
Abstract
Metaplastic breast cancers (MBCs) are characterized by complex genomes, which seem to vary according to their histologic subtype. TERT promoter hotspot mutations and gene amplification are rare in common forms of breast cancer, but present in a subset of phyllodes tumors. Here, we sought to determine the frequency of genetic alterations affecting TERT in a cohort of 60 MBCs with distinct predominant metaplastic components (squamous, 23%; spindle, 27%; osseous, 8%; chondroid, 42%), and to compare the repertoire of genetic alterations of MBCs according to the presence of TERT promoter hotspot mutations or gene amplification. Forty-four MBCs were subjected to: whole-exome sequencing (WES; n = 27) or targeted sequencing of 341-468 cancer-related genes (n = 17); 16 MBCs were subjected to Sanger sequencing of the TERT promoter, TP53 and selected exons of PIK3CA, HRAS, and BRAF. TERT promoter hotspot mutations (n = 9) and TERT gene amplification (n = 1) were found in 10 of the 60 MBCs analyzed, respectively. These TERT alterations were less frequently found in MBCs with predominant chondroid differentiation than in other MBC subtypes (p = 0.01, Fisher’s exact test) and were mutually exclusive with TP53 mutations (p < 0.001, CoMEt). In addition, a comparative analysis of the MBCs subjected to WES or targeted cancer gene sequencing (n = 44) revealed that MBCs harboring TERT promoter hotspot mutations or gene amplification (n = 6) more frequently harbored PIK3CA than TERT wild-type MBCs (n = 38; p = 0.001; Fisher’s exact test). In conclusion, TERT somatic genetic alterations are found in a subset of TP53 wild-type MBCs with squamous/spindle differentiation, highlighting the genetic diversity of these cancers.
Similar content being viewed by others

Introduction
Metaplastic breast cancer (MBC) is a rare (0.2–1%)1,2, aggressive histologic subtype of breast cancer, characterized histologically by neoplastic epithelium displaying differentiation towards squamous or mesenchymal elements, including spindle, chondroid, osseous, or rhabdoid cells. MBCs can present one (monophasic) or two or more (biphasic) components. These components can both display metaplastic histology or can be one metaplastic component and one adenocarcinoma component, which is most commonly in the form of invasive ductal carcinoma of no special type (IDC-NST). MBCs are most often of high histologic grade and display a triple-negative phenotype1,3.
The histologic diversity of MBCs is associated with distinct genomic and transcriptomic profiles4,5,6,7,8,9,10,11,12. From a genetic standpoint, MBCs are shown to frequently harbor mutations affecting TP53 and genes related to the PI3K/AKT/mTOR pathways. While TP53 mutations are found to be less frequent in MBCs with prominent spindle cell component, PIK3CA mutations are vanishingly rare in MBCs with chondroid metaplasia7,12,13. The transcriptomic features of MBCs also vary according to the predominant histologic component; for instance, MBCs with a predominant spindle cell component are preferentially classified as of claudin-low subtype, whereas MBCs with squamous or chondroid metaplasia are more frequently classified as of basal-like or even normal breast-like subtypes than as of claudin-low subtype8,9.
Somatic TERT promoter mutations (C228T and C250T), associated with telomerase activation, have been reported at a relatively high frequency in human cancers (12% overall)14 and are associated with disease progression and recurrences15,16,17. Although thought to be absent or extremely rare in common forms of breast cancer18,19,20, TERT promoter mutations and TERT gene amplifications have been reported in up to 68% of malignant phyllodes tumors of the breast, a potential differential diagnosis of MBCs, and may have a role in the malignant progression in fibroepithelial lesions15,21,22. TERT gene amplification has been reported in 13% of adenomyoepitheliomas of the breast, tumors of uncertain malignant potential which have been reported to progress to spindle cell MBCs in a minority of cases23,24. Interestingly, in one adenomyoepithelioma progressing to a triple-negative spindle cell MBC, the submodal clone that most likely gave rise to the invasive carcinoma harbored a TERT promoter hotspot mutation24. In the context of MBCs, TERT promoter hotspot mutations have been reported in up to 25% of cases, and to be associated with MBCs with spindle and/or squamous differentiation12.
Here, we sought to determine the frequency of genetic alterations affecting TERT, including TERT promoter hotspot mutations and TERT gene amplifications in MBCs with distinct types of predominant metaplastic component (e.g. squamous, spindle cell, osseous and chondroid). We have also compared the repertoire of somatic genetic alterations of MBCs harboring TERT promoter mutations or gene amplification to MBCs lacking genetic alterations targeting TERT. These analyses have revealed that 17% (10 out of 60) of MBCs harbor TERT somatic genetic alterations, and that these are associated with specific predominant metaplastic components and are seemingly mutually exclusive with TP53 mutations.
Results
Clinicopathologic characteristics
Sixty primary MBCs were included in this study (Table 1 and Supplementary Table S1), including 4 biopsies and 56 resection specimens; of these specimens, 6 (5 resections and 1 biopsy) were obtained post neoadjuvant therapy. The median age at diagnosis was 57 years old (range 34–85). Most (92%, 55/60) MBCs were of histologic grade 3 and 95% (57/60) of the MBCs analyzed were of triple-negative phenotype (Table 1). Upon central histopathological review, the MBCs included in this study were classified according to their predominant histologic type into squamous (23%; 14/60), spindle cell (27%; 16/60), osseous (8%; 5/60), or chondroid (42%, 25/60) MBCs (Fig. 1 and Supplementary Table S1). Forty-seven percent (28/60) of the MBCs were matrix-producing, including MBCs with predominant chondroid (n = 24) and osseous (n = 4; Table 1 and Supplementary Table S1) histologic components.

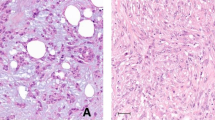
Representative hematoxylin-and-eosin photomicrographs of metaplastic breast cancers (MBCs) with predominant a squamous cell carcinoma component (MBC103T), b spindle cell component (MBC118T), c osseous metaplasia component (MBC120T), and d chondroid metaplasia component (MBC108T). Scale bars, 200 μM.
TERT genetic alterations in MBCs
We first sought to determine the frequency of TERT genetic alterations in 60 MBCs included in this study. Genetic alterations affecting TERT were identified in 17% (10/60) of the MBCs, including a recurrent hotspot mutation affecting the TERT promoter hotspot locus (C228T; 15%, 9/60) and TERT gene amplification (2%, 1/60; Fig. 2). All MBCs harboring TERT promoter alterations were of triple-negative phenotype (Fig. 2). We next performed a hypothesis-generating, exploratory analysis of the associations between the presence of TERT somatic genetic alterations and the phenotype of MBCs (Fig. 2 and Supplementary Fig. S1). This analysis revealed that TERT genetic alterations were significantly less frequently found in MBCs with a predominant chondroid component (0/25) than in the remaining MBCs (10/35; p = 0.005, Fisher’s exact test, Table 2 and Fig. 2). Nonetheless, two of these 10 MBCs harboring TERT genetic alterations including a predominant spindle cell MBC (MBC119T, TERT promoter mutation) and a predominantly osseous MBC (MT82, TERT gene amplification) displayed focal areas of chondroid differentiation (Fig. 2).

Heatmap depicting the proportion of the histologic component, frequency of TERT genetic alterations, TP53 mutations, PIK3CA mutations, HRAS mutations and BRAF genetic alterations in 60 MBCs. Mutation types are color-coded according to the legend. Cases are shown in columns, the percentage of the histological components, matrix producing, ER and HER2 status, and sequencing methods are depicted in phenobars. *Mutual exclusivity analysis, CoMEt, p < 0.001.
Comparative analysis between TERT altered MBCs and TERT wild-type MBCs
We next sought to define whether the six MBCs harboring TERT gene promoter hotspot mutations or gene amplification displayed a distinct repertoire of somatic genetic alterations as compared to the 38 TERT wild-type MBCs (Fig. 3, Supplementary Table S2) subjected to whole-exome sequencing (WES) or MSK-IMPACT targeted sequencing of 341–468 cancer-related genes. In our study, MBCs harboring TERT genetic alterations had a tumor mutation burden (median 3.1 mutations/Mb; range 0.8–6.1) comparable to that of MBCs lacking genetic alterations affecting TERT (median 3.5 mutations/Mb; range 0.8–8.7; p = 0.72, Mann–Whitney U test; Supplementary Fig. S2a). Our exploratory analysis revealed that, despite having similar tumor mutation burden, MBCs harboring TERT genetic alterations were significantly enriched for PIK3CA clonal mutations preferentially affecting hotspots (5/6, 83% TERT altered vs 5/38, 13% TERT WT; p = 0.001, Fisher’s exact test). Four of the 5 MBCs harbored clonal TERT promoter hotspot mutations co-occurring with PIK3CA mutations, and one MBC (MT45) that lacked mutations affecting PIK3CA harbored a subclonal TERT promoter mutation (Fig. 3a and Supplementary Fig. S3). TP53 mutations were significantly more frequently detected in MBCs lacking genetic alterations affecting TERT (34/38, 89% TERT wild-type vs 3/6, 50% TERT altered; p = 0.04, Fisher’s exact test; Fig. 3a). A formal mutually exclusivity analysis based on CoMEt25 in these 44 MBCs demonstrated that TP53 mutations were significantly mutually exclusive with TERT genetic alterations (p < 0.01, CoMEt). This observation was further confirmed when the entire cohort (n = 60) of MBCs was analyzed (p < 0.001, CoMEt; Fig. 2).

a Comparison of the most frequent genetic alterations affecting cancer genes identified in metaplastic breast cancers harboring TERT genetic alterations (TERT promoter hotspot mutations, n = 5; TERT gene amplification, n = 1; left) and TERT wild-type (n = 38, right), by whole-exome sequencing or MSK-IMPACT targeted sequencing. Cases are shown in columns and genes in rows. Clinicopathologic characteristics are shown on the top. Mutations are color coded according to the legend. Frequency plots and Fisher’s exact test comparison corrected for multiple testing of (b) copy number gains and losses, and (c) amplifications and homozygous deletions between TERT altered (n = 6) and TERT promoter wild-type (n = 38) MBCs. Frequency (y-axis) of gains and losses and amplifications and homozygous deletions is shown for each genomic region (x-axis). Inverse Log10 values of the two-sided Fisher’s exact test p-values are plotted according to the genomic region (lower panel). Gains and amplifications are colored in green. Losses and homozygous deletions are colored in purple. *Statistical significance was evaluated by the Fisher’s exact test (p < 0.05). **Mutual exclusivity analysis was performed using combinations of mutually exclusive alterations (CoMEt, p < 0.01). HRD, homologous recombination DNA repair defect; SNV, single nucleotide variants.
Although no other gene was significantly differentially altered between TERT altered vs wild-type MBCs (p > 0.05; Fisher’s exact test, Fig. 3a), mutations affecting PTEN, PIK3R1, chromatin remodeling genes (ARID1A, KMT2C) and tumor suppressor genes (RB1, NOTCH1, and NOTCH2) were only identified in MBCs lacking TERT genetic alterations (Fig. 3a). In addition, activating mutations affecting Ras pathway genes were detected in three MBCs harboring TERT promoter hotspot mutations, including hotspot mutations affecting KRAS (MTC01, subclonal A59G, n = 1), NRAS (MT45, clonal Q61L, n = 1) and HRAS Q61R (MT35, clonal, n = 1), which coexisted with mutations affecting TP53 (subclonal V173L and clonal E204Vfs*4) and a subclonal BRAF D594N hotspot mutation (Fig. 3a; Supplementary Fig. S3). Two TERT wild-type MBCs harbored BRAF amplification (MTC23 and MBC104T; Fig. 3a). Similar observations were made when the 16 additional MBCs were subjected to Sanger sequencing of HRAS and BRAF hotspot loci (Fig. 2). This additional analysis revealed another MBC (MBC113T) harboring an HRAS hotspot mutation (Q61K, Fig. 2) co-occurring with a TP53 hotspot mutation (D281E) but did not identify mutations affecting TERT promoter or PIK3CA hotspot locus.
Given the previous observation that HRAS Q61R hotspot mutations co-occurring with TERT promoter mutations were preferentially found in adenomyoepitheliomas24, we sought to define whether the MBCs harboring HRAS Q61 hotspot mutations would be associated with or originate from adenomyoepitheliomas. Not surprisingly, the HRAS Q61 mutant MBCs identified in our study lacked histologic features of adenomyoepithelioma, unlike TNBCs originating from adenomyoepithelioma, which have been shown to lack TP53 mutations and consistently harbor PIK3CA or PIK3R1 mutations24. Upon re-review of the MBCs, including all diagnostic slides available per case harboring HRAS Q61 mutations, only one MBC (MBC103T; Supplementary Fig. S4) was found to display features consistent with the presence of a breast adenomyoepithelioma. This MBC contained a biphasic proliferation of epithelial and myoepithelial cells (Supplementary Fig. S4), where the abluminal layer expressed p63 and calponin by immunohistochemical analysis, consistent with a diagnosis of MBC developing in the context of an adenomyoepithelioma. Sanger sequencing analysis of this case revealed mutations affecting TERT promoter (C228T) and PIK3CA hotspot locus (H1047R), but no TP53 mutations or alterations in HRAS codon Q61 (Fig. 2).
Here we demonstrate that, thirty-eight of the 44 MBCs subjected to WES or MSK-IMPACT sequencing had sufficient single nucleotide variants (SNVs) to infer accurate mutational signature (Fig. 3a, Supplementary Table S1). Based on SigMA analysis, an algorithm previously validated for the analysis of formalin-fixed paraffin-embedded (FFPE) samples subjected to the FDA-approved MSK-IMPACT sequencing assay, 23 MBCs (60%, 23/38) displayed dominant COSMIC mutational signatures 3 and 8 (associated with homologous recombination DNA repair defect; HRD; Supplementary Table S3)26,27. The aging signatures 1 and 5 were dominant in 34% (13/38) of MBCs, one case displayed a dominant APOBEC signature 2 (3%, 1/38) and one harbored a dominant signature 17 of unknown etiology (3%, 1/38). No statistically significant differences were observed in the frequency of mutational signatures between TERT altered and TERT wild-type MBCs (p > 0.05; Fisher’s exact test; Fig. 3a; Supplementary Table S3). MBCs harboring TERT genetic alterations (n = 5) displayed a lower fraction of the genome altered (FGA, median 22%; 9–51%) than MBCs lacking genetic alterations affecting TERT (median = 54%; range, 20–86%, p = 0.002, Mann–Whitney U test; Supplementary Table S1 and Supplementary Fig. S2b). Nonetheless, the patterns of gene copy number profiles of both groups were comparable (Fig. 3b, c).
Discussion
The genomic and transcriptomic diversity of MBCs has been documented by our group and others4,5,6,7,8,9,10,11. In fact, the repertoire of genetic alterations and transcriptomic features of MBCs appear to vary according to the predominant metaplastic component, consistent with the notion of likely genotypic–phenotypic correlations in these cancers. Here we demonstrate that in agreement with the results by Krings and Chen12 at variance with other forms of triple-negative breast cancer, TERT promoter hotspot mutations and gene amplification are found in substantial subset of MBCs (17%), and that these alterations are less frequently found in MBCs with a predominant chondroid component.
Previous studies7,8,9,12,13 have shown that TP53 and PIK3CA genes are the two most frequently mutated known cancer genes in MBC and that these mutations, however, vary in frequency according to the predominant metaplastic component. Consistent with previous observations7,8,9,13, the MBCs with predominant chondroid metaplasia included in this study lacked mutations affecting PIK3CA and Ras pathway genes, whereas TP53 mutations were found to be less frequent in MBCs with predominant spindle cell component compared to squamous and chondroid MBCs. These findings support the notion that a subset of MBCs harboring PIK3CA mutations may benefit from therapies targeting the PI3K/AKT/mTOR pathway. Recent studies have investigated the addition of PI3K/mTOR inhibitors to standard chemotherapy28,29,30,31, and found that patients with PI3K pathway-altered advanced triple-negative MBCs had significantly higher response rates when treated with mTOR inhibitors (temsirolimus or everolimus) in combination with liposomal doxorubicin and bevacizumab than patients with MBCs lacking PI3K/mTOR pathway alterations31. Given the enrichment of PIK3CA mutations in non-chondroid MBCs, these findings have further implications for the targeted treatment of specific histological subtypes of MBCs.
The TERT promoter hotspot mutations and TERT gene amplification described here were inversely correlated with TP53 mutations in a subset of MBCs analyzed, and significantly associated with PIK3CA hotspot mutations. It should be noted that pathogenic mutations affecting TP53 and TERT promoter hotspot mutations have also been found to be inversely correlated in other cancer types16,32, whereas TERT promoter and PIK3CA hotspot mutations have been shown to be mutually exclusive in ovarian cancers33, but to co-occur in other cancer types34,35, including breast cancer19. Whether these associations reflect epistatic interactions between TERT, TP53, and PIK3CA or whether they would result from the different prevalence of TERT alterations in different subtypes of MBC warrant further investigation.
TERT promoter hotspot mutations and TERT gene amplification have been reported in phyllodes tumors of the breast, suggesting that these genetic alterations might be the drivers of the progression from benign to malignant lesions in a subset of patients15,22,36. In addition, we have previously demonstrated that TERT somatic genetic alterations in 13% of breast adenomyoepitheliomas and in the carcinomas originating in association with or from these tumors24. The TERT promoter mutations detected in the present study are the result of an exchange of a single cytosine to a thymine at chromosome 5 base position 1,295,228 (C228T, c.-124 C > T), which results in a new binding motif for ETS transcription factors and leads to an increased transcriptional activity of the TERT promoter37,38. These mutations have been shown to constitute a mechanism of upregulated telomerase and to result in increased proliferative capacity and other oncogenic properties39. The frequency of TERT somatic alterations (i.e. in 17% of MBCs) reported here is consistent with that reported by Krings and Chen12 (i.e. 25% of MBCs), who observed an enrichment of TERT promoter mutations in MBCs with predominant spindle cell and/or squamous components. In contrast to the observations by Krings and Chen12, who reported the absence of TERT promoter mutations in chondroid matrix-producing carcinomas, in our study, TERT genetic alterations were identified in two cases displaying minor areas of chondroid differentiation, including one MBC with predominant spindle cell component and another MBC with predominant osseous component. It is possible that these discrepancies might be related to the fact that our series included MBCs with mixed histologic subtypes, in contrast to Krings and Chen12, who included only 3 MBCs with mixed components. Of these 3 cases, two had only one of the components (osseous) subjected to sequencing. The remaining cases included in their study12 were categorized as pure matrix-producing, spindle, squamous, or spindle/squamous MBCs that did not display differentiation along other metaplastic lineages.
The observations reported here as well as those made by others12 have diagnostic and taxonomic implications. First, given that TERT promoter hotspot mutations and gene amplification can also be found in MBCs, their detection should be used with caution in the differential diagnosis between MBC and malignant phyllodes tumor of the breast. Second, TERT and HRAS mutations have been shown to be vanishingly rare in primary breast cancers, including those of triple-negative phenotype; however, these alterations can be found in adenomyoepitheliomas and in a subset of MBCs, suggesting a tantalizing hypothesis that a subset of MBCs may evolve through similar genetic pathways or be etiologically related to adenomyoepithelial tumors. Further studies to investigate whether a subset of MBCs may constitute malignant myoepithelial tumors are warranted.
Our study has important limitations. Given the rarity of these tumors, the small sample size of the study and the limited amounts of DNA available for sequencing analysis in some cases, not all samples could be subjected to WES or MSK-IMPACT sequencing. Due to this limitation, our estimation of the frequency of TERT gene amplification is conservative as we cannot rule out the presence of TERT gene amplification in the 12 of the 16 MBCs subjected to Sanger sequencing that were TERT wild-type. Second, despite the multi-institutional cohort included in this study, we currently cannot define whether the mutually exclusive nature of TERT somatic genetic alterations and TP53 mutations in MBCs are derived from epistatic interactions between these genes in the context of MBC or if this mutual exclusivity is solely the result of the different frequencies of alterations affecting these genes in MBCs with distinct types of predominant metaplastic components. Hence, these observations should be interpreted with caution and warrant further investigation in larger series of MBCs. Furthermore, the multi-institutional nature of our study precludes a definitive survival analysis due to the lack of clinical follow-up information in a large subset of cases in this series. Further studies to assess survival correlations with TERT genetic alterations in MBCs patients are warranted. Despite these limitations, our study provides evidence suggesting that TERT genetic alterations may play a role in MBCs and that these are likely associated with specific subsets of the disease, emphasizing the diversity and molecular heterogeneity of MBCs.
Methods
Subjects and samples
Following approval by the Institutional Review Board (IRB) of Memorial Sloan Kettering Cancer Center (MSKCC), a retrospective series of 60 primary MBCs was selected to be included in this study. Patient consents were obtained according to the approved IRB protocol. Cases were reviewed by at least two of four pathologists (MV, FP, HZ, and/or JSR-F) following the criteria put forward by the World Health Organization3. Clinicopathologic characteristics, including age, tumor size and hormone-receptor status, were retrieved from the medical records (Supplementary Table S1). Tumors were graded according to the Nottingham grading system40. The tumor cell content and composition of the metaplastic elements were estimated (i.e., squamous cell, spindle cell, chondroid and osseous) and in each case, the metaplastic component most abundantly present was defined as described8 (Supplementary Table S1). All samples were anonymized prior to tissue processing.
Tissue preparation and DNA extraction
Ten-to-15 8-µm-thick sections from representative formalin-fixed paraffin embedded (FFPE) tumor and matched normal tissue blocks of 21 MBCs (21/60) were stained with nuclear fast red and microdissected using a sterile needle under a stereomicroscope (Olympus SZ61) to ensure a tumor content >80%, as previously described41. Genomic DNA was extracted from tumor and matched normal tissues using the DNeasy Blood and Tissue Kit (Qiagen) according to manufacturers’ instructions.
Whole-exome sequencing and MSK-IMPACT sequencing
Out of 60 MBCs included in our cohort, 44 (73%) were subjected to WES (n = 27) or to massively parallel sequencing targeting all coding regions of 341 to 468 cancer-related genes using the FDA-approved MSK Integrated Mutation Profiling of Actionable Cancer Targets assay (MSK-IMPACT, n = 17, Supplementary Table S2)42. Of the 27 MBCs subjected to WES, five MBCs were microdissected and subjected to WES at MSK’s Integrated Genomics Operations (IGO) using validated protocols, as previously described7,43, and for 22 MBCs the raw sequencing data (FASTQ files) reported in Ng et al.7 were retrieved and reanalyzed (see below). Of the 17 MBCs subjected to MSK-IMPACT sequencing, three were previously reported in Zehir et al.14. Hence, in this manuscript, we include massively parallel sequencing data from 19 previously unreported MBCs (5 subjected to WES and 14 to targeted MSK-IMPACT sequencing) as well as Sanger sequencing data from 16 previously unreported MBCs (see below). WES and MSK-IMPACT sequencing data were processed using our validated bioinformatics pipeline16,43,44. In brief, sequence reads were aligned to the reference human genome GRCh37 using the Burrows-Wheeler Aligner (BWA v0.7.15)45. Somatic single nucleotide variants (SNVs) were detected with MuTect (v1.0)46. Insertion and deletions (indels) were detected using Strelka (v2.0.15)47, VarScan2 (v2.3.7)48, Platypus (v0.8.1)49, Lancet (v1.0.0)50, and Scalpel (v0.5.3)51. Cancer cell fractions (CCFs) of each somatic mutation were computed using ABSOLUTE (v1.0.6)52, as previously described43,53. Copy number alterations (CNAs) and loss of heterozygosity were determined using FACETS54. Somatic mutations in tumor suppressor genes that were deleterious/loss-of-function or targeting a mutational hotspot in oncogenes were considered pathogenic. Mutations targeting hotspot loci were annotated using cancerhotspots.org55. Mutational signatures were defined using Signature Multivariate Analysis (SigMA) tool56, for cases with at least 5 SNVs, as previously described57. Exposure-based dominant mutational signatures obtained by SigMA56 (Supplementary Table S3) were comparable to the mutational signatures reported by Ng et al.7, which were inferred using DeconstructSigs58, in 68% (15/22) of the MBCs.
Tumour mutation burden (TMB) was calculated as the total number of non-synonymous mutations divided by the number of bases analyzed, per megabase. The fraction of genome altered (FGA) was defined as the cumulative size of copy number segments which are not copy neutral divided by the cumulative size of all copy number segments, as previously described14,59.
As part of an exploratory, hypothesis generating analysis, the repertoire of non-synonymous somatic mutations, mutational frequencies, and copy number alterations of MBCs harboring TERT somatic genetic alterations, including promoter mutations and gene amplification, were compared to MBCs lacking TERT genetic alterations. For the comparative analyses of the repertoire of non-synonymous somatic mutations, mutational frequencies, and copy number alterations of MBCs subjected to either WES or MSK-IMPACT, genes were restricted to the 341 genes included in MSK-IMPACT.
Assessment of somatic mutations by Sanger sequencing
We conducted the assessment of TERT promoter hotspot loci, TP53 (exons 2 to 11), PIK3CA (exons 9 and 20), HRAS (exon 3), and BRAF (exons 11 and 15) hotspot mutations in 16 MBCs with insufficient DNA yield by Sanger sequencing. In addition, as TERT promoter region is usually poorly covered by exome sequencing, TERT promoter hotspot mutations were assessed by Sanger sequencing in the 27 MBCs subjected to WES. PCR amplification of the selected genes was performed using the AmpliTaq Gold 360 Master Mix kit (Life Technologies, ThermoFisher Scientific) using previously described primers16,24,60 (Supplementary Table S4). PCR fragments were cleaned using ExoSAP It (ThermoFisher Scientific) and Sanger sequenced as previously described15,16.
Statistical analysis
Fisher’s exact test and Chi-Square test were used for comparison of categorical variables, and Mann–Whitney U test for continuous variables. All tests were two-tailed and p values <0.05 were considered statistically significant. A mutual exclusivity analysis was performed using combinations of mutually exclusive alterations (CoMEt) with the use of a pair-wise Fisher’s exact test to detect the presence of significant pairs of genes25.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
“The data generated and analysed during this study are described in the following data record: https://doi.org/10.6084/m9.figshare.1416048261. The whole-exome sequencing data supporting Figs. 2, 3, Supplementary Figs. S2 and S3, and Supplementary Tables S1, S2, and S3 are openly available in the Sequence Read Archive via the following accession: https://identifiers.org/ncbi/insdc.sra:SRP07369262. These data were first described in the original publication by Ng et al.7 MSK-IMPACT sequencing data of 3 samples included in the MSK-IMPACT Clinical Sequencing Cohort supporting Figs. 2, 3, Supplementary Figs. S2, S3, and Supplementary Tables S1 and S2 are publicly available at cBioPortal (https://identifiers.org/cbioportal:msk_impact_201763). These data were first described in the original publication by Zehir et al.14 Sequencing data of 19 previously unreported MBCs (5 subjected to whole-exome sequencing and 14 to MSK-IMPACT sequencing) are available at cBioPortal (https://identifiers.org/cbioportal:mbc_msk_202164). Additionally, the following data are available upon request from the corresponding authors: Histologic images supporting Fig. 1 and Supplementary Fig. S1; Sanger sequencing electropherograms supporting Fig. 2, Table 2, and Supplementary Figs. S1 and S4; Clinicopathologic data supporting Figs. 2 and 3, Table 1, and Supplementary Table S1.”
References
Weigelt, B. et al. Metaplastic breast carcinoma: more than a special type. Nat. Rev. Cancer 14, 147–148 (2014).
Reis-Filho, J. S. et al. in WHO Classification of Tumours Editorial Board., ed. Breast Tumours Vol. 2 134–138 (International Agency for Research on Cancer, 2019).
Reis-Filho J. S. et al. in WHO Classification of Tumours: Breast Tumours Vol. 2 (ed WHO Classification of Tumours Editorial Board) 134–138 (World Health Organization, 2019).
Avigdor, B. E. et al. Whole-Exome Sequencing of Metaplastic Breast Carcinoma Indicates Monoclonality with Associated Ductal Carcinoma Component. Clin. Cancer Res. 23, 4875–4884 (2017).
Tray, N. et al. Metaplastic breast cancers: Genomic profiling, mutational burden and tumor-infiltrating lymphocytes. Breast 44, 29–32 (2019).
Hennessy, B. T. et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 69, 4116–4124 (2009).
Ng, C. K. Y. et al. The Landscape of Somatic Genetic Alterations in Metaplastic Breast Carcinomas. Clin. Cancer Res. 23, 3859–3870 (2017).
Weigelt, B. et al. Metaplastic breast carcinomas display genomic and transcriptomic heterogeneity [corrected]. Mod. Pathol. 28, 340–351 (2015).
Piscuoglio, S. et al. Genomic and transcriptomic heterogeneity in metaplastic carcinomas of the breast. NPJ Breast Cancer 3, 48 (2017).
Beca, F. et al. Whole-Exome Analysis of Metaplastic Breast Carcinomas with Extensive Osseous Differentiation. Histopathology (2020).
McCart Reed, A. E. et al. Phenotypic and molecular dissection of metaplastic breast cancer and the prognostic implications. J. Pathol. 247, 214–227 (2019).
Krings, G. & Chen, Y. Y. Genomic profiling of metaplastic breast carcinomas reveals genetic heterogeneity and relationship to ductal carcinoma. Mod. Pathol. 31, 1661–1674 (2018).
Ross, J. S. et al. Genomic profiling of advanced-stage, metaplastic breast carcinoma by next-generation sequencing reveals frequent, targetable genomic abnormalities and potential new treatment options. Arch. Pathol. Lab Med. 139, 642–649 (2015).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Piscuoglio, S. et al. Massively parallel sequencing of phyllodes tumours of the breast reveals actionable mutations, and TERT promoter hotspot mutations and TERT gene amplification as likely drivers of progression. J. Pathol. 238, 508–518 (2016).
Da Cruz Paula, A. et al. Genomic profiling of primary and recurrent adult granulosa cell tumors of the ovary. Mod. Pathol. 33, 1606–1617 (2020).
Descotes, F. et al. Non-invasive prediction of recurrence in bladder cancer by detecting somatic TERT promoter mutations in urine. Br. J. Cancer 117, 583–587 (2017).
Gay-Bellile, M. et al. TERT promoter status and gene copy number gains: effect on TERT expression and association with prognosis in breast cancer. Oncotarget 8, 77540–77551 (2017).
Shimoi, T. et al. TERT promoter hotspot mutations in breast cancer. Breast Cancer 25, 292–296 (2018).
Killela, P. J. et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl Acad. Sci. USA 110, 6021–6026 (2013).
Yoshida, M. et al. TERT promoter mutations are frequent and show association with MED12 mutations in phyllodes tumors of the breast. Br. J. Cancer 113, 1244–1248 (2015).
Piscuoglio, S. et al. Massively parallel sequencing analysis of synchronous fibroepithelial lesions supports the concept of progression from fibroadenoma to phyllodes tumor. NPJ Breast Cancer 2, 16035 (2016).
Pareja, F. et al. Immunohistochemical assessment of HRAS Q61R mutations in breast adenomyoepitheliomas. Histopathology 76, 865–874 (2020).
Geyer, F. C. et al. Recurrent hotspot mutations in HRAS Q61 and PI3K-AKT pathway genes as drivers of breast adenomyoepitheliomas. Nat. Commun. 9, 1816 (2018).
Leiserson, M. D., Wu, H. T., Vandin, F. & Raphael, B. J. CoMEt: a statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol. 16, 160 (2015).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Moulder, S. et al. Inhibition of the phosphoinositide 3-kinase pathway for the treatment of patients with metastatic metaplastic breast cancer. Ann. Oncol. 26, 1346–1352 (2015).
Moulder, S. et al. Responses to liposomal Doxorubicin, bevacizumab, and temsirolimus in metaplastic carcinoma of the breast: biologic rationale and implications for stem-cell research in breast cancer. J. Clin. Oncol. 29, e572–e575 (2011).
Yang, M. H., Chen, I. C. & Lu, Y. S. PI3K inhibitor provides durable response in metastatic metaplastic carcinoma of the breast: A hidden gem in the BELLE-4 study. J. Formos. Med. Assoc. 118, 1333–1338 (2019).
Basho, R. K. et al. Targeting the PI3K/AKT/mTOR Pathway for the Treatment of Mesenchymal Triple-Negative Breast Cancer: Evidence From a Phase 1 Trial of mTOR Inhibition in Combination With Liposomal Doxorubicin and Bevacizumab. JAMA Oncol. 3, 509–515 (2017).
You, H. et al. Paradoxical prognostic impact of TERT promoter mutations in gliomas depends on different histological and genetic backgrounds. CNS Neurosci. Ther. 23, 790–797 (2017).
Wu, R. C. et al. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other major types of gynaecological malignancy. J. Pathol. 232, 473–481 (2014).
Tiedje, V. et al. NGS based identification of mutational hotspots for targeted therapy in anaplastic thyroid carcinoma. Oncotarget 8, 42613–42620 (2017).
Zacher, A. et al. Molecular Diagnostics of Gliomas Using Next Generation Sequencing of a Glioma-Tailored Gene Panel. Brain Pathol. 27, 146–159 (2017).
Pareja, F. et al. Phyllodes tumors with and without fibroadenoma-like areas display distinct genomic features and may evolve through distinct pathways. NPJ Breast Cancer 3, 40 (2017).
Horn, S. et al. TERT promoter mutations in familial and sporadic melanoma. Science 339, 959–961 (2013).
Huang, F. W. et al. Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957–959 (2013).
Yamagata, Y. et al. Changes in telomerase activity in experimentally induced atretic follicles of immature rats. Endocr. J. 49, 589–595 (2002).
Elston, C. W. & Ellis, I. O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology 19, 403–410 (1991).
Pareja, F. et al. Recurrent MED12 exon 2 mutations in benign breast fibroepithelial lesions in adolescents and young adults. J. Clin. Pathol. 72, 258–262 (2019).
Cheng, D. T. et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J. Mol. Diagn. 17, 251–264 (2015).
Weigelt, B. et al. The Landscape of Somatic Genetic Alterations in Breast Cancers From ATM Germline Mutation Carriers. J. Natl Cancer Inst. 110, 1030–1034 (2018).
Pareja, F. et al. Loss-of-function mutations in ATP6AP1 and ATP6AP2 in granular cell tumors. Nat. Commun. 9, 3533 (2018).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Saunders, C. T. et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012).
Koboldt, D. C. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012).
Rimmer, A. et al. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 46, 912–918 (2014).
Narzisi, G. et al. Genome-wide somatic variant calling using localized colored de Bruijn graphs. Commun. Biol. 1, 20 (2018).
Narzisi, G. et al. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat. Methods 11, 1033–1036 (2014).
Carter, S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012).
Pareja, F. et al. The Genomic Landscape of Mucinous Breast Cancer. J. Natl Cancer Inst. 111, 737–741 (2019).
Shen, R. & Seshan, V. E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 44, e131 (2016).
Chang, M. T. et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer Disco. 8, 174–183 (2018).
Gulhan, D. C. et al. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 51, 912–919 (2019).
Smith, E. S. et al. Endometrial Cancers in BRCA1 or BRCA2 Germline Mutation Carriers: Assessment of Homologous Recombination DNA Repair Defects. JCO Precis Oncol. 3, (2019).
Rosenthal, R. et al. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31 (2016).
Razavi, P. et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 34, 427–438.e426 (2018).
Pareja, F. et al. Immunohistochemical analysis of IDH2 R172 hotspot mutations in breast papillary neoplasms: applications in the diagnosis of tall cell carcinoma with reverse polarity. Mod. Pathol. 33, 1056–1064 (2020).
da Silva, E. M. et al. Metadata record for the manuscript: TERT promoter hotspot mutations and gene amplification in metaplastic breast cancer. figshare. https://doi.org/10.6084/m9.figshare.14160482 (2021).
Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP073692 (2016).
cBioPortal. https://identifiers.org/cbioportal:msk_impact_2017 (2017).
cBioPortal. https://identifiers.org/cbioportal:mbc_msk_2021 (2021).
Acknowledgements
This study was funded by the Breast Cancer Research Foundation (J.S.R.-F., B.W.). J.S.R.-F., B.W. and F.P. are funded in part by the National Institutes of Health/National Cancer Institute P50 CA247749 01 grant. B.W. is funded in part by Cycle for Survival and Stand Up To Cancer grants. F.P. is funded in part by a National Institutes of Health/National Cancer Institute K12 CA184746 grant. Research reported in this publication was supported in part by a Cancer Center Support Grant of the NIH/NCI (Grant No. P30CA008748).
Author information
Authors and Affiliations
Contributions
E.M.d.S., B.W., H.Z., and J.S.R.-F. conceived the study. M.V., F.P., H.Z., and J.S.R.-F. conducted pathology review. E.M.d.S. and A.B. performed sample processing. P.S., A.D.C.P., L.F., and A.G. performed bioinformatics analyses. Data acquisition, analysis, and interpretation were performed by E.M.d.S., P.S., F.P., A.D.C.P., H.D., D.S.R., N.R., S.C., P.R., L.N., H.Y.W., E.B., and H.Z. E.M.d.S., F.P., B.W. and J.S.R.-F. drafted the original manuscript, which was reviewed by all authors. The final draft of the manuscript was approved by all authors.
Corresponding authors
Ethics declarations
Competing interests
S.C. has received personal/consultancy fees from Lilly, Novartis, and Paige.AI. S.C. also reports research funds directed to MSK from Sanofi, Novartis, Daiichi-Sankyo, and Lilly, outside the scope of this study. H.Y.W. performed consulting/advisory services for Merck at one teleconference. H.Z. reports consultancy fee from Roche/Genentech, outside the scope of the submitted work. J.S.R.-F. reports receiving personal/consultancy fees from Goldman Sachs and REPARE Therapeutics, membership of the scientific advisory boards of VolitionRx, REPARE Therapeutics and Paige.AI, membership of the Board of Directors of Grupo Oncoclinicas, and ad hoc membership of the scientific advisory boards of Roche Tissue Diagnostics, Ventana Medical Systems, Novartis, Genentech and InVicro, outside the scope of this study. J.S.R.-F. and L.N. are editors with the journal. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
da Silva, E.M., Selenica, P., Vahdatinia, M. et al. TERT promoter hotspot mutations and gene amplification in metaplastic breast cancer. npj Breast Cancer 7, 43 (2021). https://doi.org/10.1038/s41523-021-00250-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-021-00250-8
This article is cited by
-
Genomic and epigenomic basis of breast invasive lobular carcinomas lacking CDH1 genetic alterations
npj Precision Oncology (2024)
-
Starfysh integrates spatial transcriptomic and histologic data to reveal heterogeneous tumor–immune hubs
Nature Biotechnology (2024)
-
Regulation and clinical potential of telomerase reverse transcriptase (TERT/hTERT) in breast cancer
Cell Communication and Signaling (2023)
-
The mixed subtype has a worse prognosis than other histological subtypes: a retrospective analysis of 217 patients with metaplastic breast cancer
Breast Cancer Research and Treatment (2023)
-
Clinicopathologic and genomic features of lobular like invasive mammary carcinoma: is it a distinct entity?
npj Breast Cancer (2023)
