
Abstract
Breast cancer is a common disease partially caused by genetic risk factors. Germline pathogenic variants in DNA repair genes BRCA1, BRCA2, PALB2, ATM, and CHEK2 are associated with breast cancer risk. FANCM, which encodes for a DNA translocase, has been proposed as a breast cancer predisposition gene, with greater effects for the ER-negative and triple-negative breast cancer (TNBC) subtypes. We tested the three recurrent protein-truncating variants FANCM:p.Arg658*, p.Gln1701*, and p.Arg1931* for association with breast cancer risk in 67,112 cases, 53,766 controls, and 26,662 carriers of pathogenic variants of BRCA1 or BRCA2. These three variants were also studied functionally by measuring survival and chromosome fragility in FANCM−/− patient-derived immortalized fibroblasts treated with diepoxybutane or olaparib. We observed that FANCM:p.Arg658* was associated with increased risk of ER-negative disease and TNBC (OR = 2.44, P = 0.034 and OR = 3.79; P = 0.009, respectively). In a country-restricted analysis, we confirmed the associations detected for FANCM:p.Arg658* and found that also FANCM:p.Arg1931* was associated with ER-negative breast cancer risk (OR = 1.96; P = 0.006). The functional results indicated that all three variants were deleterious affecting cell survival and chromosome stability with FANCM:p.Arg658* causing more severe phenotypes. In conclusion, we confirmed that the two rare FANCM deleterious variants p.Arg658* and p.Arg1931* are risk factors for ER-negative and TNBC subtypes. Overall our data suggest that the effect of truncating variants on breast cancer risk may depend on their position in the gene. Cell sensitivity to olaparib exposure, identifies a possible therapeutic option to treat FANCM-associated tumors.
Similar content being viewed by others
Introduction
The genetic architecture of inherited breast cancer is complex and involves germline pathogenic variants in high and moderate-risk genes and polygenetic factors. The major high-penetrance breast cancer risk genes include BRCA1 and BRCA2, which are key factors in the DNA double-strand break repair through homologous recombination (HR) and in the inter-strand crosslink (ICL) repair as a part of the Fanconi Anemia (FA) pathway.1,2 Recently, based on a prospective cohort of families carrying BRCA1 or BRCA2 pathogenic variants, the average cumulative risk by age 80 was estimated to be 72% and 69% for carriers of BRCA1 and BRCA2 pathogenic variants, respectively.3 PALB2 has been previously considered a moderate-risk gene, but the latest estimate of about 44% lifetime risk associated with pathogenic variants may raise this gene to the high-risk group.4 Pathogenic variants in moderate-penetrance genes ATM and CHEK2 are also associated with breast cancer, conferring a 20% average lifetime risk.5,6 Recently, BARD1, RAD51D, BRIP1, and RAD51C have been proposed as risk factors for triple-negative breast cancer (TNBC) with BARD1 and RAD51D conferring high risk, and BRIP1 and RAD51C associated with moderate risk.7 Thus, the risk associated with pathogenic variants in each gene may vary by breast tumor subtype.
Many of the BRCA/FA pathway genes when altered by biallelic mutations cause FA disease. The FANCM gene (FA complementation group M, OMIM #609644) encodes for a translocase, which is a member of the BRCA/FA molecular pathway but has been recently disqualified as a disease-causing factor for FA.8,9 Some protein-truncating variants in the FANCM gene were described as moderate breast cancer risk factors with a greater risk of TNBC. In the Finnish population, FANCM:c.5101 C > T (p.Gln1701*, rs147021911) is relatively frequent and was reported to be associated with breast cancer with odds ratio (OR) of 1.86 with 95% confidence intervals (CIs) = 1.26–2.75. A larger effect was observed in familial cases (OR = 2.11; 95% CI = 1.43–3.32), for estrogen receptor-negative (ER-negative) breast cancer (OR = 2.37; 95% CI = 1.37–4.12) and for TNBC (OR = 3.56; 95% CI = 1.81–6.98).10 We showed an increased risk (OR = 3.93; 95% CI = 1.28–12.11) of the FANCM:c.5791 C > T (rs144567652) truncating variant using familial cases and controls. In vitro analysis showed that this variant causes the skipping of the FANCM exon 22 and the creation of a downstream stop codon (p.Gly1906Alafs12*).11 However, in the present study we refer to the FANCM:c.5791 C > T base change as to FANCM:p.Arg1931*, which is the conventional amino acid annotation (consistent with the stop codon creation according to genetic code). The FANCM:p.Arg1931* was also found to be associated with TNBC risk in the Finnish population (OR = 5.14; 95% CI = 1.65–16.0).12 A burden analysis of truncating variants discovered by a re-sequencing analysis of the entire FANCM coding region in German cases and controls confirmed that FANCM pathogenic variants had a particularly high risk for TNBC (OR = 3.75; 95% CI = 1.0–12.85).13
To study the effect of FANCM on breast cancer risk further, we tested three recurrent truncating variants FANCM:p.Arg658*, p.Gln1701*, and p.Arg1931*, within the OncoArray Consortium, a collaboration of consortia established to discover germline genetic variants predisposing to different human cancers (e.g., breast, colon, lung, ovary, endometrium and prostate cancers).14 These three variants were tested for association with breast cancer risk in 67,112 breast cancer cases, 53,766 controls, and 26,662 carriers of pathogenic variants in BRCA1 or BRCA2. We also studied the functional effect of these three variants after their lentiviral transduction into a FANCM−/− patient-derived cell line in which we measured survival and chromosome fragility after exposure to diepoxybutane (DEB) or the poly (ADP-ribose) polymerase inhibitor (PARPi) olaparib.
Results
Case-control analyses
We analyzed the association of three FANCM truncating variants, p.Arg658*, p.Gln1701*, and p.Arg1931*, with breast cancer risk for each variant separately and using a burden analysis. We tested 67,112 invasive breast cancer cases and 53,766 controls collected by the Breast Cancer Association Consortium (BCAC, http://bcac.ccge.medschl.cam.ac.uk/) and 26,662 carriers of BRCA1 or BRCA2 pathogenic variants collected by the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA, http://cimba.ccge.medschl.cam.ac.uk/), of whom 13,497 were affected with breast cancer and 13,165 were unaffected.
In the BCAC dataset we assessed the breast cancer risk associated with the FANCM variants in a primary overall analysis and in a restricted analysis including only countries in which the variant carrier frequencies were higher than the median of the frequencies. In these analyses we tested association with the variants in all available invasive breast cancer cases or in the ER-positive, ER-negative and TNBC subgroups (Table 1). In the overall analysis, no evidence of association was observed, either with the presence of any FANCM variant or with any of the three variants individually. However, FANCM:p.Arg658* showed a higher heterozygote frequency in ER-negative breast cancer cases (0.093%) than in controls (0.035%) with a greater than two-fold increased breast cancer risk (OR = 2.44, 95% CI = 1.12–5.34, P = 0.034). When only TNBC cases were considered, the association was stronger (OR = 3.79, 95% CI = 1.56–9.18, P = 0.009). No association with ER-negative breast cancer or TNBC was seen for p.Gln1701* or p.Arg1931* or for all mutations combined (Table 1). In the country-restricted analyses, we confirmed the association found for p.Arg658* with risk of ER-negative disease and TNBC (OR = 2.31, 95% CI = 1.05–5.07, P = 0.047 and OR = 3.56, 95% CI = 1.46–8.69, P = 0.011, respectively). The restricted set also provided evidence for an association between p.Arg1931* and ER-negative subgroup (OR = 1.96, 95% CI = 1.24–3.10, P = 0.006), though not for TNBC. No significant association was observed for p.Gln1701* with either subgroups (Table 1).
Analyses of carriers of BRCA1 or BRCA2 pathogenic variants
We found no evidence of associations for FANCM:p.Arg658*, p.Gln1701*, and p.Arg1931* truncating variants with breast cancer risk in carriers of BRCA1 or BRCA2 pathogenic variants included in CIMBA (Supplementary Table 1). The p.Arg658* was detected with approximately four-fold higher frequencies in the BRCA1 affected individuals (0.063%) in comparison to the unaffected (0.013%), and in the BRCA2 affected individuals (0.071%) in comparison to the unaffected (0.019%). Consistently, hazard ratios (HRs) above two were estimated for BRCA1 (HR = 2.4, 95% CI = 0.52–11.12) and for BRCA2 (HR = 2.13, 95% CI = 0.41–11.14) pathogenic variant carriers. The frequencies of p.Gln1701* and p.Arg1931* were not increased in affected versus unaffected individuals carrying BRCA1 or BRCA2 pathogenic variants (Supplementary Table 1).
Functional studies
We tested the functional effect of FANCM:p.Arg658*, p.Gln1701*, and p.Arg1931* on DNA repair using genetic complementation assays (Fig. 1). These assays were based on the EGF280 cell line derived from immortalized fibroblasts from a patient who lacked the FANCM protein due to a homozygous c.1506_1507insTA (p.Ile503*, rs764743944) truncating variant.8 Complemented FANCM−/− cells were tested for sensitivity to DEB and olaparib by measuring cell survival and chromosome fragility. The FANCM protein was not detectable in the EGF280 fibroblasts. The transduction of these cells with lentiviral vectors carrying wild-type (wt) FANCM cDNA and cDNAs harboring FANCM:p.Gln1701* and p.Arg1931* variants produced, as expected, different C-terminal truncated forms of FANCM. In the EGF280 cells transduced with FANCM:p.Arg658* no visible band was observed on western blot (Fig. 1a and Supplementary Fig. 1). As we lack information on the epitope recognized by the antibody, we could not determine whether the p.Arg658*-derived truncated protein was unstable or if the epitope was lost due to the truncation. We therefore analyzed the mRNA expression of FANCM:p.Arg658* by reverse transcription and digestion of the PCR-amplified cDNAs. The c.1972C > T base substitution causing the p.Arg658* variant was expected to abolish a digestion site for the restriction enzyme TseI present in the wt sequence. TseI-digestion of wt and mutated cDNAs clearly indicated the presence of a mutated mRNA product in the EGF280 cells transduced with FANCM:p.Arg658* (Fig. 1b and Supplementary Fig. 1).

Functional studies of the FANCM:p.Arg658*, p.Gln1701* and p.Arg1931* truncating variants using the patient-derived FANCM−/− EGF280 cell line. a Western blot showing the FANCM expression in EGF280 cells complemented with lentiviral vectors harboring the three different variants. Bands corresponding to truncated FANCM protein were visible for EGF280 + p.Gln1701* and p.Arg1931*, and no bands were present for the EGF280 + p.Arg658*. b Study of the expression of the FANCM protein in EGF280 + p.Arg658*. The c.1972C > T base substitution, causing the p.Arg658* variant abrogates a digestion site for the restriction enzyme TseI that is present in the wild-type (wt) cDNA sequence. Total RNA was extracted from EGF280 + wtFANCM and from the EGF280 + p.Arg658* and subjected to reverse transcription. PCR-amplified cDNA products were digested with TseI. Digested and undigested cDNAs were loaded. In the first two lanes are shown bands of 386 bp corresponding to uncut wt cDNA, and bands of 257 and 129 bp corresponding to cut wt cDNA. In next two lanes bands of 386 bp indicate that p.Arg658* cDNA was not cut due to the c.1972C > T base substitution abrogating the TseI site. In the two lanes after the molecular weight marker (M) undigested and digested products of the two previous PCR products were mixed 1:1 and loaded as a control. c Analysis of diepoxybutane (DEB) sensitivity on cell survival. The EGF280 cells expressing p.Arg658* are significantly more sensitive to DEB than the cells expressing p.Gln1701* or p.Arg1931* (P-values from Tukey’s range test are reported in Supplementary Table 4). EGF280 and EGF280 + wtFANCM are used as controls (N = 3; error bars: standard deviation). d Chromosome fragility induced by DEB treatment (100 ng/ml). Here, the chromatidic break patterns of the cells expressing wt FANCM, of the cells harboring p.Gln1701* or p.Arg1931* variants, and of the native EGF280 cells or the cells expressing p.Arg658* were statistically different. (P-values from chi-squared test; N = 2). e Analysis of cellular sensitivity to olaparib. Contrarily to what we observed in the DEB sensitivity assays, survival rates of the different complemented cell lines were apparently not different. Human fibroblasts (BRCA2−/−) were homozygous for the c.469 A > T (p.Lys157*) truncating variant and were used as a positive control. (P-values from Tukey’s range test are reported in Supplementary Table 5; N = 3; error bars: standard deviation). All blots derive from the same experiment and were processed in parallel
In the DEB sensitivity-based assay (Fig. 1c), the EGF280 patient-derived cell line showed a high-sensitivity phenotype, that was rescued by expression of the wt FANCM. EGF280 cells expressing FANCM:p.Arg658* failed to rescue DEB sensitivity and showed survival rates overlapping with those of the native EGF280 cells. In comparison, cells expressing FANCM:p.Gln1701* and p.Arg1931* variants showed an intermediate phenotype with survival rates significantly higher than those of EGF280 cells, though significantly lower than those of the cells expressing wt FANCM (Fig. 1c and Supplementary Table 2). These results were confirmed in the chromosome fragility tests where the number of chromatidic breaks in cells harboring p.Gln1701* or p.Arg1931* variants was statistically lower than that of EGF280 cells or cells expressing the p.Arg658* and statistically higher than that of cells expressing wt FANCM (Fig. 1d). In the olaparib sensitivity-based assay, the survival rates of the cell lines transduced with the three FANCM truncating variants were not statistically different. Only at higher olaparib concentrations (>5000 nM) the survival rates of these cell lines were significantly lower than that of the wt FANCM cells and higher than that of the EGF280 cells (Fig. 1e and Supplementary Table 3).
Discussion
In this study we investigated the association of the three recurrent FANCM truncating variants p.Arg658*, p.Gln1701*, and p.Arg1931*, with breast cancer risk overall and by tumor subtype. While in non-Finnish Europeans these are the three most common FANCM truncating variants, their carrier frequency is low being 0.033, 0.21 and 0.21%, respectively (https://gnomad.broadinstitute.org/).15 We conducted large case-control studies in 67,112 unselected breast cancer cases, 53,766 controls, and 26,662 carriers of BRCA1 or BRCA2 pathogenic variants. Furthermore, we performed functional analyses based on a patient-derived FANCM−/− cell line transduced with vectors carrying the three FANCM variants and tested for sensitivity to DEB or olaparib. Our genetic data suggest that FANCM:p.Arg658* is a risk factor for ER-negative and TNBC subtypes with statistically significant ORs of 2.44 and 3.79, respectively. These associations were confirmed when we restricted the analyses to countries with higher carrier frequencies. In these restricted analyses we also found that the p.Arg1931* was associated with breast cancer risk in the ER-negative subtype with statistically significant OR = 1.96. (Table 1). These data, together with previously published genetic studies,10,11,12,13 confirm that FANCM truncating variants are risk factors for breast cancer, with a stronger association for the ER-negative and TNBC subtypes. Our functional data, obtained in a background of a FANCM null cell line, support these findings showing that all three truncating variants were deleterious; hence, it is expected that, in the heterozygous state, any of these FANCM variants have partial activity. In the functional tests, we also observed that olaparib had a greater effect on survival of the cells harboring any of the FANCM:p.Arg658*, p.Gln1701*, or p.Arg1931* variants with respect to that on EGF280 cells complemented with wt FANCM (Fig. 1e). As this is consistent with previous results,16 PARP1 inhibition might be a possible therapeutic approach to treat patients with breast tumors associated with germline FANCM pathogenic variants. On the contrary, the DEB sensitivity assays showed that FANCM:p.Arg658*, is associated with a stronger impairment of DNA repair activity, compared to p.Gln1701* and p.Arg1931*, possibly reflecting the position of protein truncation (Fig. 1c, d).
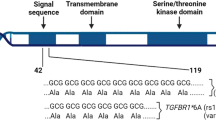
FANCM encodes for a key protein of the upstream FA/BRCA pathway mediating the assembly of the FA core complex. This protein is 2048 AA long, possesses in its N-terminal region an intrinsic ATP-dependent DNA translocase activity and, with its central region, recognizes the Bloom’s complex, which is also involved in the DNA HR repair. By interacting with its C-terminal binding partner, the FA associated protein 24 (FAAP24), the FANCM protein brings to sites of ICL DNA lesions the FA and the Bloom’s complexes initiating HR repair17 (Fig. 2). We studied FANCM:p.Arg658*, p.Gln1701*, and p.Arg1931* in the same genetic FANCM−/− background and showed that, after exposure to DEB, the N-terminal FANCM:p.Arg658* had a statistically stronger effect on cell survival and chromosome stability (presumably due to less efficient DNA repair activity) than did p.Gln1701* and p.Arg1931*. This also suggests that in human living cells the FANCM:p.Arg658* variant might impair DNA repair more severely than p.Gln1701* and p.Arg1931*. We have shown that in vitro both the p.Gln1701*- and the p.Arg1931*-derived FANCM proteins are expressed and that the p.Arg658*-mRNA is transcribed (Fig. 1a, b). An N-terminus fragment including the first 422 AA of FANCM was shown to be stable when expressed in human cell lines,17 thus supporting the possibility that the FANCM:p.Arg658*-derived protein may also be expressed and stable. Hence, we hypothesize that the observed difference in survival and chromosome fragility of cells treated with DEB may be attributable to the diverse residual function of the different truncated forms of FANCM. In fact, the p.Gln1701*- and the p.Arg1931*-derived forms are expected to lose the interaction with FAAP24, but to retain the ability of binding other FANCM interacting proteins. Hence, our data suggest that the lack of interaction between FANCM and FAAP24 has a less severe impact on the DNA damage response than when protein truncation occurs upstream the FANCM domains AA 687–1104 and AA 1027–1362 mediating the interaction with the FA core complex and the Bloom’s complex, respectively.

Schematic diagram of the 2,048 amino acid long FANCM protein. The functional or binding domains (BD) are indicated in black and as reported in Deans and West, 2009. The position of the three FANCM truncating variants c.1972C > T (p.Arg658*), c.5101 C > T (p.Gln1701*) and c.5791 C > T (p.Arg1931*) is also shown
Previously published genetic and clinical data support our hypothesis of a position effect. FANCM pathogenic variants were shown to be associated with a moderate risk of developing high-grade serous epithelial ovarian cancer, but p.Arg1931* appeared to confer a lower risk.18 Moreover, five female breast cancer probands carrying homozygous FANCM truncating variants were recently described.9 Three of these, two homozygous for p.Gln1701*, and one for p.Arg1931*, developed breast cancer at age 52 years or later and their cells did not demonstrate chromosome fragility. The other two probands were homozygous for p.Arg658* and developed early-onset breast cancer (at age 29 and 32); in addition, one developed several cancers, and the other demonstrated chromosomal fragility.9
Due to the rarity of the studied mutations in most populations, estimation of the risks is challenging. Preferably, the cases should be examined in comparison to geographically, ethnically and genetically matched controls. In the Finnish population, p.Gln1701* and p.Arg1931* are reported with carrier frequency of 1.62% and of 0.92%, respectively (https://gnomad.broadinstitute.org/).15 Case-control studies based on the Finnish population showed a strong statistical evidence of association of p.Gln1701* with ER-negative disease, with OR of 2.37 (95% CI = 1.37–4.12, P = 0.0021), and with TNBC with ORs of 3.56 (95% CI = 1.81–6.98, P = 0.0002),10 while p.Arg1931* was found associated with TNBC with an OR of 5.14 (95% CI = 1.65–16.0, P = 0.005).12 However, as our 95% CI of risk estimates for TNBC included odds ratios of 2 for both the latter mutations, the published and our results are not mutually exclusive. Risk estimates associated with rare variants may depend on their frequency and the genetic background of the population studied. Hence, pooling the data from multiple outbred and admixed populations as it was done in the present study, may yield different risk estimates than those derived from geographically, ethnically and genetically matched controls, as in the Finnish studies. Indeed, it would have been interesting to test the FANCM variant position effect in the Finnish population, but unfortunately the p.Arg658* is very rare if not absent in this population (https://gnomad.broadinstitute.org/).15
Recent attempts to identify novel, high- to moderate-risk breast cancer-predisposing genes have not been particularly fruitful. However, a few genes have emerged as potential risk factors for ER-negative disease and TNBC, with FANCM, BRIP1, and RAD51C being among those suggested to confer moderate risk of these subtypes. Other predisposing genes increasing the risk of ER-negative and TNBC may also exist. Hence, further gene discovery efforts should take into consideration that risk-associated variants may be associated with specific tumor subtypes and/or variation in risk may depend on the variant position. In addition, we provide evidence that lack of FANCM protein and truncating variants identified in breast cancer patients are associated with increased sensitivity to the PARPi olaparib suggesting a therapeutic opportunity to treat FANCM-associated breast tumors that warrants further investigation. The PARPi sensitivity test may also prove useful for preclinical investigation of further truncating or missense FANCM variants.
In summary, we have shown that FANCM:p.Arg658* is associated with risk of ER-negative breast cancer and TNBC. The outcomes of functional assays testing the DNA repair efficiency in complemented human cells support the hypothesis that breast cancer risk may be greater for N-terminal than C-terminal FANCM truncating variants. Further genetic studies and meta-analyses are warranted to derive more precise risk estimates for the different FANCM variants.
Methods
Study participants
The individuals included in this study were women of genetically confirmed European ancestry who were originally ascertained in 73 case-control studies from 19 countries participating in the BCAC or in 59 studies enrolling BRCA1 or BRCA2 pathogenic variants carrier from 30 countries participating in the CIMBA.
Ethics
All participating studies, listed in Supplementary Table 4 and Supplementary Table 5, were approved by their ethics review boards and followed national guidelines for informed consent. However, due to the retrospective nature of the majority of the studies, not all participant individuals have provided written informed consent to take part in the present analysis. The Milan Breast Cancer Study Group (MBCSG) was approved by ethics committee from Istituto Nazionale dei Tumori di Milano and Istituto Europeo di Oncologia, in Milan.
The BCAC studies contributed 67,112 invasive breast cancer cases and 53,766 controls. The majority of these studies were population-based, hospital-based or case-control studies nested within population-based cohorts (86%); few were family-clinic-based studies (14%; Supplementary Table 4). For each study subject, information on the disease status and the age at diagnosis or at interview were provided. Data on lifestyle risk factors were available for most subjects and clinical and pathological data were available for most cases. All these data were incorporated in the BCAC dataset (version 10). A total of 44,565 (66%) cases were ER-positive, 10,770 (16%) were ER-negative, and 4,805 (7%) were TNBC; 13,743 (20%) had a positive first-degree family history of breast cancer.
The CIMBA studies contributed 15,679 carriers of a pathogenic BRCA1 variant and 10,983 carriers of a pathogenic BRCA2 variant to this analysis (Supplementary Table 5). Nearly all (98%) of these carriers were ascertained through cancer genetic clinics; few carriers were recruited by population-based sampling of cases or by community recruitment. In some instances, multiple members of the same family were included. For each pathogenic variant carrier, the information on the type of the BRCA1 or BRCA2 variant, disease status, and censoring variables (see below, Statistical analyses) were collected and included in the CIMBA database.
Genotyping
Genotyping of FANCM:p.Arg658*, p.Gln1701*, and p.Arg1931* truncating variants was conducted using a custom-designed Illumina genotyping array (the “OncoArray”, Illumina, Inc. San Diego, CA, USA) at six independent laboratories. To ensure consistency of the genotype data, all laboratories used the same genotype-clustering file and genotyped the same set of reference-samples selected from the HapMap project. Samples with a call rate <95% and those with heterozygosity <5% or >40% were excluded. Further details of the genotype-calling and quality control have been described previously.14 The cluster plots of the three FANCM truncating variants were curated manually to confirm the automatic calls (Supplementary Fig. 2).
Statistical analyses
The BCAC data were analyzed to test the association between FANCM:p.Arg658*, p.Gln1701*, and p.Arg1931* and breast cancer risk. Logistic regression analyses were performed to estimate ORs with 95% CIs for variant carriers versus non-carriers, adjusting for country and the first ten principal components, as previously described.19 P-values were calculated by applying the likelihood ratio test (LRT) comparing the model containing the variant carrier status as a covariate to a model without the variant carrier status. The primary analyses were performed including all invasive breast cancer cases and controls and subgrouping cases based on tumor hormonal status. We then performed a country-restricted analysis including the 50% of the countries with the higher variant carrier frequencies. Specifically, we included only countries in which the carrier frequencies in cases and controls combined were higher than the median of the carrier frequencies observed in all countries. Median frequencies were 0.007, 0.114 and 0.163 for p.Arg658*, p.Gln1701* and p.Arg1931* carriers, respectively.
The CIMBA data were analyzed to evaluate the association between each FANCM truncating variant and breast cancer risk in carriers of BRCA1 or BRCA2 pathogenic variant. A survival analyses framework was applied. Briefly, each variant carrier was followed from the age of 18 years until the first breast cancer diagnosis, or censored as unaffected at ovarian cancer diagnosis, bilateral prophylactic mastectomy, or age at last follow-up. The analyses were performed by modelling the retrospective likelihood of the observed genotypes conditional on the disease phenotype as detailed previously.20 All analyses were stratified for country. The per-allele hazard ratio (HR), 95% CIs were estimated separately for each variant. A score test was used to derive P-values for the associations. The analyses of the BCAC data were performed using STATA version 15 (StataCorp LLC, College Station, Texas, USA). The analyses of the CIMBA data were carried out using custom-written code in Python and Fortran. All statistical tests were two-sided and P-values <0.05 were considered statistically significant.
Cell lines, plasmids, and lentiviral particles production and transduction
The immortalized patient-derived FANCM−/− cell line EGF2808 was transduced with pLenti CMV rtTA3 Blast, a gift from E. Campeau (Addgene plasmid #26429). The doxycycline-inducible lentiviral vector pLVX-TRE3G-FANCM, a gift from N. Ameziane (Vrije Universiteit Medical Center, Amsterdam) was mutated by site-directed mutagenesis using the QuickChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies) and the following PAGE purified mutagenic primers. FANCM c.1972C > T primer 1: 5’-GCCTTCTCGGAACTTGCAGTGAAAGTCATCTATCTTTTCC-3’ and primer 2: 5’-GGAAAAGATAGATGACTTTCACTGCAAGTTCCGAGAAGGC-3’ for the p.Arg658*; FANCM c.5101 C > T primer 1: 5’-TTAAACAATGGTCCTATTGTTTGTTCTTCTTAACAGTGCTTGGGT-3’ and primer 2: 5’-ACCCAAGCACTGTTAAGAAGAACAAACAATAGGACCATTGTTTAA-3’ for the p.Gln1701*. Generation of the lentiviral vector containing the FANCM:c.5791 C > T (p.Arg1931*) and transduction of the EGF280 cells were already described.11 Expression of exogenous FANCM protein was achieved supplementing cell culture medium with doxycycline (1 μg/ml, final concentration). All the cell lines used in this study were routinely checked for mycoplasma contamination using the MycoAlert™ Mycoplasma Detection Kit (Lonza).
Western blot and mRNA expression studies
Cell lysis and western blot assays were performed as previously described.8 The following primary antibodies were used: mouse monoclonal anti-FANCM antibody, clone CV5.1 diluted 1:100 (ref: MABC545, MERCK Millipore), mouse monoclonal anti-Vinculin diluted 1:3000 (ref: ab18058, abcam). Western blotting detection was achieved with LuminataTM Classico (Millipore) (Vinculin) and LuminataForteTM (Millipore) (FANCM). We used RT PCR to test the expression of the mutant FANCM:p.Arg658*. Total RNA was extracted (RNeasy Mini Kit Qiagen) from the wtFANCM and FANCM:p.Arg658* transduced EGF280 cell lines. Reverse transcription was performed using High-Capacity RNA-to-cDNA Kit (Thermofisher); a cDNA region corresponding to the FANCM sequence containing the amino acid (AA) position Arg658 was amplified by PCR using the forward: 5’-AGTAACAGGCAGGTCCTTCA-3´and reverse: 5’-TGATCTTGCCACAGTCTCCA-3’ primers. The 386 bp PCR products were then digested with TseI restriction enzyme (New England Biolabs) for two hours at 65 °C and analyzed by standard agarose gel electrophoresis.
Cell survival assay
The effect of the different FANCM variants on cell survival was measured with a Sulforhodamine B (SRB) assay.21 One-thousand cells were seeded in 96-well plates and treated constantly with DEB or PARPi olaparib at the indicated concentrations until untreated cells reached confluency. Cell monolayers were fixed overnight at 4 °C with 75 μl of 20% trichloroacetic acid (TCA). TCA was aspirated, and cells washed with tap water. Once dried, 50 μl of SRB was added to the wells and plates were incubated on a shaker at room temperature for 30 min. The excess of SRB dye was removed by washing repeatedly with 1% acetic acid, the plates were dried for 20 min, and the protein-bound dye was dissolved in 10 mM Tris for OD determination at 492 nm using a microplate reader (Tecan Sunrise™, Tecan Group Ltd. Männedorf, Switzerland). At least three independent experiments were performed for each cell line and in each experiment, 12 wells were measured per concentration point. These results were statistically analyzed using the Prism (GraphPad) software. Two-Way ANOVA test was used for single comparisons between different cell lines and statistical significance was assessed with the Tukey’s range test. A P-value < 0.05 was considered statistically significant.
Chromosome fragility test
Chromosome fragility test was performed as previously described.11 Twenty-five metaphases were scored for chromosome breakages using the Metafer Slide Scanning Platform from Metasystems. Results were graphed as distributions of metaphases presenting 0, 1, and >1 chromatid break. Statistical analysis was performed applying chi-squared test.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary
Data availability
A subset of the genotype data analysed in this study is publicly available from the dbGaP repository and can be accessed at https://identifiers.org/dbgap:phs001265.v1.p1 (data generated as part of the BCAC studies) and at https://identifiers.org/dbgap:phs001321.v1.p1 (data generated as part of the CIMBA studies). The remaining genotype data analysed in this study (and generated as part of the BCAC and CIMBA studies listed in Supplementary Tables 4 and 5 of the related article, respectively) are not publicly available due to restraints imposed by the ethics committees of individual studies, but can be accessed from the corresponding author on reasonable request as described at https://doi.org/10.6084/m9.figshare.8982296.22 Additional datasets generated during this study (and supporting Fig. 1 and Supplementary Tables 2 and 3 in the published article) are available on request as described above. The data generated and analyzed during this study are described in the following data record: https://doi.org/10.6084/m9.figshare.8982296.22
References
Bogliolo, M. & Surralles, J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet Dev. 33, 32–40 (2015).
Michl, J., Zimmer, J. & Tarsounas, M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. 35, 909–923 (2016).
Kuchenbaecker, K. B. et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 317, 2402–2416 (2017).
Antoniou, A. C. et al. Breast-cancer risk in families with mutations in PALB2. N. Engl. J. Med. 371, 497–506 (2014).
Easton, D. F. et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 372, 2243–2257 (2015).
Schmidt, M. K. et al. Age- and tumor subtype-specific breast cancer risk estimates for CHEK2*1100delC carriers. J. Clin. Oncol. 34, 2750–2760 (2016).
Shimelis, H. et al. Triple-negative breast cancer risk genes identified by multigene hereditary cancer panel testing. J. Natl Cancer Inst. 110, 855–862 (2018).
Bogliolo, M. et al. Biallelic truncating FANCM mutations cause early-onset cancer but not Fanconi anemia. Genet Med. 20, 458–463 (2018).
Catucci, I. et al. Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genet Med. 20, 452–457 (2018).
Kiiski, J. I. et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl Acad. Sci. USA 111, 15172–15177 (2014).
Peterlongo, P. et al. FANCM c.5791C>T nonsense mutation (rs144567652) induces exon skipping, affects DNA repair activity and is a familial breast cancer risk factor. Hum. Mol. Genet 24, 5345–5355 (2015).
Kiiski, J. I. et al. FANCM mutation c.5791C>T is a risk factor for triple-negative breast cancer in the Finnish population. Breast Cancer Res. Treat. 166, 217–226 (2017).
Neidhardt, G. et al. Association between loss-of-function mutations within the FANCM gene and early-onset familial breast cancer. JAMA Oncol. 3, 1245–1248 (2017).
Amos, C. I. et al. The OncoArray consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemiol. Biomark. Prev. 26, 126–135 (2017).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291 (2016).
Stoepker, C. et al. DNA helicases FANCM and DDX11 are determinants of PARP inhibitor sensitivity. DNA Repair (Amst.) 26, 54–64 (2015).
Deans, A. J. & West, S. C. FANCM connects the genome instability disorders Bloom’s Syndrome and Fanconi Anemia. Mol. Cell 36, 943–953 (2009).
Dicks, E. et al. Germline whole exome sequencing and large-scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget 8, 50930–50940 (2017).
Michailidou, K. et al. Association analysis identifies 65 new breast cancer risk loci. Nature 551, 92–94 (2017).
Antoniou, A. C. et al. RAD51 135G->C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. Am. J. Hum. Genet 81, 1186–1200 (2007).
Vichai, V. & Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 1, 1112–1116 (2006).
Figlioli, G. et al. Metadata supporting data files of the related manuscript: The FANCM:p.Arg658* truncating variant is associated with risk of triple-negative breast cancer. figshare. https://doi.org/10.6084/m9.figshare.8982296 (2019).
Acknowledgements
Peterlongo laboratory is supported by Associazione Italiana Ricerca sul Cancro (AIRC; IG2015 no.16732) to P. Peterlongo and by a fellowship from Fondazione Umberto Veronesi to G. Figlioli. Surrallés laboratory is supported by the ICREA-Academia program, the Spanish Ministry of Health (projects FANCOSTEM and FANCOLEN), the Spanish Ministry of Economy and Competiveness (projects CB06/07/0023 and RTI2018-098419-B-I00), the European Commission (EUROFANCOLEN project HEALTH-F5-2012-305421 and P-SPHERE COFUND project), the Fanconi Anemia Research Fund Inc, and the “Fondo Europeo de Desarrollo Regional, una manera de hacer Europa” (FEDER). CIBERER is an initiative of the Instituto de Salud Carlos III, Spain. BCAC: we thank all the individuals who took part in these studies and all the researchers, clinicians, technicians and administrative staff who have enabled this work to be carried out. ABCFS thank Maggie Angelakos, Judi Maskiell, Tu Nguyen-Dumont is a National Breast Cancer Foundation (Australia) Career Development Fellow. ABCS thanks the Blood bank Sanquin, The Netherlands. Samples are made available to researchers on a non-exclusive basis. BCEES thanks Allyson Thomson, Christobel Saunders, Terry Slevin, BreastScreen Western Australia, Elizabeth Wylie, Rachel Lloyd. The BCINIS study would not have been possible without the contributions of Dr. Hedy Rennert, Dr. K. Landsman, Dr. N. Gronich, Dr. A. Flugelman, Dr. W. Saliba, Dr. E. Liani, Dr. I. Cohen, Dr. S. Kalet, Dr. V. Friedman, Dr. O. Barnet of the NICCC in Haifa, and all the contributing family medicine, surgery, pathology and oncology teams in all medical institutes in Northern Israel. The BREOGAN study would not have been possible without the contributions of the following: Manuela Gago-Dominguez, Jose Esteban Castelao, Angel Carracedo, Victor Muñoz Garzón, Alejandro Novo Domínguez, Maria Elena Martinez, Sara Miranda Ponte, Carmen Redondo Marey, Maite Peña Fernández, Manuel Enguix Castelo, Maria Torres, Manuel Calaza (BREOGAN), José Antúnez, Máximo Fraga and the staff of the Department of Pathology and Biobank of the University Hospital Complex of Santiago-CHUS, Instituto de Investigación Sanitaria de Santiago, IDIS, Xerencia de Xestion Integrada de Santiago-SERGAS; Joaquín González-Carreró and the staff of the Department of Pathology and Biobank of University Hospital Complex of Vigo, Instituto de Investigacion Biomedica Galicia Sur, SERGAS, Vigo, Spain. BSUCH thanks Peter Bugert, Medical Faculty Mannheim. CBCS thanks study participants, co-investigators, collaborators and staff of the Canadian Breast Cancer Study, and project coordinators Agnes Lai and Celine Morissette. CCGP thanks Styliani Apostolaki, Anna Margiolaki, Georgios Nintos, Maria Perraki, Georgia Saloustrou, Georgia Sevastaki, Konstantinos Pompodakis. CGPS thanks staff and participants of the Copenhagen General Population Study. For the excellent technical assistance: Dorthe Uldall Andersen, Maria Birna Arnadottir, Anne Bank, Dorthe Kjeldgård Hansen. The Danish Cancer Biobank is acknowledged for providing infrastructure for the collection of blood samples for the cases. Investigators from the CPS-II cohort thank the participants and Study Management Group for their invaluable contributions to this research. They also acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention National Program of Cancer Registries, as well as cancer registries supported by the National Cancer Institute Surveillance Epidemiology and End Results program. The CTS Steering Committee includes Leslie Bernstein, Susan Neuhausen, James Lacey, Sophia Wang, Huiyan Ma, and Jessica Clague DeHart at the Beckman Research Institute of City of Hope, Dennis Deapen, Rich Pinder, and Eunjung Lee at the University of Southern California, Pam Horn-Ross, Peggy Reynolds, Christina Clarke Dur and David Nelson at the Cancer Prevention Institute of California, Hoda Anton-Culver, Argyrios Ziogas, and Hannah Park at the University of California Irvine, and Fred Schumacher at Case Western University. DIETCOMPLYF thanks the patients, nurses and clinical staff involved in the study. The DietCompLyf study was funded by the charity Against Breast Cancer (Registered Charity Number 1121258) and the NCRN. We thank the participants and the investigators of EPIC (European Prospective Investigation into Cancer and Nutrition). ESTHER thanks Hartwig Ziegler, Sonja Wolf, Volker Hermann, Christa Stegmaier, Katja Butterbach. FHRISK thanks NIHR for funding. GC-HBOC thanks Stefanie Engert, Heide Hellebrand, Sandra Kröber and LIFE - Leipzig Research Centre for Civilization Diseases (Markus Loeffler, Joachim Thiery, Matthias Nüchter, Ronny Baber). The GENICA Network: Dr. Margarete Fischer-Bosch-Institute of Clinical Pharmacology, Stuttgart, and University of Tübingen, Germany [HB, Wing-Yee Lo], German Cancer Consortium (DKTK) and German Cancer Research Center (DKFZ) [HB], Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2180 - 390900677 [HB], Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany [Yon-Dschun Ko, Christian Baisch], Institute of Pathology, University of Bonn, Germany [Hans-Peter Fischer], Molecular Genetics of Breast Cancer, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, Germany [Ute Hamann], Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, Germany [TB, Beate Pesch, Sylvia Rabstein, Anne Lotz]; and Institute of Occupational Medicine and Maritime Medicine, University Medical Center Hamburg-Eppendorf, Germany [Volker Harth]. HABCS thanks Michael Bremer. HEBCS thanks Heidi Toiminen, Kristiina Aittomäki, Irja Erkkilä and Outi Malkavaara. HMBCS thanks Peter Hillemanns, Hans Christiansen and Johann H. Karstens. HUBCS thanks Shamil Gantsev. KARMA thanks the Swedish Medical Research Counsel. KBCP thanks Eija Myöhänen, Helena Kemiläinen. LMBC thanks Gilian Peuteman, Thomas Van Brussel, EvyVanderheyden and Kathleen Corthouts. MABCS thanks Milena Jakimovska (RCGEB “Georgi D. Efremov), Katerina Kubelka, Mitko Karadjozov (Adzibadem-Sistina” Hospital), Andrej Arsovski and Liljana Stojanovska (Re-Medika” Hospital) for their contributions and commitment to this study. MARIE thanks Petra Seibold, Dieter Flesch-Janys, Judith Heinz, Nadia Obi, Alina Vrieling, Sabine Behrens, Ursula Eilber, Muhabbet Celik, Til Olchers and Stefan Nickels. MBCSG (Milan Breast Cancer Study Group) thanks Daniela Zaffaroni, Irene Feroce, and the personnel of the Cogentech Cancer Genetic Test Laboratory. We thank the coordinators, the research staff and especially the MMHS participants for their continued collaboration on research studies in breast cancer. MSKCC thanks Marina Corines and Lauren Jacobs. MTLGEBCS would like to thank Martine Tranchant (CHU de Québec Research Center), Marie-France Valois, Annie Turgeon and Lea Heguy (McGill University Health Center, Royal Victoria Hospital; McGill University) for DNA extraction, sample management and skillful technical assistance. J.S. is Chairholder of the Canada Research Chair in Oncogenetics. NBHS thanks study participants and research staff for their contributions and commitment to the studies. We would like to thank the participants and staff of the Nurses’ Health Study and Nurses’ Health Study II for their valuable contributions as well as the following state cancer registries for their help: AL, AZ, AR, CA, CO, CT, DE, FL, GA, ID, IL, IN, IA, KY, LA, ME, MD, MA, MI, NE, NH, NJ, NY, NC, ND, OH, OK, OR, PA, RI, SC, TN, TX, VA, WA, WY. The study protocol was approved by the institutional review boards of the Brigham and Women’s Hospital and Harvard T.H. Chan School of Public Health, and those of participating registries as required. The authors assume full responsibility for analyses and interpretation of these data. OFBCR thanks Teresa Selander and Nayana Weerasooriya. ORIGO thanks E. Krol-Warmerdam, and J. Blom for patient accrual, administering questionnaires, and managing clinical information. PBCS thanks Louise Brinton, Mark Sherman, Neonila Szeszenia-Dabrowska, Beata Peplonska, Witold Zatonski, Pei Chao and Michael Stagner. The ethical approval for the POSH study is MREC /00/6/69, UKCRN ID: 1137. We thank staff in the Experimental Cancer Medicine Centre (ECMC) supported Faculty of Medicine Tissue Bank and the Faculty of Medicine DNA Banking resource. PREFACE thanks Sonja Oeser and Silke Landrith. PROCAS thanks NIHR for funding. RBCS thanks Petra Bos, Jannet Blom, Ellen Crepin, Elisabeth Huijskens, Anja Kromwijk-Nieuwlaat, Annette Heemskerk, the Erasmus MC Family Cancer Clinic. We thank the SEARCH and EPIC teams. SKKDKFZS thanks all study participants, clinicians, family doctors, researchers and technicians for their contributions and commitment to this study. We thank the SUCCESS Study teams in Munich, Duessldorf, Erlangen and Ulm. SZBCS thanks Ewa Putresza. UCIBCS thanks Irene Masunaka. UKBGS thanks Breast Cancer Now and the Institute of Cancer Research for support and funding of the Breakthrough Generations Study, and the study participants, study staff, and the doctors, nurses and other health care providers and health information sources who have contributed to the study. We acknowledge NHS funding to the Royal Marsden/ICR NIHR Biomedical Research Centre. CIMBA: we are grateful to all the families and clinicians who contribute to the studies; Sue Healey, in particular taking on the task of mutation classification with the late Olga Sinilnikova; Maggie Angelakos, Judi Maskiell, Helen Tsimiklis; members and participants in the New York site of the Breast Cancer Family Registry; members and participants in the Ontario Familial Breast Cancer Registry; Vilius Rudaitis and Laimonas Griškevičius; Yuan Chun Ding and Linda Steele for their work in participant enrollment and biospecimen and data management; Bent Ejlertsen and Anne-Marie Gerdes for the recruitment and genetic counseling of participants; Alicia Barroso, Rosario Alonso and Guillermo Pita; all the individuals and the researchers who took part in CONSIT TEAM (Consorzio Italiano Tumori Ereditari Alla Mammella), thanks in particular: Giulia Cagnoli, Roberta Villa, Irene Feroce, Mariarosaria Calvello, Riccardo Dolcetti, Giuseppe Giannini, Laura Papi, Gabriele Lorenzo Capone, Liliana Varesco, Viviana Gismondi, Maria Grazia Tibiletti, Daniela Furlan, Antonella Savarese, Aline Martayan, Stefania Tommasi, Brunella Pilato, Isabella Marchi, Elena Bandieri, Antonio Russo, Daniele Calistri and the personnel of the Cogentech Cancer Genetic Test Laboratory, Milan, Italy. FPGMX: members of the Cancer Genetics group (IDIS): Ana Blanco, Miguel Aguado, Uxía Esperón and Belinda Rodríguez. We thank all participants, clinicians, family doctors, researchers, and technicians for their contributions and commitment to the DKFZ study and the collaborating groups in Lahore, Pakistan (Noor Muhammad, Sidra Gull, Seerat Bajwa, Faiz Ali Khan, Humaira Naeemi, Saima Faisal, Asif Loya, Mohammed Aasim Yusuf) and Bogota, Colombia (Diana Torres, Ignacio Briceno, Fabian Gil). Genetic Modifiers of Cancer Risk in BRCA1/2 Mutation Carriers (GEMO) study is a study from the National Cancer Genetics Network UNICANCER Genetic Group, France. We wish to pay a tribute to Olga M. Sinilnikova, who with Dominique Stoppa-Lyonnet initiated and coordinated GEMO until she sadly passed away on the 30th June 2014. The team in Lyon (Olga Sinilnikova, Mélanie Léoné, Laure Barjhoux, Carole Verny-Pierre, Sylvie Mazoyer, Francesca Damiola, Valérie Sornin) managed the GEMO samples until the biological resource centre was transferred to Paris in December 2015 (Noura Mebirouk, Fabienne Lesueur, Dominique Stoppa-Lyonnet). We want to thank all the GEMO collaborating groups for their contribution to this study. Drs.Sofia Khan, Irja Erkkilä and Virpi Palola; The Hereditary Breast and Ovarian Cancer Research Group Netherlands (HEBON) consists of the following Collaborating Centers: Netherlands Cancer Institute (coordinating center), Amsterdam, NL: M.A. Rookus, F.B.L. Hogervorst, F.E. van Leeuwen, M.A. Adank, M.K. Schmidt, N.S. Russell, D.J. Jenner; Erasmus Medical Center, Rotterdam, NL: J.M. Collée, A.M.W. van den Ouweland, M.J. Hooning, C.M. Seynaeve, C.H.M. van Deurzen, I.M. Obdeijn; Leiden University Medical Center, NL: C.J. van Asperen, P. Devilee, T.C.T.E.F. van Cronenburg; Radboud University Nijmegen Medical Center, NL: C.M. Kets, A.R. Mensenkamp; University Medical Center Utrecht, NL: M.G.E.M. Ausems, M.J. Koudijs; Amsterdam Medical Center, NL: C.M. Aalfs, H.E.J. Meijers-Heijboer; VU University Medical Center, Amsterdam, NL: K. van Engelen, J.J.P. Gille; Maastricht University Medical Center, NL: E.B. Gómez-Garcia, M.J. Blok; University of Groningen, NL: J.C. Oosterwijk, A.H. van der Hout, M.J. Mourits, G.H. de Bock; The Netherlands Comprehensive Cancer Organisation (IKNL): S. Siesling, J.Verloop; The nationwide network and registry of histo- and cytopathology in The Netherlands (PALGA): A.W. van den Belt-Dusebout. HEBON thanks the study participants and the registration teams of IKNL and PALGA for part of the data collection. Overbeek; the Hungarian Breast and Ovarian Cancer Study Group members (Janos Papp, Aniko Bozsik, Zoltan Matrai, Miklos Kasler, Judit Franko, Maria Balogh, Gabriella Domokos, Judit Ferenczi, Department of Molecular Genetics, National Institute of Oncology, Budapest, Hungary) and the clinicians and patients for their contributions to this study; HVH (University Hospital Vall d’Hebron) the authors acknowledge the Oncogenetics Group (VHIO) and the High Risk and Cancer Prevention Unit of the University Hospital Vall d’Hebron, Miguel Servet Progam (CP10/00617), and the Cellex Foundation for providing research facilities and equipment; the ICO Hereditary Cancer Program team led by Dr. Gabriel Capella; the ICO Hereditary Cancer Program team led by Dr. Gabriel Capella; Dr Martine Dumont for sample management and skillful assistance; Catarina Santos and Pedro Pinto; members of the Center of Molecular Diagnosis, Oncogenetics Department and Molecular Oncology Research Center of Barretos Cancer Hospital; Heather Thorne, Eveline Niedermayr, all the kConFab investigators, research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (which has received funding from the NHMRC, the National Breast Cancer Foundation, Cancer Australia, and the National Institute of Health (USA)) for their contributions to this resource, and the many families who contribute to kConFab; the investigators of the Australia New Zealand NRG Oncology group; members and participants in the Ontario Cancer Genetics Network; Kevin Sweet, Caroline Craven, Julia Cooper, Amber Aielts, and Michelle O’Conor; Christina Selkirk; Helena Jernström, Karin Henriksson, Katja Harbst, Maria Soller, Ulf Kristoffersson; from Gothenburg Sahlgrenska University Hospital: Anna Öfverholm, Margareta Nordling, Per Karlsson, Zakaria Einbeigi; from Stockholm and Karolinska University Hospital: Anna von Wachenfeldt, Annelie Liljegren, Annika Lindblom, Brita Arver, Gisela Barbany Bustinza; from Umeå University Hospital: Beatrice Melin, Christina Edwinsdotter Ardnor, Monica Emanuelsson; from Uppsala University: Hans Ehrencrona, Maritta Hellström Pigg, Richard Rosenquist; from Linköping University Hospital: Marie Stenmark-Askmalm, Sigrun Liedgren; Cecilia Zvocec, Qun Niu; Joyce Seldon and Lorna Kwan; Dr. Robert Nussbaum, Beth Crawford, Kate Loranger, Julie Mak, Nicola Stewart, Robin Lee, Amie Blanco and Peggy Conrad and Salina Chan; Carole Pye, Patricia Harrington and Eva Wozniak. OSUCCG thanks Kevin Sweet, Caroline Craven, Julia Cooper, Michelle O’Conor and Amber Aeilts. BCAC is funded by Cancer Research UK [C1287/A16563, C1287/A10118], the European Union’s Horizon 2020 Research and Innovation Programme (grant numbers 634935 and 633784 for BRIDGES and B-CAST respectively), and by the European Community´s Seventh Framework Programme under grant agreement number 223175 (grant number HEALTH-F2-2009-223175) (COGS). The EU Horizon 2020 Research and Innovation Programme funding source had no role in study design, data collection, data analysis, data interpretation or writing of the report. Genotyping of the OncoArray was funded by the NIH Grant U19 CA148065, and Cancer UK Grant C1287/A16563 and the PERSPECTIVE project supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research (grant GPH-129344) and, the Ministère de l’Économie, Science et Innovation du Québec through Genome Québec and the PSRSIIRI-701 grant, and the Quebec Breast Cancer Foundation. The Australian Breast Cancer Family Study (ABCFS) was supported by grant UM1 CA164920 from the National Cancer Institute (USA). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The ABCFS was also supported by the National Health and Medical Research Council of Australia, the New South Wales Cancer Council, the Victorian Health Promotion Foundation (Australia) and the Victorian Breast Cancer Research Consortium. J.L.H. is a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellow. M.C.S. is a NHMRC Senior Research Fellow. The ABCS study was supported by the Dutch Cancer Society [grants NKI 2007-3839; 2009 4363]. The Australian Breast Cancer Tissue Bank (ABCTB) was supported by the National Health and Medical Research Council of Australia, The Cancer Institute NSW and the National Breast Cancer Foundation. The AHS study is supported by the intramural research program of the National Institutes of Health, the National Cancer Institute (grant number Z01-CP010119), and the National Institute of Environmental Health Sciences (grant number Z01-ES049030). The work of the BBCC was partly funded by ELAN-Fond of the University Hospital of Erlangen. The BBCS is funded by Cancer Research UK and Breast Cancer Now and acknowledges NHS funding to the NIHR Biomedical Research Centre, and the National Cancer Research Network (NCRN). The BCEES was funded by the National Health and Medical Research Council, Australia and the Cancer Council Western Australia. For the BCFR-NY, BCFR-PA, BCFR-UT this work was supported by grant UM1 CA164920 from the National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the BCFR. BCINIS study was funded by the BCRF (The Breast Cancer Research Foundation, USA). The BREast Oncology GAlician Network (BREOGAN) is funded by Acción Estratégica de Salud del Instituto de Salud Carlos III FIS PI12/02125/Cofinanciado FEDER; Acción Estratégica de Salud del Instituto de Salud Carlos III FIS Intrasalud (PI13/01136); Programa Grupos Emergentes, Cancer Genetics Unit, Instituto de Investigacion Biomedica Galicia Sur. Xerencia de Xestion Integrada de Vigo-SERGAS, Instituto de Salud Carlos III, Spain; Grant 10CSA012E, Consellería de Industria Programa Sectorial de Investigación Aplicada, PEME I + D e I + D Suma del Plan Gallego de Investigación, Desarrollo e Innovación Tecnológica de la Consellería de Industria de la Xunta de Galicia, Spain; Grant EC11-192. Fomento de la Investigación Clínica Independiente, Ministerio de Sanidad, Servicios Sociales e Igualdad, Spain; and Grant FEDER-Innterconecta. Ministerio de Economia y Competitividad, Xunta de Galicia, Spain. The BSUCH study was supported by the Dietmar-Hopp Foundation, the Helmholtz Society and the German Cancer Research Center (DKFZ). Sample collection and processing was funded in part by grants from the National Cancer Institute (NCI R01CA120120 and K24CA169004). CBCS is funded by the Canadian Cancer Society (grant # 313404) and the Canadian Institutes of Health Research. CCGP is supported by funding from the University of Crete. The CECILE study was supported by Fondation de France, Institut National du Cancer (INCa), Ligue Nationale contre le Cancer, Agence Nationale de Sécurité Sanitaire, de l’Alimentation, de l’Environnement et du Travail (ANSES), Agence Nationale de la Recherche (ANR). The CGPS was supported by the Chief Physician Johan Boserup and Lise Boserup Fund, the Danish Medical Research Council, and Herlev and Gentofte Hospital. The American Cancer Society funds the creation, maintenance, and updating of the CPS-II cohort. The CTS was initially supported by the California Breast Cancer Act of 1993 and the California Breast Cancer Research Fund (contract 97-10500) and is currently funded through the National Institutes of Health (R01 CA77398, K05 CA136967, UM1 CA164917, and U01 CA199277). Collection of cancer incidence data was supported by the California Department of Public Health as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885. The University of Westminster curates the DietCompLyf database funded by Against Breast Cancer Registered Charity No. 1121258 and the NCRN. The coordination of EPIC is financially supported by the European Commission (DG-SANCO) and the International Agency for Research on Cancer. The national cohorts are supported by: Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), Federal Ministry of Education and Research (BMBF) (Germany); the Hellenic Health Foundation, the Stavros Niarchos Foundation (Greece); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); Health Research Fund (FIS), PI13/00061 to Granada, PI13/01162 to EPIC-Murcia, Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, ISCIII RETIC (RD06/0020) (Spain); Cancer Research UK (14136 to EPIC-Norfolk; C570/A16491 and C8221/A19170 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk, MR/M012190/1 to EPIC-Oxford) (United Kingdom). The ESTHER study was supported by a grant from the Baden Württemberg Ministry of Science, Research and Arts. Additional cases were recruited in the context of the VERDI study, which was supported by a grant from the German Cancer Aid (Deutsche Krebshilfe). FHRISK is funded from NIHR grant PGfAR 0707-10031. The GC-HBOC (German Consortium of Hereditary Breast and Ovarian Cancer) is supported by the German Cancer Aid (grant no 110837, coordinator: Rita K. Schmutzler, Cologne). This work was also funded by the European Regional Development Fund and Free State of Saxony, Germany (LIFE - Leipzig Research Centre for Civilization Diseases, project numbers 713-241202, 713-241202, 14505/2470, 14575/2470). The GENICA was funded by the Federal Ministry of Education and Research (BMBF) Germany grants 01KW9975/5, 01KW9976/8, 01KW9977/0 and 01KW0114, the Robert Bosch Foundation, Stuttgart, Deutsches Krebsforschungszentrum (DKFZ), Heidelberg, the Institute for Prevention and Occupational Medicine of the German Social Accident Insurance, Institute of the Ruhr University Bochum (IPA), Bochum, as well as the Department of Internal Medicine, Evangelische Kliniken Bonn gGmbH, Johanniter Krankenhaus, Bonn, Germany. The GEPARSIXTO study was conducted by the German Breast Group GmbH. The GESBC was supported by the Deutsche Krebshilfe e. V. [70492] and the German Cancer Research Center (DKFZ). The HABCS study was supported by the Claudia von Schilling Foundation for Breast Cancer Research, by the Lower Saxonian Cancer Society, and by the Rudolf Bartling Foundation. The HEBCS was financially supported by the Helsinki University Central Hospital Research Fund, Academy of Finland (266528), the Finnish Cancer Society, and the Sigrid Juselius Foundation. The HMBCS was supported by a grant from the German Research Foundation (Do 761/10-1). The HUBCS was supported by a grant from the German Federal Ministry of Research and Education (RUS08/017), and by the Russian Foundation for Basic Research and the Federal Agency for Scientific Organizations for support the Bioresource collections and RFBR grants 14-04-97088, 17-29-06014 and 17-44-020498. E.K was supported by the program for support the bioresource collections №007-030164/2 and study was performed as part of the assignment of the Ministry of Science and Higher Education of Russian Federation (№АААА-А16-116020350032-1). Financial support for KARBAC was provided through the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and Karolinska Institutet, the Swedish Cancer Society, The Gustav V Jubilee foundation and Bert von Kantzows foundation. The KARMA study was supported by Märit and Hans Rausings Initiative Against Breast Cancer. The KBCP was financially supported by the special Government Funding (EVO) of Kuopio University Hospital grants, Cancer Fund of North Savo, the Finnish Cancer Organizations, and by the strategic funding of the University of Eastern Finland. LMBC is supported by the ‘Stichting tegen Kanker’. DL is supported by the FWO. The MABCS study is funded by the Research Centre for Genetic Engineering and Biotechnology “Georgi D. Efremov” and supported by the German Academic Exchange Program, DAAD. The MARIE study was supported by the Deutsche Krebshilfe e.V. [70-2892-BR I, 106332, 108253, 108419, 110826, 110828], the Hamburg Cancer Society, the German Cancer Research Center (DKFZ) and the Federal Ministry of Education and Research (BMBF) Germany [01KH0402]. MBCSG is supported by grants from the Italian Association for Cancer Research (AIRC) and by funds from the Italian citizens who allocated the 5/1000 share of their tax payment in support of the Fondazione IRCCS Istituto Nazionale Tumori, according to Italian laws (INT-Institutional strategic projects “5 × 1000”). The MCBCS was supported by the NIH grants CA192393, CA116167, CA176785 an NIH Specialized Program of Research Excellence (SPORE) in Breast Cancer [CA116201], and the Breast Cancer Research Foundation and a generous gift from the David F. and Margaret T. Grohne Family Foundation. MCCS cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further supported by Australian NHMRC grants 209057 and 396414, and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry (VCR) and the Australian Institute of Health and Welfare (AIHW), including the National Death Index and the Australian Cancer Database. The MEC was support by NIH grants CA63464, CA54281, CA098758, CA132839 and CA164973. The MISS study is supported by funding from ERC-2011-294576 Advanced grant, Swedish Cancer Society, Swedish Research Council, Local hospital funds, Berta Kamprad Foundation, Gunnar Nilsson. The MMHS study was supported by NIH grants CA97396, CA128931, CA116201, CA140286 and CA177150. MSKCC is supported by grants from the Breast Cancer Research Foundation and Robert and Kate Niehaus Clinical Cancer Genetics Initiative. The work of MTLGEBCS was supported by the Quebec Breast Cancer Foundation, the Canadian Institutes of Health Research for the “CIHR Team in Familial Risks of Breast Cancer” program – grant # CRN-87521 and the Ministry of Economic Development, Innovation and Export Trade – grant # PSR-SIIRI-701. The NBHS was supported by NIH grant R01CA100374. Biological sample preparation was conducted the Survey and Biospecimen Shared Resource, which is supported by P30 CA68485. The Northern California Breast Cancer Family Registry (NC-BCFR) and Ontario Familial Breast Cancer Registry (OFBCR) were supported by grant UM1 CA164920 from the National Cancer Institute (USA). The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the USA Government or the BCFR. The Carolina Breast Cancer Study was funded by Komen Foundation, the National Cancer Institute (P50 CA058223, U54 CA156733, U01 CA179715), and the North Carolina University Cancer Research Fund. The NHS was supported by NIH grants P01 CA87969, UM1 CA186107, and U19 CA148065. The NHS2 was supported by NIH grants UM1 CA176726 and U19 CA148065. The ORIGO study was supported by the Dutch Cancer Society (RUL 1997-1505) and the Biobanking and Biomolecular Resources Research Infrastructure (BBMRI-NL CP16). The PBCS was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. Genotyping for PLCO was supported by the Intramural Research Program of the National Institutes of Health, NCI, Division of Cancer Epidemiology and Genetics. The PLCO is supported by the Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, National Institutes of Health. The POSH study is funded by Cancer Research UK (grants C1275/A11699, C1275/C22524, C1275/A19187, C1275/A15956 and Breast Cancer Campaign 2010PR62, 2013PR044. PROCAS is funded from NIHR grant PGfAR 0707-10031. The RBCS was funded by the Dutch Cancer Society (DDHK 2004-3124, DDHK 2009-4318). SEARCH is funded by Cancer Research UK [C490/A10124, C490/A16561] and supported by the UK National Institute for Health Research Biomedical Research Centre at the University of Cambridge. The University of Cambridge has received salary support for PDPP from the NHS in the East of England through the Clinical Academic Reserve. The Sister Study (SISTER) is supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01-ES044005 and Z01-ES049033). The Two Sister Study (2SISTER) was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01-ES044005 and Z01-ES102245), and, also by a grant from Susan G. Komen for the Cure, grant FAS0703856. SKKDKFZS is supported by the DKFZ. The SMC is funded by the Swedish Cancer Foundation and the Swedish Research Council [grant 2017-00644 for the Swedish Infrastructure for Medical Population-based Life-course Environmental Research (SIMPLER)]. The SZBCS is financially supported under the program of Minister of Science and Higher Education “Regional Initiative of Excellence” in years 2019-2022, Grant No 002/RID/2018/19. The TNBCC was supported by: a Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), a grant from the Breast Cancer Research Foundation, a generous gift from the David F. and Margaret T. Grohne Family Foundation. The UCIBCS component of this research was supported by the NIH [CA58860, CA92044] and the Lon V Smith Foundation [LVS39420]. The UKBGS is funded by Breast Cancer Now and the Institute of Cancer Research (ICR), London. ICR acknowledges NHS funding to the NIHR Biomedical Research Centre. The UKOPS study was funded by The Eve Appeal (The Oak Foundation) and supported by the National Institute for Health Research University College London Hospitals Biomedical Research Centre. The USRT Study was funded by Intramural Research Funds of the National Cancer Institute, Department of Health and Human Services, USA. CIMBA CIMBA: The CIMBA data management and data analysis were supported by Cancer Research – UK grants C12292/A20861, C12292/A11174. ACA is a Cancer Research -UK Senior Cancer Research Fellow. GCT and ABS are NHMRC Research Fellows. The PERSPECTIVE project was supported by the Government of Canada through Genome Canada and the Canadian Institutes of Health Research, the Ministry of Economy, Science and Innovation through Genome Québec, and The Quebec Breast Cancer Foundation. BCFR: UM1 CA164920 from the National Cancer Institute. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Breast Cancer Family Registry (BCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the BCFR. BFBOCC: Lithuania (BFBOCC-LT): Research Council of Lithuania grant SEN-18/2015 and Nr. P-MIP-19-164. BIDMC: Breast Cancer Research Foundation. BMBSA: Cancer Association of South Africa (PI Elizabeth J. van Rensburg). CNIO: Spanish Ministry of Health PI16/00440 supported by FEDER funds, the Spanish Ministry of Economy and Competitiveness (MINECO) SAF2014-57680-R and the Spanish Research Network on Rare diseases (CIBERER). COH-CCGCRN: Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under grant number R25CA112486, and RC4CA153828 (PI: J. Weitzel) from the National Cancer Institute and the Office of the Director, National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. CONSIT TEAM: Associazione Italiana Ricerca sul Cancro (AIRC; IG2014 no.15547) to P. Radice. Funds from Italian citizens who allocated the 5 × 1000 share of their tax payment in support of the Fondazione IRCCS Istituto Nazionale Tumori, according to Italian laws (INT-Institutional strategic projects ‘5 × 1000’) to S. Manoukian. UNIROMA1: Italian Association for Cancer Research (AIRC; grant no. 21389) to L. Ottini. DFKZ: German Cancer Research Center. EMBRACE: Cancer Research UK Grants C1287/A10118 and C1287/A11990. D. Gareth Evans and Fiona Lalloo are supported by an NIHR grant to the Biomedical Research Centre, Manchester (IS-BRC-1215-20007). The Investigators at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust are supported by an NIHR grant to the Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust. Ros Eeles and Elizabeth Bancroft are supported by Cancer Research UK Grant C5047/A8385. Ros Eeles is also supported by NIHR support to the Biomedical Research Centre at The Institute of Cancer Research and The Royal Marsden NHS Foundation Trust. FCCC: NIH/NCI grant P30-CA006927. The University of Kansas Cancer Center (P30 CA168524) and the Kansas Bioscience Authority Eminent Scholar Program. A.K.G. was funded by R0 1CA140323, R01 CA214545, and by the Chancellors Distinguished Chair in Biomedical Sciences Professorship. Ana Vega is supported by the Spanish Health Research Foundation, Instituto de Salud Carlos III (ISCIII), partially supported by FEDER funds through Research Activity Intensification Program (contract grant numbers: INT15/00070, INT16/00154, INT17/00133), and through Centro de Investigación Biomédica en Red de Enferemdades Raras CIBERER (ACCI 2016: ER17P1AC7112/2018); Autonomous Government of Galicia (Consolidation and structuring program: IN607B), and by the Fundación Mutua Madrileña (call 2018). GC-HBOC: German Cancer Aid (grant no 110837, Rita K. Schmutzler) and the European Regional Development Fund and Free State of Saxony, Germany (LIFE - Leipzig Research Centre for Civilization Diseases, project numbers 713-241202, 713-241202, 14505/2470, 14575/2470). GEMO: Ligue Nationale Contre le Cancer; the Association “Le cancer du sein, parlons-en!” Award, the Canadian Institutes of Health Research for the “CIHR Team in Familial Risks of Breast Cancer” program, the French National Institute of Cancer (INCa) (grants AOR 01 082, 2013-1-BCB-01-ICH-1 and SHS-E-SP 18-015) and the Fondation ARC pour la recherche sur le cancer (grant PJA 20151203365). GEORGETOWN: the Survey, Recruitment and Biospecimen Shared Resource at Georgetown University (NIH/NCI grant P30-CA051008) and the Fisher Center for Hereditary Cancer and Clinical Genomics Research. HCSC: Spanish Ministry of Health PI15/00059, PI16/01292, and CB-161200301 CIBERONC from ISCIII (Spain), partially supported by European Regional Development FEDER funds. HEBCS: Helsinki University Hospital Research Fund, Academy of Finland (266528), the Finnish Cancer Society and the Sigrid Juselius Foundation. HEBON: the Dutch Cancer Society grants NKI1998-1854, NKI2004-3088, NKI2007-3756, the Netherlands Organization of Scientific Research grant NWO 91109024, the Pink Ribbon grants 110005 and 2014-187.WO76, the BBMRI grant NWO 184.021.007/CP46 and the Transcan grant JTC 2012 Cancer 12-054. HUNBOCS: Hungarian Research Grants KTIA-OTKA CK-80745 and NKFI_OTKA K-112228. HVH (University Hospital Vall d’Hebron) This work was supported by Spanish Instituto de Salud Carlos III (ISCIII) funding, an initiative of the Spanish Ministry of Economy and Innovation partially supported by European Regional Development FEDER Funds: FIS PI12/02585 and PI15/00355. ICO: The authors would like to particularly acknowledge the support of the Asociación Española Contra el Cáncer (AECC), the Instituto de Salud Carlos III (organismo adscrito al Ministerio de Economía y Competitividad) and “Fondo Europeo de Desarrollo Regional (FEDER), una manera de hacer Europa” (PI10/01422, PI13/00285, PIE13/00022, PI15/00854, PI16/00563, P18/01029, and CIBERONC) and the Institut Català de la Salut and Autonomous Government of Catalonia (2009SGR290, 2014SGR338, 2017SGR449, and PERIS Project MedPerCan), and CERCA program. IHCC: PBZ_KBN_122/P05/2004. ILUH: Icelandic Association “Walking for Breast Cancer Research” and by the Landspitali University Hospital Research Fund. INHERIT: Canadian Institutes of Health Research for the “CIHR Team in Familial Risks of Breast Cancer” program – grant # CRN-87521 and the Ministry of Economic Development, Innovation and Export Trade – grant # PSR-SIIRI-701. IOVHBOCS: Ministero della Salute and “5 × 1000” Istituto Oncologico Veneto grant. IPOBCS: Liga Portuguesa Contra o Cancro. kConFab: The National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. MAYO: NIH grants CA116167, CA192393 and CA176785, an NCI Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA116201), and a grant from the Breast Cancer Research Foundation. MCGILL: Jewish General Hospital Weekend to End Breast Cancer, Quebec Ministry of Economic Development, Innovation and Export Trade. Marc Tischkowitz is supported by the funded by the European Union Seventh Framework Program (2007Y2013)/European Research Council (Grant No. 310018). MSKCC: the Breast Cancer Research Foundation, the Robert and Kate Niehaus Clinical Cancer Genetics Initiative, the Andrew Sabin Research Fund and a Cancer Center Support Grant/Core Grant (P30 CA008748). NCI: the Intramural Research Program of the US National Cancer Institute, NIH, and by support services contracts NO2-CP-11019-50, N02-CP-21013-63 and N02-CP-65504 with Westat, Inc, Rockville, MD. NNPIO: the Russian Foundation for Basic Research (grants 17-54-12007, 17-00-00171 and 18-515-45012). NRG Oncology: U10 CA180868, NRG SDMC grant U10 CA180822, NRG Administrative Office and the NRG Tissue Bank (CA 27469), the NRG Statistical and Data Center (CA 37517) and the Intramural Research Program, NCI. OSUCCG: was funded by the Ohio State University Comprehensive Cancer Center. PBCS: Italian Association of Cancer Research (AIRC) [IG 2013 N.14477] and Tuscany Institute for Tumors (ITT) grant 2014-2015-2016. SMC: the Israeli Cancer Association. SWE-BRCA: the Swedish Cancer Society. UCHICAGO: NCI Specialized Program of Research Excellence (SPORE) in Breast Cancer (CA125183), R01 CA142996, 1U01CA161032 and by the Ralph and Marion Falk Medical Research Trust, the Entertainment Industry Fund National Women’s Cancer Research Alliance and the Breast Cancer research Foundation. UCSF: UCSF Cancer Risk Program and Helen Diller Family Comprehensive Cancer Center. UKFOCR: Cancer Researc h UK. UPENN: National Institutes of Health (NIH) (R01-CA102776 and R01-CA083855; Breast Cancer Research Foundation; Susan G. Komen Foundation for the cure, Basser Research Center for BRCA. UPITT/MWH: Hackers for Hope Pittsburgh. VFCTG: Victorian Cancer Agency, Cancer Australia, National Breast Cancer Foundation. WCP: Dr Karlan is funded by the American Cancer Society Early Detection Professorship (SIOP-06-258-01-COUN) and the National Center for Advancing Translational Sciences (NCATS), Grant UL1TR000124.
Author information
Authors and Affiliations
Consortia
Contributions
G.F. and M.B. contributed equally to this work as first author. J.S. and P.P. contributed equally to this work as last author. Conceived the study: G.F., M.B., I.C., P.R., H.N., J.S., P.P. Wrote the manuscript: G.F., P.P. Contributed to manuscript writing: M.B., T.A.M., A.J.S., M.H.G., W.K.C., R.L.M., G.C-T., T.D., M.K.S., D.F.E., P.R., H.N. Conceived, designed or obtained financial support for the OncoArray: P.K., J.S., D.F.E., A.C. Data management: D.R., J.D., K.M., M.K.B., G.L., A.M.D. M.L., Q.W., L.M., M.T.P., P.D.P.P., A.C.A. Statistical analyses: G.F., J.I.K., H.N., P.P. Functional analyses: M.B., L.C., S.V.L., R.P., J.S. Provided DNA samples and/or phenotypic data: C.M.A., M.A.A., J.A., S.A., C.A.A., B.A.A., T.A., K.A., C.B.A., L.A., H.A-C., N.N.A., V.A., N.A., K.J.A., B.K.A., E.A., B.A., P.A., J.Az., J.B., R.B.B., D.B., J.B., L.E.B.F., C.J.B., M.W.B., S.B., J.Bar., R.B., M.B., A.M.B., C.B., N.V.B., A.B., S.E.B., B.B., A,Bor., A.F.B., H.B., H.Br., T.B., B.Bu., S.S.B., T.C., A.C., M.A.C., D.C., I.G.C., F.C., J.E.C., J.C-C., S.J.C., K.B.M.C., C.L.C., A.Co., T.A.C., D.G.C., C.C., K.C., M.B.D., M.dlH., P.D., O.D., Y.C.D., G.S.D., N.D., S.M.D., C.M.D., I.d-S-S., K.D., M.D., D.M.E., A.B.E., A.H.E., C.E., M.E., D.G.E., P.A.F., J.F., H.F., W.D.F., T.M.F., E.F., M.G., P.G., M.G-D., C.G., S.M.G., J.G., M.G-C., J.A.G-S., M.M.G., S.A.G., G.G.G., G.G., A.K.G., M.S.G., D.E.G., P.Gu., A.G-B., L.H., C.A.H., N.H., P.H., U.H., P.A.H., A.H., J.H., P.Hi., A.Ho., J.L.H., H.D.H-III, A.How., C.H., P.J.H., D.J.H., E.N.I., C.I., M.J., A.J., P.J., R.J., W.J., E.M.J., M.E.J., A.Ju., R.K., B.Y.K., E.K., C.M.K., I.K., S.K., P.K., D.L., C.L., L.LM., J.L., F.L., J.Li., J.T.L., K.H.L., R.N.L., J.Lu., A.M., M.M., S.M., S.Ma., J.W.M.M., T.M., D.M., N.M., A.Me., U.M., A.Mi., M.Mo., K.L.N., S.L.N., W.G.N., T.N-D., F.C.N., S.N., L.N-Z., K.O., E.O., O.I.O., A.F.O., J.E.O., H.O., A.O., L.O., B.P., A.P., J.P., D.P-K., T.P., N.P., M.A.P., K.P., B.R., J.R., M.U.R., R.R-M., G.R., H.S.R., V.R., A.R., M.A.R., E.A.R., M.R., V.Ru., M.Ru., E.S., K.S., M.S., M.T.S., R.K.S., M.Sc., C.S., L.S., M.Sh., P.S., X-O.S., J.S., C.F.S., C.So., P.So., M.C.S., J.J.S., L.St., D.S-L., W.J.T., M.R.T., M.B.T., M.T., J.T., D.L.T., M.Ti., R.A.E.M.T., D.T., M.A.T., T.T., N.T., M.U., C.M.V., E.J.vR., E.M.vV., A.V., A.Vi., B.W., J.N.W., C.W., G.W., A.W., X.R.Y., W.Z., A.Z., K.K.Z., A.M.D., M.L., Q.W., L.M., M.T.P. P.D.P.P., F.F., A.E.T., I.L.A., S.J.R., A.J.S., M.H.G., W.K.C., R.L.M., G.C-T., T.D., M.K.S. D.F.E., P.R., E.H., A.C.A., F.J.C., H.N., P.P. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
C.I. reports consulting with AstraZeneca and Pfizer and her institution receives research support for Tesaro, AstraZeneca, and Pfizer. B.Y.K. served on Invitae Corporation’s Advisory Board from 2017 to 2018. K.P. reports receiving fee, paid to his institution, for lectures and/or participation in advisory board of AstraZeneca and Pfizer and travel support from AstraZeneca and Pfizer. Jordi Surrallés’ institution received research support from Rocket Pharmaceuticals. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Figlioli, G., Bogliolo, M., Catucci, I. et al. The FANCM:p.Arg658* truncating variant is associated with risk of triple-negative breast cancer. npj Breast Cancer 5, 38 (2019). https://doi.org/10.1038/s41523-019-0127-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-019-0127-5
