials (Figures S3–S5, Tables S1–S4), and also in our repository (https://github.com/LiQianxiao/CloudPoint-MachineLearning) along with details of our code implementation.
References
Garcia, S. J. Effect of polymer architecture on the intrinsic self-healing character of polymers. Eur. Polym. J. 53, 118–125 (2014).
Rinkenauer, A. C., Schubert, S., Traeger, A. & Schubert, U. S. The influence of polymer architecture on in vitro pDNA transfection. J. Mater. Chem. B 3, 7477–7493 (2015).
Paramelle, D., Gorelik, S., Liu, Y. & Kumar, J. Photothermally responsive gold nanoparticle conjugated polymer-grafted porous hollow silica nanocapsules. Chem. Commun. 52, 9897–9900 (2016).
Mannodi-Kanakkithodi, A., Pilania, G., Huan, T. D., Lookman, T. & Ramprasad, R. Machine learning strategy for accelerated design of polymer dielectrics. Sci. Rep. 6, 20952 (2016).
Wei, J. N., Duvenaud, D. & Aspuru-Guzik, A. Neural networks for the prediction of organic chemistry reactions. ACS Cent. Sci. 2, 725–732 (2016).
Gómez-Bombarelli, R. et al. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 4, 268–276 (2018).
Sanchez-Lengeling, B. et al. A Bayesian approach to predict solubility parameters. Adv. Theory Simul. https://doi.org/10.1002/adts.201800069 (2018).
Ye, W., Chen, C., Wang, Z., Chu, I.-H. & Ong, S. P. Deep neural networks for accurate predictions of crystal stability. Nat. Commun. 9, 3800 (2018).
Gómez-Bombarelli, R. et al. Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat. Mater. 15, 1120 (2016).
Brandt, R. E. et al. Rapid photovoltaic device characterization through bayesian parameter estimation. Joule 1, 843–856 (2017).
Raccuglia, P. et al. Machine-learning-assisted materials discovery using failed experiments. Nature 533, 73 (2016).
Bejagam, K. K., An, Y., Singh, S. & Deshmukh, S. A. Machine-learning enabled new insights into the coil-to-globule transition of thermosensitive polymers using a coarse-grained model. J. Phys. Chem. Lett. 9, 6480–6488 (2018).
Jiang, R., Jin, Q., Li, B., Ding, D. & Shi, A.-C. Phase diagram of poly(ethylene oxide) and poly(propylene oxide) triblock copolymers in aqueous solutions. Macromolecules 39, 5891–5896 (2006).
Ashbaugh, H. S. & Paulaitis, M. E. Monomer hydrophobicity as a mechanism for the LCST behavior of poly(ethylene oxide) in water. Ind. Eng. Chem. Res 45, 5531–5537 (2006).
Aseyev, V., Tenhu, H. & Winnik, F. M. in Self Organized Nanostructures of Amphiphilic Block Copolymers II (eds Müller, A. H. E. & Borisov, O.) 29–89 (Springer, Berlin Heidelberg, 2011).
Hoogenboom, R. et al. Tuning the LCST of poly(2-oxazoline)s by varying composition and molecular weight: alternatives to poly(N-isopropylacrylamide)? Chem. Commun. 0, 5758–5760 (2008).
Huan, T. D. et al. A polymer dataset for accelerated property prediction and design. Sci. Data 3, 160012 (2016).
Mannodi-Kanakkithodi, A. et al. Scoping the polymer genome: a roadmap for rational polymer dielectrics design and beyond. Mater. Today 21, 785–796 (2018).
Kim, C., Chandrasekaran, A., Huan, T. D., Das, D. & Ramprasad, R. Polymer genome: a data-powered polymer informatics platform for property predictions. J. Phys. Chem. C. 122, 17575–17585 (2018).
Kutzner, C. et al. Best bang for your buck: GPU nodes for GROMACS biomolecular simulations. J. Comput. Chem. 36, 1990–2008 (2015).
Dünweg, B. & Kremer, K. Molecular dynamics simulation of a polymer chain in solution. J. Chem. Phys. 99, 6983–6997 (1993).
Stuart, M. A. C. et al. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 9, 101–113 (2010).
Halperin, A., Kröger, M. & Winnik, F. M. Poly(N-isopropylacrylamide) phase diagrams: fifty years of research. Angew. Chem. Int Ed. 54, 15342–15367 (2015).
Contreras, M. M., Mattea, C., Rueda, J. C., Stapf, S. & Bajd, F. Synthesis and characterization of block copolymers from 2-oxazolines. Des. Monomers Polym. 18, 170–179 (2015).
Glassner, M., Lava, K., de la Rosa, V. R. & Hoogenboom, R. Tuning the LCST of poly(2-cyclopropyl-2-oxazoline) via gradient copolymerization with 2-ethyl-2-oxazoline. J. Polym. Sci. A 52, 3118–3122 (2014).
Diab, C., Akiyama, Y., Kataoka, K. & Winnik, F. M. Microcalorimetric study of the temperature-induced phase separation in aqueous solutions of poly(2-isopropyl-2-oxazolines). Macromolecules 37, 2556–2562 (2004).
Park, J.-S., Akiyama, Y., Winnik, F. M. & Kataoka, K. Versatile synthesis of end-functionalized thermosensitive poly(2-isopropyl-2-oxazolines). Macromolecules 37, 6786–6792 (2004).
Park, J.-S. & Kataoka, K. Precise control of lower critical solution temperature of thermosensitive poly(2-isopropyl-2-oxazoline) via gradient copolymerization with 2-ethyl-2-oxazoline as a hydrophilic comonomer. Macromolecules 39, 6622–6630 (2006).
Park, J.-S. & Kataoka, K. Comprehensive and accurate control of thermosensitivity of poly(2-alkyl-2-oxazoline)s via well-defined gradient or random copolymerization. Macromolecules 40, 3599–3609 (2007).
Zhang, Q., Weber, C., Schubert, U. S. & Hoogenboom, R. Thermoresponsive polymers with lower critical solution temperature: from fundamental aspects and measuring techniques to recommended turbidimetry conditions. Mater. Horiz. 4, 109–116 (2017).
Cortes, C. & Vapnik, V. Support-vector networks. Mach. Learn 20, 273–297 (1995).
Rokach, L. & Maimon, O. Data Mining With Decision Trees: Theory and Applications (World Scientific Publishing Co., Inc., Singapore, 2014).
LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436 (2015).
Hastie, T., Tibshirani, R. & Friedman, J. The Elements of Statistical Learning, Vol. 1 (Springer, New York, 2001).
Acknowledgements
We thank Kedar Hippalgaonkar for scientific and framing discussions. J.N.K., Q.L. and T.B. are supported by the AME Programmatic Fund by the Agency for Science, Technology, and Research under Grant no. A1898b0043.
Author information
Authors and Affiliations
Contributions
J.N.K. was responsible for the (1) ideation; (2) experiments in polymer synthesis and characterization; (3) data curation and application of machine learning; (4) collation of information and representation of findings. Q.L. developed the machine-learning methodology, was involved in data curation, algorithm development, and was the architect of the multi-method strategy, including particle-swarm optimization. K.Y.T.T. carried out experiments in polymer synthesis and characterization and data curation. T.B. was involved in the ideation, collation, and representation of findings. A.L.G-O. developed the machine-learning methodology and algorithm development, as well as the application of inverse design. J.Y. was involved in ideation, data curation, and the representation of findings.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kumar, J.N., Li, Q., Tang, K.Y.T. et al. Machine learning enables polymer cloud-point engineering via inverse design. npj Comput Mater 5, 73 (2019). https://doi.org/10.1038/s41524-019-0209-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-019-0209-9
This article is cited by
-
Sizing up feature descriptors for macromolecular machine learning with polymeric biomaterials
npj Computational Materials (2023)
-
Applied machine learning as a driver for polymeric biomaterials design
Nature Communications (2023)
-
Artificial intelligence driven design of catalysts and materials for ring opening polymerization using a domain-specific language
Nature Communications (2023)
-
Polymer graph neural networks for multitask property learning
npj Computational Materials (2023)
-
Materials informatics approach using domain modelling for exploring structure–property relationships of polymers
Scientific Reports (2022)
