
Abstract
Population-based estimates of breast cancer risk for carriers of pathogenic variants identified by gene-panel testing are urgently required. Most prior research has been based on women selected for high-risk features and more data is needed to make inference about breast cancer risk for women unselected for family history, an important consideration of population screening. We tested 1464 women diagnosed with breast cancer and 862 age-matched controls participating in the Australian Breast Cancer Family Study (ABCFS), and 6549 healthy, older Australian women enroled in the ASPirin in Reducing Events in the Elderly (ASPREE) study for rare germline variants using a 24-gene-panel. Odds ratios (ORs) were estimated using unconditional logistic regression adjusted for age and other potential confounders. We identified pathogenic variants in 11.1% of the ABCFS cases, 3.7% of the ABCFS controls and 2.2% of the ASPREE (control) participants. The estimated breast cancer OR [95% confidence interval] was 5.3 [2.1–16.2] for BRCA1, 4.0 [1.9–9.1] for BRCA2, 3.4 [1.4–8.4] for ATM and 4.3 [1.0–17.0] for PALB2. Our findings provide a population-based perspective to gene-panel testing for breast cancer predisposition and opportunities to improve predictors for identifying women who carry pathogenic variants in breast cancer predisposition genes.
Similar content being viewed by others


Introduction
Data from breast cancer predisposition gene panel testing are accumulating rapidly as it becomes more affordable and more accessible in different settings, including clinical care. Large studies based in clinical and commercial testing laboratories have demonstrated that gene panel testing has increased the number of clinically actionable variants identified in women undergoing testing, compared with previous testing that included only BRCA1 and BRCA2. This increase in actionable findings is in the order of 5–10% depending on the setting, inclusion criteria for actionable variants and study design1,2,3,4,5.
Most of this work has been based on women selected for high-risk features, such as personal or family history of breast cancer, who underwent gene panel testing for cancer susceptibility at commercial laboratories6,7,8. Far fewer data are available to make inference about breast cancer risk for women unselected for family history, which is important to consider for population screening of affected and unaffected women. The value of population-based case-control studies and gene panel testing have recently been illustrated by Hu et al.9 who reported the outcome of a US-based study (CARRIERS consortium), involving over 32,000 affected and 32,000 unaffected women and Dorling et al.10 who reported the outcome of an international study (BRIDGES) involving 60,000 women affected and over 53,000 women unaffected by breast cancer. These studies provided improved estimates of the prevalence and the magnitude of breast cancer risk associated with pathogenic variants in known breast cancer predisposition genes to guide genetic counselling.
Several studies have only included women affected by breast cancer (case only) and have reported variant prevalence1,2,3,4. Kurian et al.11 linked cancer registries from Georgia and California (USA) to the gene panel testing outcomes from four key clinical testing laboratories. Their study linked 24.1% of the 77,085 women with breast cancer to genetic testing results and reported that panel testing increased the frequency of actionable genetic findings by 1.5%. Several large studies have reported estimates of breast cancer risk associated with carrying a rare germline variant in a gene included in these gene panel tests7,8,9,10,12,13,14,15,16.
Here, we report the prevalence and breast cancer risk estimates associated with pathogenic rare variants identified in breast cancer predisposition gene panel tests, conducted in an Australian population-based case-control study of breast cancer (with an emphasis on early age at disease onset), involving both (i) age-matched population-based controls and (ii) a healthy older group of Australian women as controls.
Results
Study subjects
Table 1 and Fig. 1 give descriptive statistics for the study subjects. Regarding the six potential confounders that we used as adjustment variables in our main analyses, the ABCFS cases and controls were very similar to each other, and both were similar to the ASPREE controls except that the ASPREE controls were older, as expected due to study design differences.

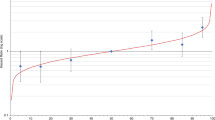
Boxplots of potential confounders, used as adjustment variables in the analyses, for controls from the ASPirin in Reducing Events in the Elderly (ASPREE; in green) study, and for cases and controls from the Australian Breast Cancer Family Study (ABCFS; in purple and blue). Panels are for age (years), height (m), body mass index (kg/m2), number of children, education (years) and alcohol consumption (drinks per week), as indicated. For each boxplot, the horizontal lines are at the potential confounder’s median (bold line), 25th and 75th percentiles (horizontal bounds of the box) and most extreme data points within a distance, from the box, of 1.5 times the interquartile range (shorter horizontal lines).
Gene panel testing
There were 162 (11%) ABCFS cases with a pathogenic variant, compared with 32 (4%) of the ABCFS controls and 145 (2%) of the ASPREE controls (Table 2). Further details of pathogenic variants detected are provided in Supplementary Tables 1–3 for the ABCFS cases, ABCFS controls and ASPREE participants, respectively. The number of carriers of pathogenic variants in genes other than BRCA1 or BRCA2 in the ABCFS cases, ABCFS controls and ASPREE controls were 73 (5%), 22 (3%) and 128 (2%) respectively.
Statistical analyses
We found evidence of association with breast cancer risk for four genes, with estimated adjusted ORs of 5.3 [95% CI: 2.1–16.2] for BRCA1, 4.0 [95% CI: 1.9–9.1] for BRCA2, 3.4 [95% CI: 1.4–8.4] for ATM and 4.3 [95% CI: 1.0–17.0] for PALB2 (Table 2, Fig. 2). CHEK2 was the gene that carried the highest number of pathogenic variants after BRCA1 and BRCA2. We observed no evidence of an association between pathogenic variants in this gene and breast cancer risk in our main analyses (OR 1.3 [95% CI: 0.53–3, p = 0.6]), though there was evidence of an association from unadjusted analyses (p = 0.0009; Supplementary Table 4), which give biased estimates of risk but valid tests of association. Our study also found no statistical difference between c.1100delC (OR 1.1 [95% CI: 0.29–3.8]) and all other CHEK2 pathogenic variants (OR 1.4 [95% CI: 0.44–5.1]), p = 0.75. For the other genes included in our analysis, the evidence for an association between breast cancer and pathogenic variants was weak or absent, and did not reach statistical significance (Table 2). Adjustment for age had a large influence on the ORs for some genes, as expected, but further adjustment for the remaining potential confounders had a relatively small effect (Supplementary Table 4).

Adjusted odds ratios (large dots) and corresponding 95% confidence intervals (vertical lines) for the association between breast cancer and pathogenic variants in various genes, sorted by p-value.
Breast cancer case subjects with pathogenic variants in BRCA1 or BRCA2 had, on average, a younger age at diagnosis than case subjects with pathogenic variants in genes other than BRCA1 or BRCA2 (p = 0.002), with average ages at diagnosis of 38.2 [95% CI: 36.7–39.8] years and 42.4 [95% CI: 40.3–44.6] years, respectively. Carriers with pathogenic variants in BRCA1 or BRCA2 were also more likely to have a family history of breast cancer than carriers of pathogenic variants in the other genes (p < 0.0001).
Pathogenic variants in BRCA1 were strongly associated with an increased risk of ER-negative breast cancer (Supplementary Table 5). There was also weak evidence that pathogenic variants in CHEK2 were associated with risk of ER-negative breast cancer, though only after age adjustment. Pathogenic variants in ATM, BRCA1, BRCA2 and PALB2 were all associated with the risk of ER-positive breast cancer (Supplementary Table 6). Carriers of pathogenic variants in BRCA1 were less likely than non-carriers to have ER-positive breast cancer, presumably as a consequence of the strong association between pathogenic variants in BRCA1 and ER-negative breast cancer, since ER-positive and ER-negative breast cancer were treated as separate diseases for these analyses.
Sensitivity analysis showed that excluding the ASPREE controls gave broadly similar adjusted OR estimates as the main analyses, though with wider confidence intervals (Supplementary Table 7), validating our adjustment for age. Another sensitivity analysis showed that our results were almost unaffected by the exclusion of subjects with pathogenic variants in two or more genes (Supplementary Table 8).
Discussion
The OR for breast cancer risk associated with a pathogenic variant in BRCA1 and BRCA2 in this study is consistent with other estimates8. These estimates are lower than reports that involve cases selected via criteria targeting breast and ovarian cancer syndromes and triple-negative breast cancer12,13. The breast cancer risk estimates reported here for BRCA1 and BRCA2 are less than the point estimates published by Dorling et al. and Hu et al. but are not statistically significantly different9,10.
Consistent with many other studies we found that, after pathogenic variants in BRCA1 or BRCA2, pathogenic variants in CHEK2 were the most frequently identified. The prevalence of CHEK2 c.1100delC in some populations makes it possible for analyses to consider the risk associated with this variant individually. Although there is some evidence that this risk may be higher than that associated with all other CHEK2 pathogenic variants, our estimates did not reach statistical significance. For instance, for breast cancer risk, Lu et al. estimated an OR of 4.00 [95% CI: 2.04–8.73] for CHEK2 c.1100delC and an OR of 1.42 [95% CI: 0.76–2.81] for all other CHEK2 pathogenic variants in their study of 11,416 affected women16 and Dorling et al. estimated an OR of 2.66 [95% CI: 2.27–3.11] for CHEK2 c.1100delC and an OR of 2.13 [95% CI: 1.60–2.84] for all other CHEK2 pathogenic variants in the population-based setting of their study10. Our study also found no statistical difference between c.1100delC (OR 1.1 [95% CI: 0.29–3.8]) and all other CHEK2 pathogenic variants (p = 0.75). CHEK2 pathogenic variant carriers are an important group of women to identify as they are at an increased risk of contralateral breast cancer and have a lower survival compared with non-CHEK2 pathogenic variant carriers (these data are predominantly informed by information about CHEK2 c.1100delC17,18,19,20,21,22) and could benefit from specific screening modalities such as magnetic resonance imaging23.
We identified 17 carriers of pathogenic variants in ATM, only one of which was ATM c.7271T > G, a pathogenic variant that is well described in the Australian population and has an established association with a substantially increased risk of breast cancer24,25,26. Breast cancer risk estimates for women carrying other pathogenic variants in ATM have been consistently reported to be in the order of 2–3-fold8,9,10,12,13,14,16.
The literature has consistently reported a prevalence of germline pathogenic variants in genes other than BRCA1 and BRCA2 of ~4% when affected women attending high-risk clinics are the study subjects and gene panel tests are applied1,2. The prevalence of pathogenic variants in genes other than BRCA1 or BRCA2 are also similar in reports from clinical series of affected women (unselected for family history)4. Our findings are consistent with previous work in this setting and illustrate that, in contrast to pathogenic variants in BRCA1 and BRCA2 which are less prevalent in women diagnosed at older ages, the prevalence of pathogenic variants in other breast cancer genes is independent of age at diagnosis4,9.
In our Australian population-based case subjects, the frequency of pathogenic variants in genes other than BRCA1 or BRCA2 was 5%, as high as that reported from groups of women from high-risk clinical settings. This frequency may be surprising given the population-based ABCFS recruited participants unselected for family history. However, other attributes of the ABCFS participants and the nature of the breast cancer risks associated with pathogenic variants in genes other than BRCA1 and BRCA2 may partially provide an explanation. Predictive factors used to identify families appropriate to refer to high-risk genetics clinics, which include family history, have not been found to be as predictive for carrying pathogenic variants in genes other than BRCA1 and BRCA24. In addition, the average penetrance of pathogenic variants in these genes, although not precisely estimated beyond PALB227,28, is anticipated to be lower than for pathogenic variants in BRCA1 and BRCA2. The relative risks estimated here and elsewhere support this expectation. Therefore, by not selecting for affected women with a family history, yet having a focus on early onset disease, the ABCFS has a prevalence of pathogenic variants in other breast cancer susceptibility genes similar to that of highly selected women attending genetics services. Improved predictors for identifying women and families who carry pathogenic variants in breast cancer predisposition genes other than BRCA1 and BRCA2 are urgently needed.
A significant challenge for studies in this setting, and the assessment of very rare variants, is the availability of suitable datasets to use as reference controls. Few have had resources that provide population-specific reference control datasets of suitable size to incorporate into risk estimation methodology29,30. Large publicly available databases have recently become available, including ExAC and gnomAD. Although they constitute invaluable resources as variant frequency databases and can be used to filter out “common” variants that are unlikely to be associated with increased risk of disease, these databases have important limitations when used as controls in case-control studies7,12. These limitations include (i) potential technical artefacts resulting from the aggregation of data generated by different sequencing platforms, (ii) differences in the call sets due to the cases and controls not being jointly processed or annotated, (iii) the absence or limited lifestyle and ancestry information for the control subjects and (iv) the absence of genetic information available at the individual subject level as only variant-based data is available. In the context of gene-burden analysis for rare conditions, these public databases can serve as reasonable control datasets with additional computational precautions to mitigate the above-mentioned issues, as described by Guo et al.31. For common diseases including cancer, they are still likely to contain affected individuals, even when excluding the TCGA sample set of gnomAD and ExAC. By using 862 population-based age-match controls from the ABCFS and 6549 older healthy Australian women participating in ASPREE, our study overcame some of these limitations.
The inclusion of older controls from ASPREE in this study means that our unadjusted OR estimates are biased, so we adjusted all ORs in our main analyses for age and other potential confounders (though we note that our unadjusted p-values are valid, since ascertainment bias disappears under the null hypothesis in our case, and super-cases and super-controls can validly be used for gene discovery). A sensitivity analysis based just on the age-matched cases and controls from the ABCFS gave broadly similar ORs as the main analyses, validating our adjustment methods and our adjusted OR estimates.
In our study, although different sequencing platforms have been used to generate the raw sequencing data, we aimed to reduce potential artefactual variant calls by utilising the processing pipeline that was the most appropriate for the sequencing technology used to produce the raw sequencing data for the case and the control subjects, then harmonising the variant calls by (i) restricting calls to regions that are equally able to be called across the three targeted regions and (ii) applying the same filtering and annotation pipelines. Our study used ClinVar to select pathogenic variants to include, as a group, in our association analysis. Although the level of confidence in ClinVar calls can be variable, as demonstrated by the star rating system or the “Conflicting evidence of pathogenicity” label, this approach allowed us to harmonise our pathogenicity calls with other studies, e.g., from Ambry8 or Myriad11 who regularly deposit their classification calls into ClinVar. For genes such as BRCA1, BRCA2, TP53 or the mismatch repair genes, we were also able to keep our pathogenicity assessment contemporary with regular updates from the genes respective expert panels.
A limitation of our approach is the potential to underestimate the contribution of missense variants, as they are very challenging to classify. Functional assays can provide important additional information for variant classification but are currently less well developed for breast cancer predisposition genes other than BRCA1 and BRCA2, although some recent and promising progress has been made for PALB232. A large number of unclassified variants (n = 924) were identified in the case subjects of the ABCFS in this study. It is likely that an extremely small number of these variants will be classified as pathogenic in the future. Recent data from Dorling et al.10, provided further evidence for breast cancer risk for missense variants in a number of breast cancer susceptibility genes, most notably CHEK210,33. Considerable effort has been invested by the ENIGMA consortium to understand the effect of deleterious variants in these genes and keep the variant classification up-to-date and publicly available.
Our data provide a population perspective to gene panel testing for breast cancer predisposition and contribute to international efforts to refine the breast cancer risk estimates for genetic variants identified in panel testing in women enriched for early age at breast cancer diagnosis and unselected for family history.
Methods
Subjects
The present study includes cases and controls from the Australian Breast Cancer Family Study (ABCFS) and participants from the ASPirin in Reducing Events in the Elderly (ASPREE) study.
Aspects of the ABCFS relevant to this study are the population-based probands and corresponding data collected at baseline. Briefly, the ABCFS probands were either breast cancer cases (identified through population-complete cancer registries) or age-matched controls. All probands completed interviewer-administered risk factor questionnaires and verification of cancers was sought through pathologist reviews of cancer tissue, pathology reports, cancer registries, medical records, and death certificates34,35.
The ASPREE study is a randomized, placebo-controlled trial for daily low-dose aspirin. We selected Australian participants aged 70 years or older at enrolment, without a previous diagnosis or current symptoms of atherothrombotic cardiovascular disease, physical disability, or dementia. Study design, recruitment, baseline characteristics and outcomes have been previously described36,37. Our statistical analysis only used ASPREE data that were collected at baseline. ASPREE female participants who reported at baseline a personal history of breast cancer were excluded from the statistical analysis.
Written informed consent was obtained from all individual participants included in the study. This study was approved by the Human Research Ethics Committee of the University of Melbourne and Monash University.
Gene panel testing
We analysed rare genetic variants identified in the blood-derived germline DNA of 1,451 women diagnosed with breast cancer and 857 age-matched controls participating in the ABCFS, and 13,197 individuals (6549 women) enroled in the ASPREE trial.
Genes included in the gene panel
Our analysis was restricted to the coding region and proximal intron-exon junctions of 24 genes; ATM: NM_000051, BARD1: NM_000465.2, BRCA1: NM_007294.3, BRCA2: NM_000059.3, BRIP1: NM_032043.2, CDH1: NM_004360.3, CHEK2: NM_007194.3, FANCM: NM_020937.2, MLH1: NM_000249.3, MRE11A: NM_005591.3, MSH2: NM_000251.2, MSH6: NM_000179.2, MUTYH: NM_001128425.1, NBN: NM_002485.4, NF1: NM_000267.3, PALB2: NM_024675.3, PMS2: NM_000535.5, PTEN: NM_000314.4, RAD50: NM_005732.3, RAD51C: NM_058216.2, RAD51D: NM_002878.3, RECQL: NM_002907.3, STK11: NM_000455.4, TP53: NM_000546.5.
Only selected regions of PMS2 were targeted as described previously38. Gene-panel testing and raw DNA sequencing reads alignment to the reference genome GRCh37 were performed as described in Nguyen-Dumont et al. and Lacaze et al. for the ABCFS and ASPREE subjects, respectively38,39. Briefly, the ABCFS subjects were sequenced in-house, using either a Hi-Plex panel on the NextSeq55040 or a HaloPlexHS panel on the HiSeq3000 (both Illumina, San Diego, CA, USA). The ASPREE subjects were sequenced using an AmpliSeq panel on the Ion Torrent S5TM XL (Thermo Fisher Scientific, Waltham, MA, USA) and aligned sequencing files (BAMs) were provided for variant calling in this study.
Variant calling and filtering
Variant calling was performed using VarDict 1.741 and restricted to the overlap of the regions targeted by the three panels. For ASPREE controls sequenced on the Ion Torrent platform, variant calling had also been performed using the Torrent Variant Calling Suite v1.5 as previously described42 and the intersection with the variant calls from VarDict was used in downstream analyses. Subsequent genetic analyses were restricted to variants: (i) with the following read depth and variant allele frequency: 50X and 0.2 for Hi-Plex and AmpliSeq samples, and 30X and 0.15 for HaloPlexHS samples. In addition, for the ASPREE samples, we determined a conservative but high-confidence call set by filtering out (i) variants present in more than 0.05% of all ASPREE participants (n = 65), under the assumption that common variants were either sequencing artefacts or too common to be associated with disease risk, and (ii) variants that had passed our quality filters described above in <95% of the genotype calls at a given genomic location, to ensure that variants that progressed to the next analysis stage were adequately covered.
Variant annotation was performed using VarSeq VSClinical v2.2 (Golden Helix Inc., Bozeman, MT, USA) and included ClinVar annotations from July 2020. This study focused on rare predicted protein-truncating variants (PTVs) and pathogenic (including likely pathogenic) variants. Rare variants were defined as those identified in ExAC v.0.3 with a minor allele frequency ≤0.01 in the non-Finnish European population (NFE non-TCGA). Genetic variants were considered pathogenic if they were annotated as “Pathogenic” or “Likely Pathogenic” in ClinVar. Mono-allelic pathogenic MUTYH variant carriers are reported in Supplementary Table 1 but not included in our analysis. Predicted PTVs that were classified as “Conflicting” in ClinVar with annotations tending towards pathogenicity (e.g., CHEK2 c.1100delC) were included in this analysis. Also included were PTVs that were absent (unreported) in ClinVar, except if they were located in the last coding exon. Further details can be found in Supplementary Table 1.
Statistical methods
For each of the genes considered, pathogenic variants were combined and an odds ratio (OR) for their association with breast cancer was estimated using unconditional multivariate logistic regression. These analyses were adjusted for the following potential confounders (or, where indicated, a subset of these): age at enrolment, height, body mass index, number of children, number of years of education and number of alcoholic drinks per week. These potential confounders are the known breast cancer risk factors that are available in both the ABCFS and ASPREE datasets, and data harmonisation was performed to make the relevant variables from these two studies comparable.
Excluded from all statistical analyses were males, ASPREE females with a previous diagnosis of breast cancer, and females with no gene-panel testing data. Women with missing data were excluded from analyses involving the relevant variables, though <1% had missing values for each variable (except for number of children, which was missing for 4.3% of ASPREE). Women with pathogenic variants in two or more genes (other than MUTYH, see above) were excluded from the main analyses.
The effect of exclusions and other analytical choices was investigated with sensitivity analyses. Wald confidence intervals were calculated for each OR, and the likelihood ratio test was used to generate p-values for comparing nested models. All p-values were two-sided and a p-value threshold of 0.05 was used to define statistical significance. All analyses were performed using R version 3.4.2.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All rare pathogenic variants identified and the corresponding phenotype data are reported in the Supplementary Tables.
References
Kurian, A. W. et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J. Clin. Oncol. 32, 2001–2009 (2014).
Desmond, A. et al. Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol. 1, 943–951 (2015).
Maxwell, K. N. et al. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet. Med. 17, 630–638 (2015).
Tung, N. et al. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J. Clin. Oncol. 34, 1460–1468 (2016).
Lerner-Ellis, J. et al. Retesting of women who are negative for a BRCA1 and BRCA2 mutation using a 20-gene panel. J. Med. Genet. 57, 380–384 (2020).
Buys, S. S. et al. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 123, 1721–1730 (2017).
Shimelis, H. et al. Triple-negative breast cancer risk genes identified by multigene hereditary cancer panel testing. J. Natl. Cancer Inst. 110, 855–862 (2018).
LaDuca, H. et al. A clinical guide to hereditary cancer panel testing: evaluation of gene-specific cancer associations and sensitivity of genetic testing criteria in a cohort of 165,000 high-risk patients. Genet. Med. 22, 407–415 (2020).
Hu, C. et al. A population-based study of genes previously implicated in breast cancer. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2005936 (2021).
Dorling, L. et al. Breast cancer risk genes - association analysis in more than 113,000 women. N. Engl. J. Med. 384, 428–439 (2021).
Kurian, A. W. et al. Genetic testing and results in a population-based cohort of breast cancer patients and ovarian cancer patients. J. Clin. Oncol. 37, 1305–1315 (2019).
Slavin, T. P. et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer 3, 22 (2017).
Hauke, J. et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 7, 1349–1358 (2018).
Castera, L. et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 20, 1677–1686 (2018).
Suszynska, M., Klonowska, K., Jasinska, A. J. & Kozlowski, P. Large-scale meta-analysis of mutations identified in panels of breast/ovarian cancer-related genes - Providing evidence of cancer predisposition genes. Gynecol. Oncol. 153, 452–462 (2019).
Lu, H. M. et al. Association of breast and ovarian cancers with predisposition genes identified by large-scale sequencing. JAMA Oncol. 5, 51–57 (2019).
Zhang, S. et al. Frequency of the CHEK2 1100delC mutation among women with breast cancer: an international study. Cancer Res. 68, 2154–2157 (2008).
de Bock, G. H. et al. Tumour characteristics and prognosis of breast cancer patients carrying the germline CHEK2*1100delC variant. J. Med. Genet. 41, 731–735 (2004).
Nagel, J. H. et al. Gene expression profiling assigns CHEK2 1100delC breast cancers to the luminal intrinsic subtypes. Breast Cancer Res. Treat. 132, 439–448 (2012).
Fletcher, O. et al. Family history, genetic testing, and clinical risk prediction: pooled analysis of CHEK2 1100delC in 1,828 bilateral breast cancers and 7,030 controls. Cancer Epidemiol. Biomark. Prev. 18, 230–234 (2009).
Schmidt, M. K. et al. Breast cancer survival and tumor characteristics in premenopausal women carrying the CHEK2*1100delC germline mutation. J. Clin. Oncol. 25, 64–69 (2007).
Meyer, A., Dork, T., Sohn, C., Karstens, J. H. & Bremer, M. Breast cancer in patients carrying a germ-line CHEK2 mutation: outcome after breast conserving surgery and adjuvant radiotherapy. Radiother. Oncol. 82, 349–353 (2007).
Weischer, M. et al. CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer-specific death, and increased risk of a second breast cancer. J. Clin. Oncol. 30, 4308–4316 (2012).
Goldgar, D. E. et al. Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res 13, R73 (2011).
Southey, M. C. et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J. Med. Genet. https://doi.org/10.1136/jmedgenet-2016-103839 (2016).
Reiner, A. S. et al. Radiation treatment, ATM, BRCA1/2, and CHEK2*1100delC pathogenic variants, and risk of contralateral breast cancer. J. Natl. Cancer Inst. https://doi.org/10.1093/jnci/djaa031 (2020).
Southey, M. C. et al. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res. 12, R109 (2010).
Yang, X. et al. Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J. Clin. Oncol. 38, 674–685 (2020).
Ramus, S. J. et al. Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J. Natl. Cancer Inst. https://doi.org/10.1093/jnci/djv214 (2015).
Girard, E. et al. Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int. J. Cancer 144, 1962–1974 (2019).
Guo, M. H., Plummer, L., Chan, Y.-M., Hirschhorn, J. N. & Lippincott, M. F. Burden testing of rare variants identified through exome sequencing via publicly available control data. Am. J. Hum. Genet. 103, 522–534 (2018).
Southey, M. C., Rewse, A. & Nguyen-Dumont, T. PALB2 genetic variants: can functional assays assist translation? Trends Cancer 6, 263–265 (2020).
Le Calvez-Kelm, F. et al. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: results from a breast cancer family registry case-control mutation-screening study. Breast Cancer Res. 13, R6 (2011).
Hopper, J. L. et al. Design and analysis issues in a population-based, case-control-family study of the genetic epidemiology of breast cancer and the Co-operative Family Registry for Breast Cancer Studies (CFRBCS). J. Natl. Cancer Inst. Monogr. https://doi.org/10.1093/oxfordjournals.jncimonographs.a024232 (1999).
Dite, G. S. et al. Familial risks, early-onset breast cancer, and BRCA1 and BRCA2 germline mutations. J. Natl. Cancer Inst. 95, 448–457 (2003).
Aspree Investigator Group. Study design of ASPirin in Reducing Events in the Elderly (ASPREE): a randomized, controlled trial. Contemp. Clin. Trials 36, 555–564 (2013).
McNeil, J. J. et al. Baseline characteristics of participants in the ASPREE (ASPirin in reducing events in the elderly) study. J. Gerontol. A Biol. Sci. Med. Sci. 72, 1586–1593 (2017).
Nguyen-Dumont, T. et al. Mismatch repair gene pathogenic germline variants in a population-based cohort of breast cancer. Fam. Cancer 19, 197–202 (2020).
Lacaze, P. et al. Medically actionable pathogenic variants in a population of 13,131 healthy elderly individuals. Genet. Med. 22, 1883–1886 (2020).
Hammet, F. et al. Hi-Plex2: a simple and robust approach to targeted sequencing-based genetic screening. Biotechniques https://doi.org/10.2144/btn-2019-0026 (2019).
Lai, Z. et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 44, e108 (2016).
Lacaze, P. et al. Medically actionable pathogenic variants in a population of 13,131 healthy elderly individuals. Genet. Med. 22, 1883–1886 (2020).
Acknowledgements
We thank all the participants in this study, the entire team of the Australian Breast Cancer Family Study/Registry (BCFR-AU) and past and current investigators. This work was supported by the U.S. National Institute of Health (grant number RO1CA159868). The ABCFS was supported in Australia by the National Health and Medical Research Council, the New South Wales Cancer Council, the Victorian Health Promotion Foundation, the Victorian Breast Cancer Research Consortium, Cancer Australia, and the National Breast Cancer Foundation. The six sites of the Breast Cancer Family Registry (BCFR) were supported by grant UM1 CA164920 from the U.S. National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the BCFR, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government or the BCFR. T.N-.D is a National Breast Cancer Foundation (Australia) Career Development Fellow (ECF-17–001), B.J.P. is the recipient of a Victorian Health and Medical Research Fellowship and MCS is a National Health and Medical Research Council (NMHRC, Australia) Senior Research Fellow (APP1155163). This work was supported by an NHMRC Program grant (APP1074383), The National Breast Cancer Foundation (BRA-STRAP; NT-15-016), NHMRC European Union Collaborative Research Grant (APP1101400) and Monash University, Melbourne, Australia.
Author information
Authors and Affiliations
Contributions
Conceptualization; M.C.S., J.L.H., Data Curation; T.N.-D., J.A.S., M.R., Formal Analysis; M.C.S., J.G.D., M.R., J.A.S., R.S., E.S., P.L., T.N.-D., Funding acquisition; M.C.S., J.L.H., J.J.M., P.L., K.T., J.K., P.J., I.W., N. P., N. P., Investigation; D.J.P., B.J.P., K.M., F.H., M.M., H.T., D.T., A.R., Methodology M.C.S., T.N.-D., D.J.P., B.J.P., K.M., F.H.; Project Administration; M.C.S., Resources; M.C.S., J.L.H., J.J.M., P.L., G.G.G., R.L.M., Supervision; M.C.S., T.N.-D., J.J.M., J.L., Validation; D.J.P., B.J.P., K.M., F.H., M.M., H.T., D.T., A.R., Writing—original draft; M.C.S., T.N.-D., J.G.D., Writing—review and editing; M.C.S., J.G.D., M.R., J.A.S., A.-L.R., K.T., J.K., P.J., I.W., N. Poplawski, N. Pachter, S.G., D.J.P., B.J.P., K.M., F.H., M.M., H.T., D.T., A.R., A.W., A.M., C.S., R.H., R.S., E.S., P.L., J.J.M., G.G.G., R.L.M., J.L.H., T.N.-D. All authors have made substantial contributions to the conception or design of the work or acquisition, analysis or interpretation of the data; drafting the work or revisiting it critically for important intellectual content; have approved the final version of the completed manuscript and have ensured that questions related to the accuracy or integrity and any part of the work have been appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Southey, M.C., Dowty, J.G., Riaz, M. et al. Population-based estimates of breast cancer risk for carriers of pathogenic variants identified by gene-panel testing. npj Breast Cancer 7, 153 (2021). https://doi.org/10.1038/s41523-021-00360-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-021-00360-3
