
Abstract
Metaplastic breast cancer (MBC) is a rare special histologic type of triple-negative breast cancer, characterized by the presence of neoplastic cells showing differentiation towards squamous epithelium and/or mesenchymal elements. Here we sought to define whether histologically distinct subgroups of MBCs would be underpinned by distinct genomic and/or transcriptomic alterations. Microarray-based copy number profiling identified limited but significant differences between the distinct MBC subtypes studied here, despite the limited sample size (n = 17). In particular, we found that, compared to MBCs with chondroid or squamous cell metaplasia, MBCs with spindle cell differentiation less frequently harbored gain of 7q11.22-23 encompassing CLDN3 and CLDN4, consistent with their lower expression of claudins and their association with the claudin-low molecular classification. Microarray-based and RNA-sequencing-based gene expression profiling revealed that MBCs with spindle cell differentiation differ from MBCs with chondroid or squamous cell metaplasia on the expression of epithelial-to-mesenchymal transition-related genes, including down-regulation of CDH1 and EPCAM. In addition, RNA-sequencing revealed that the histologic patterns observed in MBCs are unlikely to be underpinned by a highly recurrent expressed fusion gene or a pathognomonic expressed mutation in cancer genes. Loss of PTEN expression or mutations affecting PIK3CA or TSC2 observed in 8/17 MBCs support the contention that PI3K pathway activation plays a role in the development of MBCs. Our data demonstrate that despite harboring largely similar patterns of gene copy number alterations, MBCs with spindle cell, chondroid and squamous differentiation are distinct at the transcriptomic level but are unlikely to be defined by specific pathognomonic genetic alterations.
Similar content being viewed by others

Introduction
Metaplastic breast carcinoma (MBC) is a rare special histologic type of breast cancer, accounting for 0.2–5% of invasive breast cancers.1,2 MBCs comprise a heterogeneous group of tumors characterized by the presence of malignant epithelial cells showing differentiation towards squamous epithelium and/or mesenchymal elements, such as spindle, chondroid, osseous or rhabdoid cells.1,2 Most MBCs display a triple-negative (TN) phenotype (i.e., lack of expression of estrogen receptor (ER), progesterone receptor (PR), and HER2), and are classified as basal-like or claudin-low molecular subtypes.3,4,5 Akin to the basal-like subtype, the claudin-low subtype is enriched for triple-negative breast cancers (TNBCs), but is further characterized by a gene expression profile associated with epithelial-to-mesenchymal transition (EMT) and cancer stem cells. MBCs have an aggressive clinical behavior, and unlike other forms of TNBCs, these tumors seem not to respond to conventional chemotherapy regimens.6
Despite their distinctive histologic and clinical features, relatively few molecular alterations that discriminate MBCs from histologic grade- and ER-matched invasive ductal carcinomas of no special type (IDC-NSTs) have been found. Overall, MBCs and histologic grade-matched and ER-matched IDC-NSTs display similar transcriptomic and copy number profiles.4,7 Akin to TN IDC-NSTs, MBCs also frequently harbor TP53 somatic mutations,8,9 loss of CDKN2A, and EGFR overexpression and amplification.10 Compared to TN IDC-NSTs, MBCs frequently display an EMT-like gene signature, display downregulation of DNA damage response pathways,4 and harbor frequent genetic alterations affecting the PI3K and Wnt pathways.3,9,11 Importantly, however, different histologic subtypes of or components within MBCs are associated with specific transcriptomic subtypes,5 and may be underpinned by distinct copy number alterations (CNAs)8 and mutational profiles.9 These data provided evidence to suggest that stratification of MBCs according to their histologic subtypes may be required to identify molecular alterations and targets specific for this disease.
There is burgeoning evidence to suggest that some special histologic subtypes of TNBC may be driven by recurrent fusion genes. For example, secretory carcinomas are characterized by the ETV6-NTRK3 fusion gene,12 whereas adenoid cystic carcinomas are underpinned the MYB-NFIB fusion gene13 or rearrangements of the MYBL1 gene.14 Genomic analyses investigating the presence of fusion genes in MBCs have yet to be described.
Given that MBCs of different histologic subtypes are likely to have distinct molecular features, here we sought to define whether histologically distinct subgroups of MBCs would be underpinned by distinct copy number or gene expression profiles. Furthermore, we sought to determine whether MBCs, akin to other special histologic types of TNBC, would be underpinned by a recurrent fusion gene. To this end, MBCs of distinct histologic subtypes (spindle cell, squamous, and chondroid) were subjected to array-based gene expression, gene copy number profiling, and RNA-sequencing.
Results
Histopathologic and immunophenotypic characterization of MBCs
Of the 17 MBCs included in this study (Supplementary Fig. 1), seven (41%), five (29.5%) and five (29.5%) cases were classified as chondroid, squamous or spindle cell subtypes, respectively, and the vast majority were of high histologic grade (15/17, 88% grade 3; 2/17, 12% grade 2). Immunophenotypic assessment revealed that all MBCs, irrespective of histologic subtype, were of TN phenotype and expressed at least one basal marker, either high molecular weight cytokeratins (CKs), including CK14, CK17, CK5/6, or EGFR10 (Supplementary Tables 1 and 2). Additionally, seven (41%) MBCs showed strong overexpression of p53, a surrogate marker for the presence of TP53 missense mutations,15 and four cases lacked PTEN expression by immunohistochemistry (Fig. 1 and Supplementary Table 1).

Immunohistochemical analysis of p53 and PTEN in MBCs. Representative micrographs of metaplastic breast carcinomas (MBCs), a with spindle cell (META32) and b-d with squamous metaplasia (META41, META37, and META42, respectively). Immunohistochemical analysis showed loss of PTEN expression in e META32 and f META41 and p53 overexpression in g META37 and h META42. In e and f, note the positive internal control for PTEN in blood vessels. Scale bars, 100 µm
Histologic subtypes of MBCs harbor similar patterns of gene copy number alterations
Compared to a set of TN IDC-NSTs16 matched in a 2:1 ratio with MBCs, both MBCs and TN IDC-NSTs exhibited similar patterns of gene CNAs, with frequent gains of 1q and 8q, and losses of 5q and 12q (Supplementary Fig. 2 and Supplementary Table 3). In the MBCs analyzed, homozygous deletions of PTEN (10q23) were found in two cases (META32 and META41; Supplementary Table 4), and were associated with loss of PTEN protein expression as assessed by immunohistochemistry (Fig. 1). Additional two cases harbored homozygous deletions of the CDKN2A/CDKN2B and MTAP gene loci (9p21, META42, and META49; Supplementary Table 4). Recurrent focal amplification of ZNF703/FGFR1/BRF2 (8p11.23-11.22) was found in two cases (META31 and META39). Additional recurrent focal amplifications included those affecting WNT9A/WNT3A (1q42.12-42.2, META41, and META52) and TNIK (3q26.2-26.31, META39 and META42), a Wnt target gene activator.17
Given that MBCs of distinct histologic subtypes have been reported to display different gene expression profiles5 and mutational repertoires9, we performed an exploratory analysis to define whether the histologic diversity would be underpinned by distinct patterns of gene CNAs. Hierarchical clustering of the categorical CNA states revealed that the three MBC histologic subtypes did not form distinct clusters (Fig. 2a). We observed, however, two stable clusters, one composed predominantly of MBCs with squamous differentiation (cluster 2, consisting of META37, META40, META53, and META62), and the other composed mainly of MBCs with mesenchymal differentiation (chondroid or spindle cell differentiation; cluster 1, consisting of all other samples), with a significant enrichment of MBCs with squamous differentiation in cluster 2 (p-value = 0.004, Fisher’s exact test). The clusters differed predominantly by gains of 1p and losses of chromosome 2 and 9q (Fig. 2b, Supplementary Table 5). Indeed, direct comparisons between the distinct histologic MBC subtypes revealed that MBCs with squamous differentiation had higher fractions of the genome altered compared to non-squamous MBCs (p-value = 0.02, Mann–Whitney U test, Supplementary Fig. 3), but few CNAs were significantly different between them. MBCs with chondroid differentiation were characterized by 21q11.2 gains and high level gain/ amplification of 8q21.11-24.3 compared to non-chondroid MBCs (both p-value < 0.05, Fisher’s exact tests, Supplementary Fig. 3a, Supplementary Table 6). MBCs with spindle cells harbored similar frequencies of gene CNAs as MBCs with squamous or chondroid differentiation, apart from the less frequently observed 7q11.22-23 gain encompassing CLDN3 and CLDN4 (p-value < 0.05, Fisher’s exact test, Supplementary Fig. 3b, Supplementary Table 7 ). Compared to MBCs with spindle cell or chondroid differentiation, MBCs with squamous differentiation infrequently harbored amplification/ high-level gain of 8q24, but harbored more frequent losses on 7q and 12q and gains on 11p and 12q (all p-value < 0.05, Fisher’s exact tests, Supplementary Fig. 3c–d, Supplementary Table 8). Furthermore, we noticed that two cases harbored very few or no CNAs (META36 and META47). The tumor cell content for META36, a chondroid MBC, was estimated to be 87 and 80% by ABSOLUTE and pathology review, respectively, whereas META47, a spindle cell MBC, did not harbor any CNA and its tumor cell content was estimated to be 70% by pathology review but could not be estimated by ABSOLUTE (Supplementary Table 1). It should be noted, however, that META47 was classified as claudin-low, as opposed to normal breast-like, based on intrinsic subtyping.5 These analyses support the contention that the lack of CNAs was not a result of insufficient tumor cell content. In fact, gene copy number analysis of the samples of the METABRIC study has revealed the existence of a subset of TNBCs lacking CNAs despite the adequate tumor cell content.18

Landscape of gene copy number alterations in MBCs. a Hierarchical cluster analysis performed with SNP 6.0 defined copy number alterations (i.e., gains, losses, amplifications/high-level gains and deletions) using Euclidean distance metric and Ward’s algorithm. Histologic subtypes of MBCs are color-coded according to the legend. b Frequency plot of (top) copy number gains and losses and (bottom) amplifications/high-level gains and deletions in copy number cluster 1 and copy number cluster 2 identified by hierarchical clustering. The proportion of tumors in which each probe is gained/ amplified (green bars) or lost/ deleted (red bars) is plotted (y-axis) for each probe according to its genomic position (x-axis). Inverse Log10 values of Fisher’s exact p value are plotted according to genomic position (x-axis) at the bottom of each graph
Taken together, MBCs seem not to be underpinned by a pathognomonic CNA. In fact, these tumors display CNAs similar to those observed in conventional forms of high-grade TNBCs, and, regardless of the histologic subtype, MBCs harbor similar CNA profiles. Significant differences in the CNAs according to subtype were identified, however these are unlikely to account for the histologic diversity seen in these cancers. Further studies, with a larger sample size, are however required to refine the boundaries of the regions differentially gained or lost in the histologically distinct subtypes of MBC.
MBCs with spindle cell differentiation show distinct transcriptomic profiles
Consistent with our previous observations,5 using the PAM50 and claudin-low intrinsic gene lists,19,20,21,22 here we observed that the five MBCs with spindle cell morphology were classified as of claudin-low intrinsic subtype, compared to only three of the 12 MBCs with squamous or chondroid metaplasia (Fig. 3a and Supplementary Tables 1 and 2). By contrast, MBCs with chondroid and squamous differentiation were variably classified as of normal-like, basal-like or claudin-low intrinsic subtypes. Given the heterogeneity observed in the intrinsic subtypes between MBCs of different morphologic subtypes,2 we further explored the overall gene expression profiles of these tumors using unsupervised and supervised methods.

MBCs of spindle subtype have a unique transcriptomic profile amongst MBCs. a Unsupervised hierarchical clustering of MBCs based on gene expression arrays using Pearson’s correlation and Ward’s algorithm. Histologic subtypes of MBCs are color-coded according to the legend. Zoomed-in heatmap on the right shows genes differentially expressed between MBCs with spindle cells as compared to chondroid and squamous MBCs, including tight-junction related genes, such as CLDN3, and MYH11, and EMT-related genes, such as CDH1 and EPCAM. b Validation of significantly differentially expressed genes between spindle and non-spindle MBCs identified by microarray-based gene expression analysis was performed using RNA-sequencing data. For each gene, normalized expression value is plotted for each MBC, grouped by histologic subtype. p-values were calculated from the differential expression analysis using limma46 (see Methods)
Unsupervised hierarchical clustering of gene expression data revealed that the five MBCs with spindle cells formed a stable cluster distinct from the remaining MBCs (p < 0.05, Fisher’s exact test, Fig. 3a and Supplementary Fig. 4). The second cluster comprised two sub-clusters, with one encompassing most MBCs with chondroid differentiation and the other comprising most MBCs with squamous differentiation (Fig. 3a). These two sub-clusters were, however, unstable, and the separation between the two histologic subtypes was not statistically significant (p = 0.2424, Fisher’s exact test, Supplementary Fig. 4). Consistent with our findings that spindle cell MBCs are more frequently of claudin-low subtype5, these data provide evidence that MBCs with spindle cell metaplasia harbor distinct transcriptomic profiles compared to other forms of MBCs.
Significance analysis of microarrays (SAM) revealed that 190 transcripts were downregulated in spindle cell compared to non-spindle cell MBCs (Supplementary Table 9). Functional annotation and pathway analysis of these transcripts using the Ingenuity Pathway Analysis (IPA) software revealed that MBCs with spindle cells displayed lower expression of tight-junction-related genes, including CLDN3, CLDN8, CGN, MYH11, and MYH14 (p-value < 0.05, Supplementary Fig. 5) than other forms of MBCs. Additionally, spindle cell MBCs differentially expressed EMT-related genes compared to squamous and chondroid MBCs, including reduced CDH1 and EPCAM expression (Supplementary Table 9). The down-regulation of EMT-related genes in spindle cell MBCs, including claudins, is consistent with their claudin-low intrinsic subtype. Consistent with the enrichment of tight-junction and EMT-related genes found by IPA, g:Profiler23 pathway analysis of the differentially expressed genes revealed an enrichment for genes involved in cell–cell junction, tight junction and cell junction organization (p-value < 0.05) and genes enriched in extracellular matrix organization and cell adhesion (p-value < 0.05, Supplementary Table 10).
The comparison between MBCs with and without chondroid differentiation identified 48 differentially expressed transcripts (Supplementary Table 11). Interestingly, chondroid tumors showed over-expression of EXTL1, a gene encoding exostosin-like glycosyltransferase 1 and involved in chain elongation of heparan sulfate,24 an integral proteoglycan of cartilage, and genes involved in chondrocyte differentiation, including COL9A1, COL11A2, and ACAN. Aggrecan (encoded by ACAN) is a proteoglycan and major extracellular matrix component of cartilaginous tissues.25 One could posit that overexpression of these genes may contribute to their cartilaginous histologic appearance; however, further studies are warranted to define the basis of their overexpression in MBCs with chondroid differentiation.
Only 19 transcripts were found to be differentially expressed between MBCs with and without squamous differentiation, including the upregulation of MAPK13, a gene involved in cell cycle, as well as two genes that regulate proliferation, EHF and MMP7 (Supplementary Table 12).
To validate our gene expression results using an orthogonal method, we performed hierarchical clustering and differential gene expression analysis using the data generated from RNA-sequencing. Transcriptomic profiling by RNA-sequencing generally recapitulated the unsupervised clustering analysis made using gene expression arrays with the MBCs with spindle cells preferentially clustering together (Supplementary Fig. 6). Supervised analysis of gene expression based on the RNA-sequencing data further confirmed the gene expression microarray analysis findings in that CLDN3 and CDH1 were downregulated in MBCs with spindle cells; MAPK13, EHF, and MMP7 were upregulated in squamous MBCs; and ACAN, COL9A1, COL11A2, and EXTL1 were upregulated in chondroid MBCs (Fig. 3b, Supplementary Fig. 7 and Supplementary Tables 13–15). These findings confirm our previous observations,5 and demonstrate that the gene expression profiles of MBCs vary according to the type of metaplastic elements present.
Genes amplified and overexpressed in MBCs
Integration of copy number and gene expression data for all 17 MBCs included in this study identified 3071 copy number regulated probes, encoding 2605 genes (Supplementary Table 16). To identify potential amplicon drivers, we interrogated for genes significantly overexpressed when amplified. Pathway analysis of the 153 genes overexpressed when amplified (Supplementary Table 17) using IPA and g:Profiler demonstrated an enrichment of genes involved in oxidative phosphorylation, cellular metabolic pathways and cell cycle (Supplementary Fig. 8 and Supplementary Table 18). Interestingly, several of these genes were present on 8q, a recurrently amplified region in MBCs and TNBCs, including COX6C, YWHAZ, and ATP6V1C1 on 8q22 (Fig. 4a) and RAD21, NDUFB9, and MYC on 8q24 (Fig. 4b). As an exploratory, hypothesis-generating analysis, we sought to define whether the repertoire of genes overexpressed when amplified would vary according to the histologic subtype of MBCs. This analysis did not reveal genes significantly overexpressed when amplified when the three subtypes were analyzed separately; it should be noted, however, that this negative finding may stem from the small sample size.

Genes significantly overexpressed when amplified in MBCs. Composite heatmaps of copy number (left) and gene expression (middle) of genes mapping to a the 8q22 amplicon and b the 8q24.12-24.2 amplicon. Microarray-based gene expression and SNP 6.0 copy number values are depicted in two matching heatmaps, with SNP 6.0 states on the left and expression values on the right, in which the genes are ordered according to their chromosomal positions. Bar plots on the right show the results of Mann–Whitney U tests for expression as a continuous variable and gene amplification as the grouping variable. Bars in red show adjusted p-values < 0.05. SNP 6.0: copy number loss (green), no copy number change (black), copy number gain (dark red), amplification (bright red). Gene expression: downregulation (green), upregulation (red). AMP amplified, MWU Mann–Whitney U test, NA not amplified
MBCs are not driven by a recurrent fusion gene or an expressed pathognomonic mutation in cancer genes
To determine whether MBCs are driven by a highly recurrent fusion gene, we subjected the 17 MBCs to paired-end RNA-sequencing. Analysis using defuse26 and ChimeraScan27 identified 43 fusion transcripts (Supplementary Table 19). Akin to the gain-of-function oncogenic fusion genes identified in other special histologic types of breast cancer,12,13 we prioritized fusion transcripts that comprised open reading frames with known associated functions, and that harbored intact functional domains. Using the above criteria, we identified and validated by RT-PCR nine in-frame fusion transcripts in the index cases (Figs. 5a–i and Supplementary Table 19), though none was recurrent in our series. Importantly, however, we identified several interesting chimeric transcripts including a promoter swap of TBL1XR1-PIK3CA that has previously been described in breast and prostate cancers,28 and additional fusions of WAPAL-CDHR1, MAP2K3-HMGCLL1, PARG-BMS1, FN1-ICAM1, AAK1-ARNT2, TNKS1BP1-SPARC, MBTPS1-TCEANC2, and PSMA6-SHMT1 (Figs. 5a–i and Supplementary Fig. 9).

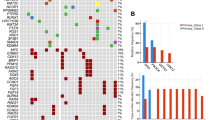
Schematic representation of nine validated expressed in-frame fusion transcripts and repertoire of expressed non-synonymous mutations defined using RNA-sequencing in MBCs. Reverse transcription (RT)-PCR was used to validate in-frame fusion transcripts identified by RNA-sequencing to comprise open reading frames with known associated functions and that harbored intact functional domains. a TBL1XR1-PIK3CA, b FN1-ICAM1, c MAP2K3-HMGCLL1, d MBTPS1-TCEANC2, e PARG-BMS1, f PSMA6-SHMT1, g TNKS1BP1- SPARC, h AAK1-ARNT2, i WAPAL-CDHR1. In each panel, chromosomes are indicated in alternating black and gray boxes, with the 5′ and the 3′ marked at the corresponding end of each gene. Light blue boxes above the schematics of the wild-type genes and below the schematics of the fusion genes indicate the protein domains present. Fusion junctions with respective exon numbers are shown. j Expressed non-synonymous mutations identified in 17 MBCs subjected to RNA-sequencing. Mutations affecting genes included in the cancer gene lists29,30,31 are reported. The effects of the mutations are color-coded according to the color key, with hotspot48 mutations colored in red. The presence of multiple non-synonymous mutations in the same gene is represented by an asterisk. The metaplastic subtype of each MBC is indicated below the heatmap according to the color key
To define the repertoire of expressed mutations in MBCs, we performed a mutational analysis of the RNA-sequencing data. Given the challenges for de novo identification of mutations based on RNA-sequencing analysis, we rigorously curated the putative mutations to exclude likely germline polymorphisms and likely passenger mutations and focused on previously described cancer genes included in Kandoth et al. (127 significantly mutated genes),29 the Cancer Gene Census30 or Lawrence et al. (Cancer5000-S gene set).31 The mutational analysis revealed that TP53 (9/17, 53%), PIK3CA, DDX5, and TSC2 (each 2/17, 12%, Fig. 5j and Supplementary Table 20) were recurrently mutated, including TP53 hotspot mutations C176F, N239S, R249G, R273C, C275R, P278L, and c.673-1G>T splice site, each found in one MBC. Both PIK3CA mutations found were the hotspot H1047R mutation. Mutations in other cancer genes including CBFB, IDH1, JAK1, KMT2A, SMAD4, and SMARCA4 were found in one MBC each. As a hypothesis-generating analysis, we compared the mutational frequency of TP53 between MBC subtypes. All MBCs with squamous metaplasia harbored TP53 mutation, compared to 33% of MBCs of other subtypes (p-value = 0.03, Fisher’s exact test, Fig. 5j), similar to our recent report based on whole-exome sequencing.9
These data suggest that although MBCs are not driven by a pathognomonic gene fusion or a pathognomonic expressed mutation in previously described cancer genes29,30,31, some of these genetic alterations may play a role in individual MBCs.
Discussion
Here we have characterized the patterns of CNAs, gene expression, expressed fusion genes and expressed mutations affecting previously described cancer genes in 17 MBCs. Our results demonstrate that MBCs are highly heterogeneous tumors at the genomic and transcriptomic levels, and that they are unlikely to be underpinned by a highly recurrent fusion gene or a pathognomonic expressed mutation in previously described cancer genes29,30,31 (Table 1).
We have previously demonstrated that based on PAM50/claudin-low molecular subtyping, MBCs with spindle metaplasia are preferentially classified as of claudin-low subtype, whereas MBCs with squamous or chondroid metaplasia are more frequently classified as of basal-like or even normal breast-like subtypes than as of claudin-low subtype (Table 1).5 Here we expand on our previous observations, and demonstrate that MBCs with spindle cell metaplasia display transcriptomic features characterized by an EMT-like gene expression profile, which is not as conspicuous in MBCs with other forms of metaplasia. MBCs with chondroid or squamous metaplasia, but not MBCs with spindle metaplasia, were found to harbor recurrent copy number gains of 7q11.22-23, a region that encompasses CLDN3 and CLDN4. Given that the lack of 7q11.22-23 gain was the only CNA feature found to distinguish MBCs with spindle cells and the remaining MBCs, the vast majority of the differences in gene expression between these subsets of MBCs are therefore unlikely to be copy number-driven. Although MBCs with spindle cell metaplasia more frequently displayed PIK3CA mutations than MBCs with other types of metaplasia, PIK3CA mutations were also found in MBCs with squamous differentiation.9 These results suggest that the differences in gene expression between MBCs with or without spindle cell metaplasia may stem from other types of somatic genetic alterations or epigenetic modifications or distinct cells of origin. Our findings also contextualize specific aspects related to the molecular classification of MBCs; the characteristic EMT-like transcriptomic profiles and high prevalence of claudin-low subtype reported in studies on MBCs3,21,22 may actually stem from the fact that those studies primarily focused on MBCs that displayed a spindle cell/sarcomatoid histologic subtype.
In terms of CNAs, MBCs as a group have been shown to be remarkably similar to other TNBCs (Table 1).7 When we compared the copy number profiles of the 17 MBCs in the current cohort to those of 34 grade-matched TNBCs,16 the frequency of CNAs was similar between MBCs and TNBCs. We did identify, however, some regions that were altered significantly more frequently in MBCs, including losses of 6q13-14.1, 6q21, 10p13-15.3, 10q11.21-21.1, and 12p13.31 (Supplementary Fig. 2 and Supplementary Table 3). Furthermore, integration of genomic and transcriptomic information yielded the amplification of 8q as a CNA that significantly affects gene expression in MBCs. Amplification of 8q, however, is a relatively frequent finding in other forms of TNBCs.
At variance with other rare forms of TNBC,12,13,14,32 MBCs were found not to harbor a highly recurrent expressed fusion gene or a highly recurrent expressed mutation affecting previously described cancer genes. In addition, the histologic differences observed in distinct subtypes of MBCs appear not to be underpinned by specific expressed fusion genes or expressed mutations affecting previously described cancer genes. Potentially pathogenic fusion genes private to individual tumors were, however, identified, some of which comprised partner genes involved in the PI3K pathway and/or DNA damage repair mechanisms. Specifically, the TBL1XR1-PIK3CA fusion gene, which was detected in two ER-negative invasive breast cancers from the TCGA breast cancer study and may result in PIK3CA over-expression,28 was detected in META52; however, in this case, the PIK3CA expression levels were not significantly higher than in other MBCs studied here (Supplementary Figs. 10a and 10b). The fusion genes PARG-BMS1 and WAPAL-CDHR1 contain partners whose protein products are involved in genomic integrity; disruption of these genes by a rearrangement could, theoretically, contribute to the complex patterns of genomic rearrangements observed in MBCs. The WAPAL-CDHR1 fusion transcript contains the first 16 exons of the Wings apart-like protein homolog (WAPAL) gene and the last seven exons of the Cadherin Related Family Member 1 (CDHR1) gene. WAPAL encodes a cohesin binding protein necessary for sister chromatid resolution which is truncated at residue 1107 in the chimeric transcript. Double mutations in WAPAL at M1116A/I1120A have been found to result in a functionally defective protein that can still bind cohesin.33 These data suggest that this chimeric transcript may potentially result in a defective form of the WAPAL gene product that may affect chromatid cohesion and subsequently the rate of segregation errors and aneuploidy. Of the other fusion genes identified and validated in our study, PARG-BMS1 and TNKS1BP1-SPARC may also have an impact on genomic integrity, given that the Poly(ADP-ribose) glycohydrolase (PARG) protein hydrolyzes PAR (poly(ADP-ribose)) and plays an important role in DNA damage repair,34 whereas the Tankyrase 1 Binding Protein 1 (TNKS1BP1) protein is a binding partner of TANK1,35 a member of the PARP superfamily that regulates telomere length, DNA repair and WNT signaling.36
Here, we found that 53% of MBCs expressed TP53 mutations. Apart from frequent TP53 mutations, we have recently demonstrated by whole-exome sequencing analysis that PIK3CA, PIK3R1, and PTEN are significantly mutated in MBCs and that up to 60% of MBCs harbor somatic mutations in the PI3K/AKT/mTOR pathway (Table 1).9 These observations are in agreement with a previous study, in which sequencing analyses of known pathogenic PIK3CA and PTEN mutations revealed their involvement in 53% of MBCs.3 Here we have also detected homozygous deletions of PTEN in two MBCs (META32 and META41) and loss of expression by immunohistochemistry in additional two cases (META49 and META62), suggesting that 24% of MBCs likely displayed PTEN loss-of-function. In an additional four cases (META31, META47, META59, and META64), we identified either bona fide activating PIK3CA mutation or TSC2 missense mutations predicted to be likely pathogenic by mutation function predictors.37 These findings are consistent with the notion that dysregulation of the PI3K/AKT pathway is likely to play a pivotal role in MBCs. Of note, however, PIK3CA and PTEN mutations in TNBCs are not restricted to MBCs. In fact, of the TNBCs from The Cancer Genome Atlas breast cancer study, approximately 10% of cases harbor amplification or somatic mutations in PIK3CA and 8% harbor homozygous deletions or somatic mutations in PTEN.38 Therefore, these recurrent alterations affecting PI3K pathway-related genes are unlikely to explain the unique histologic features of MBCs. We also reported that MBCs frequently harbored mutations in the Wnt pathway.9 Consistent with our observations from whole-exome sequencing analysis of MBCs,9 we did not identify expressed mutations affecting CTNNB1. We did, however, detect an expressed frameshift SMAD4 mutation and an expressed missense TCF7L2 mutation. Of note, most of the Wnt pathway-related genes previously found to be mutated in MBCs are not in the cancer gene lists employed for the RNA-sequencing-based mutation analysis29,30,31 and were not within the scope of the mutational analysis performed in this study, given the challenges in identifying somatic mutations using RNA-sequencing alone.
This study has several limitations. Owing to the rarity of MBCs and the challenges in having frozen blocks representative of the metaplastic elements of MBC, our sample size was small. It should be noted, however, that despite the small sample size, we could detect transcriptomic features and CNAs that varied between the subtypes of MBC. Importantly, our study was sufficiently powered to have detected a pathognomonic fusion gene, a pathognomonic expressed mutation affecting a previously reported cancer gene, or a pathognomonic CNA in at least two cases. This suggests that, unlike other rare forms of TNBC,12,13,14,32 this rare and aggressive histologic special type of TNBCs is unlikely to be driven by a highly recurrent fusion gene, expressed mutation in a known cancer gene or CNA. Second, we have focused on the repertoire of CNAs, transcriptomic changes, fusion genes and expressed mutations in MBCs, and were unable to identify pathognomonic genetic alterations that define each histologic subtype of MBCs. Although mutations were identified using RNA-sequencing, defining the repertoire of somatic mutations using RNA-sequencing data has proven to be fraught with difficulties, with high rates of false-negative and false-positive results. Through a series of rigorous filters, we were able to identify recurrently expressed mutations in MBCs, which were consistent with the observations we have made by whole-exome sequencing of 35 MBCs.9 Further studies are warranted to define whether the distinctive features of MBCs and of each subtype of the disease would be driven by somatic genetic alterations affecting non-coding regions of the genome or by specific repertoires of epigenetic alterations.
Despite these limitations, our study demonstrates that MBCs resemble other high-grade TN IDC-NSTs at the genomic level. We have provided direct evidence that MBCs are unlikely to be driven by a highly-recurrent/ pathognomonic expressed fusion gene or expressed mutation affecting known cancer genes, and that transcriptomic profiles and, to a lesser extent, gene CNAs varied between the metaplastic elements present in MBCs, corroborating the notion that MBCs comprise a heterogeneous group of breast cancers whose common denominator is the presence of metaplastic elements. Further studies are warranted to define whether the histologic diversity of MBCs stems from distinct non-coding somatic genetic alterations and/or epigenetic events, distinct cells of origin, or the interactions between specific genetic/ epigenetic alterations and cell of origin.
Methods
Case selection and nucleic acids extraction
Cases diagnosed as MBCs were selected from a previously described cohort.5 In this study we included cases for which both gene expression microarray profiling and SNP 6.0 copy number profiling were available from our previous study,5 and for which sufficient RNA was available for RNA-sequencing (see below, Supplementary Fig. 1 and Supplementary Table 1). All cases were independently reviewed by three pathologists with an interest and expertize in breast cancer (FCG, AV-S and JSR-F), who classified the tumors into three groups: MBCs with spindle cell metaplasia (n = 5), squamous metaplasia (n = 5) or chondroid metaplasia (n = 7) according to the most prevalent component in the sample subjected to molecular analyses (Supplementary Table 1), given that the most abundant metaplastic component has been shown to have a substantial impact on the transcriptomic profiles of each tumor.5 Tumors were graded according to the Nottingham grading system39 and tumor cell content for each lesion was defined semi-quantitatively (AV-S and JSR-F). Samples were anonymized prior to analysis. This study was approved by the local research ethics committees of the authors’ institutions. Written consent was obtained from patients alive and contactable prior to the start of the study. Methods were performed in accordance with relevant regulations and guidelines. Nucleic acid extraction and quality control were performed as previously described.5
Immunohistochemistry
Immunohistochemical analysis was performed for ER, PR, and HER2, CK5/6, CK14, CK17, PTEN, p53, p63, c-KIT, and EGFR on representative sections from formalin-fixed paraffin-embedded (FFPE) tissue blocks (Supplementary Table 21 and Supplementary Methods).
Gene copy number analysis
Copy number profiling data using the human SNP Array 6.0 (Affymetrix)5 were re-analyzed (Supplementary Methods). Focal amplifications were defined as amplifications/high-level gains that were smaller than 25% of the respective chromosome arm and visually inspected using genome plots. Homozygous deletions were further defined using ASCAT40 and ABSOLUTE41 (Supplementary Methods). Tumor cell content was estimated using ABSOLUTE.41 Statistically significant differences between CNAs of different morphologic subtypes of MBCs were defined using multi-Fisher’s exact tests and unsupervised hierarchical cluster analysis was performed as previously described (Supplementary Methods).7 For the comparison of gene copy number profiles of MBCs with those of common-type TNBCs, SNP 6.0 array data were obtained from triple-negative IDC-NSTs16 and matched in a 2:1 ratio with MBCs and processed using the aroma.affymetrix and DNAcopy packages as described above.
Gene expression profiling
Gene expression profiling data using the HumanHT-12 v4 Expression BeadChip Kit (Illumina)5 were re-normalized and annotated (Supplementary Methods). Unsupervised hierarchical clustering analysis was performed as previously described42 (Supplementary Methods). Differentially expressed genes were determined by significance analysis of microarrays (SAM),43 adopting a false discovery rate of 1% after 100 permutations.
Integration of copy number and expression data
To identify genes whose expression levels correlate with CNAs and genes that were up-regulated when gained, down-regulated when lost or over-expressed when amplified, Pearson’s correlations and Mann–Whitney U tests were used as previously described7 (Supplementary Methods).
Pathway analysis
Significantly regulated pathways and networks in the gene expression data were determined using Ingenuity Pathway Analysis (IPA, http://www.ingenuity.com,) and g:Profiler23 (Supplementary Methods).
RNA-sequencing and fusion transcript identification
RNA-sequencing (2 × 54 bp) was performed for all 17 MBCs included in this study using the standard Illumina mRNA library protocol on a Genome Analyzer II (Illumina) as previously described.42 deFuse26 and ChimeraScan27 were used to identify mate-pairs supporting novel chimeric transcripts as previously described42 (Supplementary Methods). Candidates that resulted in open reading frames were annotated using OncoFuse.44 Nominated in-frame fusion gene candidates, candidates identified by both deFuse and ChimeraScan, as well as those with known associated functions, and those that harbored intact functional domains were prioritized for validation in the index cases by reverse transcription (RT)-PCR (Supplementary Table 22 and Supplementary Methods). Fusion genes validated in the index cases were further screened in all cases in the cohort for which RNA samples were available.
RNA-sequencing gene expression and mutation analysis
For gene expression analysis, RNA-sequencing data were aligned to the transcriptome (GRCh37) using STAR45 and differential expression analyses were performed using limma (Supplementary Methods).46 Mutation analysis was performed according to the Genome Analysis Toolkit47 Best Practices workflow for single nucleotide variant (SNVs) and small insertion and deletion (indel) calling on RNA-seq data (http://gatkforums.broadinstitute.org/gatk/discussion/3891/calling-variants-in-rnaseq). Variants affecting hotspots48 were white-listed, and additional mutations were defined using GATK HaplotypeCaller, removing likely polymorphisms and likely passenger variants (Supplementary Methods). Only variants covered by at least five reads, with at least two reads supporting the variant and at least two reads supporting the reference allele were included, as those devoid of a reference allele were highly enriched for germline variations.42 Mutations affecting cancer genes included in the lists described by Kandoth et al., (127 significantly mutated genes),29 the Cancer Gene Census30 and/or Lawrence et al., (Cancer5000-S gene set)31 and their functional effects (Supplementary Methods) were reported.
Power calculations
The binomial probability was used to estimate the statistical power of identifying a pathognomonic alteration, focusing on those that would be detected in at least two cases (i.e., recurrent). To calculate the statistical power, we made the assumption that a pathognomonic alteration in MBC would be present in ≥70% of cases, which is a conservative estimate and one that is substantially lower than the observed >95% frequency of ETV6-NTRK3 fusion in breast secretory carcinomas (originally identified in a cohort of six)12 and >90% frequency of MYB-NFIB fusion in adenoid cystic carcinoma (originally identified in a cohort of ten, of which only four were breast adenoid cystic carcinomas).13 Thus by sequencing five samples (the smallest number of cases for any MBC histologic subtype), we would have 97% statistical power to detect a pathognomonic alteration in at least two cases. Even if we lower the assumed frequency of a pathognomonic driver alteration to be present in 50% of the cases, we would still have 80% statistical power to detect the pathognomonic alteration in at least two cases.
Code availability
The R script of all microarray-based gene expression profiling and gene copy number analyses performed is available at GitHub (https://github.com/charlottekyng/piscuoglio-et-al-metaplastic-breast-carcinoma).
Data availability
The raw and processed gene expression and SNP 6.0 data are available on Gene Expression Omnibus (GSE57549). RNA-sequencing data have been deposited to the Sequence Read Archive (SRP070780).
References
Lakhani, S. R., Ellis, I. O., Schnitt, S. J., Tan, P. H. & van de Vijver, M. J. WHO Classification of Tumours of the Breast. (IARC Press, Lyon, 2012).
Weigelt, B., Eberle, C., Cowell, C. F., Ng, C. K. & Reis-Filho, J. S. Metaplastic breast carcinoma: more than a special type. Nat. Rev. Cancer 14, 147–148 (2014).
Hennessy, B. T. et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 69, 4116–4124 (2009).
Weigelt, B., Kreike, B. & Reis-Filho, J. S. Metaplastic breast carcinomas are basal-like breast cancers: a genomic profiling analysis. Breast Cancer Res. Treat. 117, 273–280 (2009).
Weigelt, B. et al. Metaplastic breast carcinomas display genomic and transcriptomic heterogeneity [corrected]. Mod. Pathol. 28, 340–351 (2015).
Hennessy, B. T. et al. Biphasic metaplastic sarcomatoid carcinoma of the breast. Ann. Oncol. 17, 605–613 (2006).
Horlings, H. M. et al. Genomic profiling of histological special types of breast cancer. Breast Cancer Res. Treat. 142, 257–269 (2013).
Geyer, F. C. et al. Molecular analysis reveals a genetic basis for the phenotypic diversity of metaplastic breast carcinomas. J. Pathol. 220, 562–573 (2010).
Ng, C. K. Y. et al. The landscape of somatic genetic alterations in metaplastic breast carcinomas. Clin. Cancer. Res. 23, 3859–3870 (2017).
Reis-Filho, J. S. et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. J. Pathol. 209, 445–453 (2006).
Moulder, S. et al. Inhibition of the phosphoinositide 3-kinase pathway for the treatment of patients with metastatic metaplastic breast cancer. Ann. Oncol. 26, 1346–1352 (2015).
Tognon, C. et al. Expression of the ETV6-NTRK3 gene fusion as a primary event in human secretory breast carcinoma. Cancer Cell. 2, 367–376 (2002).
Persson, M. et al. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl Acad. Sci. USA 106, 18740–18744 (2009).
Gonda, T. J. & Ramsay, R. G. Adenoid cystic carcinoma can be driven by MYB or MYBL1 rearrangements: new insights into MYB and tumor biology. Cancer Discov. 6, 125–127 (2016).
Bartek, J., Iggo, R., Gannon, J. & Lane, D. P. Genetic and immunochemical analysis of mutant p53 in human breast cancer cell lines. Oncogene 5, 893–899 (1990).
Popova, T. et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 72, 5454–5462 (2012).
Mahmoudi, T. et al. The kinase TNIK is an essential activator of Wnt target genes. EMBO J. 28, 3329–3340 (2009).
Curtis, C. et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012).
Parker, J. S. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 27, 1160–1167 (2009).
Weigelt, B. et al. Breast cancer molecular profiling with single sample predictors: a retrospective analysis. Lancet Oncol. 11, 339–349 (2010).
Herschkowitz, J. I. et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 8, R76 (2007).
Prat, A. et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 12, R68 (2010).
Reimand, J. et al. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res. 44, W83–W89 (2016).
Busse-Wicher, M., Wicher, K. B. & Kusche-Gullberg, M. The exostosin family: proteins with many functions. Matrix. Biol. 35, 25–33 (2014).
Kiani, C., Chen, L., Wu, Y. J., Yee, A. J. & Yang, B. B. Structure and function of aggrecan. Cell. Res. 12, 19–32 (2002).
McPherson, A. et al. deFuse: an algorithm for gene fusion discovery in tumor RNA-Seq data. PLoS Comput. Biol. 7, e1001138 (2011).
Iyer, M. K., Chinnaiyan, A. M. & Maher, C. A. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics 27, 2903–2904 (2011).
Stransky, N., Cerami, E., Schalm, S., Kim, J. L. & Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 5, 4846 (2014).
Kandoth, C. et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013).
Futreal, P. A. et al. A census of human cancer genes. Nat. Rev. Cancer 4, 177–183 (2004).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014).
Chiang, S. et al. IDH2 mutations define a unique subtype of breast cancer with altered nuclear polarity. Cancer Res. 76, 7118–7129 (2016).
Ouyang, Z. et al. Structure of the human cohesin inhibitor Wapl. Proc. Natl Acad. Sci. USA 110, 11355–11360 (2013).
Mortusewicz, O., Fouquerel, E., Ame, J. C., Leonhardt, H. & Schreiber, V. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res. 39, 5045–5056 (2011).
Seimiya, H. & Smith, S. The telomeric poly(ADP-ribose) polymerase, tankyrase 1, contains multiple binding sites for telomeric repeat binding factor 1 (TRF1) and a novel acceptor, 182-kDa tankyrase-binding protein (TAB182). J. Biol. Chem. 277, 14116–14126 (2002).
Riffell, J. L., Lord, C. J. & Ashworth, A. Tankyrase-targeted therapeutics: expanding opportunities in the PARP family. Nat. Rev. Drug Discov. 11, 923–936 (2012).
Carter, H. et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 69, 6660–6667 (2009).
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Elston, C. W. & Ellis, I. O. Pathological prognostic factors in breast cancer. I. The value of histological grade in breast cancer: experience from a large study with long-term follow-up. Histopathology 19, 403–410 (1991).
Van Loo, P. et al. Allele-specific copy number analysis of tumors. Proc. Natl Acad. Sci. USA 107, 16910–16915 (2010).
Carter, S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012).
Piscuoglio, S. et al. Integrative genomic and transcriptomic characterization of papillary carcinomas of the breast. Mol. Oncol. 8, 1588–1602 (2014).
Tusher, V. G., Tibshirani, R. & Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl Acad. Sci. USA 98, 5116–5121 (2001).
Shugay, M., Ortiz de Mendibil, I., Vizmanos, J. L. & Novo, F. J. Oncofuse: a computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics 29, 2539–2546 (2013).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
McKenna, A. et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Chang, M. T. et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat. Biotechnol. 34, 155–163 (2016).
Acknowledgements
The authors acknowledge Leticia Fuhrmann for her assistance with retrieving clinical information. This study was funded in part by Breast Cancer Research Foundation (BCRF). S.P. is funded by Swiss National Science Foundation (Ambizione grant number PZ00P3_168165). R.N. is funded by a Breast Cancer Campaign Career Development Fellowship (2011MaySF01). Research reported in this publication was supported in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (Grant No. P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
L.N., A.V.-S., B.W. and J.S.R.-F. conceived the study. O.M., A.V.-S. provided samples. S.P., F.C.G., C.F.C., L.G.M., R.N. and T.P. performed experiments. F.C.G., A.V.-S. and J.S.R.-F. performed the histologic review. C.K.Y.N., K.A.B., C.A.M., R.S.L., and I.d.B. performed bioinformatics analyses. S.P., C.K.Y.N., F.C.G. and C.F.C. wrote the manuscript that was initially edited by B.W. and J.S.R.-F. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Piscuoglio, S., Ng, C.K.Y., Geyer, F.C. et al. Genomic and transcriptomic heterogeneity in metaplastic carcinomas of the breast. npj Breast Cancer 3, 48 (2017). https://doi.org/10.1038/s41523-017-0048-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-017-0048-0
This article is cited by
-
Starfysh integrates spatial transcriptomic and histologic data to reveal heterogeneous tumor–immune hubs
Nature Biotechnology (2024)
-
Transcriptomic alterations underlying metaplasia into specific metaplastic components in metaplastic breast carcinoma
Breast Cancer Research (2023)
-
Rare subtypes of triple negative breast cancer: Current understanding and future directions
npj Breast Cancer (2023)
-
Epithelial to mesenchymal transition (EMT) in metaplastic breast cancer and phyllodes breast tumors
Medical Oncology (2023)
-
Breast carcinomas with osteoclast-like giant cells: a comprehensive clinico-pathological and molecular portrait and evidence of RANK-L expression
Modern Pathology (2022)
