
Abstract
Veliparib combined with carboplatin (VC) was an experimental regimen evaluated in the biomarker-rich neoadjuvant I-SPY 2 trial for breast cancer. VC showed improved efficacy in the triple negative signature. However, not all triple negative patients achieved pathologic complete response and some HR+HER2− patients responded. Pre-specified analysis of five DNA repair deficiency biomarkers (BRCA1/2 germline mutation; PARPi-7, BRCA1ness, and CIN70 expression signatures; and PARP1 protein) was performed on 116 HER2− patients (VC: 72 and concurrent controls: 44). We also evaluated the 70-gene ultra-high risk signature (MP1/2), one of the biomarkers used to define subtype in the trial. We used logistic modeling to assess biomarker performance. Successful biomarkers were combined using a simple voting scheme to refine the ‘predicted sensitive’ group and Bayesian modeling used to estimate the pathologic complete response rates. BRCA1/2 germline mutation status associated with VC response, but its low prevalence precluded further evaluation. PARPi-7, BRCA1ness, and MP1/2 specifically associated with response in the VC arm but not the control arm. Neither CIN70 nor PARP1 protein specifically predicted VC response. When we combined the PARPi-7 and MP1/2 classifications, the 42% of triple negative patients who were PARPi7-high and MP2 had an estimated pCR rate of 75% in the VC arm. Only 11% of HR+/HER2− patients were PARPi7-high and MP2; but these patients were also more responsive to VC with estimated pathologic complete response rates of 41%. PARPi-7, BRCA1ness and MP1/2 signatures may help refine predictions of VC response, thereby improving patient care.
Similar content being viewed by others

Introduction
Poly(ADP-ribose) polymerase (PARP) inhibitors belong to an emerging class of drugs that operate on the principle known as ‘synthetic lethality’.1, 2 PARP is involved in single strand break (SSB) DNA repair; and upon PARP inhibition, some SSBs are converted into double strand breaks (DSBs) at replication forks. In homologous repair competent cells, DSBs are repaired so that the cells can survive.3 However, in homologous repair-deficient cells, DSBs are repaired via the less accurate non-homologous end joining pathway or by single strand annealing, resulting in aberrant chromatids that trigger cell death.4 Thus, cells with BRCA mutations or other homologous repair defects5,6,7 are preferentially sensitive to PARP inhibitors.
The PARP inhibitor, olaparib, was recently FDA approved for BRCA1/2 mutation positive patients with advanced ovarian cancer. There are at least five PARP inhibitors under clinical investigation, including olaparib (AstraZeneca, London), veliparib (ABT-888; Abbott Laboratories, IL), rucaparib (AG014699; Clovis Oncology), niraparib (MK4827; Merck), and talazoparib (BMN673; Medivation, CA)8,9,10,11,12,13,14,15 for BRCA-associated, triple negative (TN) and/or basal breast cancers and other solid tumors, either as monotherapy in DNA repair deficient patients or as potentiating agents for radiation or DNA-damaging anticancer agents.16,17,18 Results from clinical trials in breast cancer in the advanced disease setting appear to vary depending on patient characteristics and the therapeutic regimen under investigation.19 Phase 2 trials showed that olaparib as monotherapy led to objective response rates in 31–41% of BRCA1/2 mutation carriers who had previously received several courses of chemotherapy.14, 20, 21 However, results for BRCA1/2 wild-type breast cancer patients have been inconsistent. TN breast cancer patients without BRCA mutations largely did not respond to olaparib monotherapy in a phase 2 trial,22 whereas preclinical studies and phase 1 trials suggested that PARP inhibitors can be efficacious in these patients when combined with DNA damaging cytotoxic agents.17
Veliparib in combination with carboplatin (VC) was recently evaluated in I-SPY 2, a multi-center Phase 2 adaptive standing platform trial for women with early stage, locally advanced, aggressive breast cancer.23 I-SPY 2 is designed to screen multiple experimental regimens in addition to standard neoadjuvant chemotherapy (Fig. 1a). Women with HER2+ disease also receive trastuzumab or other HER2-targeted therapy. This trial is adaptive, in that a patient randomized to receive experimental treatment is assigned preferentially to the arm where her cancer subtype is most likely to respond. Subtype is defined by hormone receptor (HR) status, HER2 status, and MammaPrint (MP) High 1/High 2 risk status (MP1/2), essentially a further stratification of the MP poor prognosis group (MP High) into high and ultra-high risk groups. The primary endpoint is pathologic complete response (pCR), i.e., no invasive cancer left in the breast or lymph nodes. The goal of I-SPY 2 is to identify (graduate) regimens with >85% predicted probability of succeeding in a 1:1 randomized 300-patient phase 3 trial where pCR is the endpoint, in the signatures defined by HR, HER2, and MP where the drug is most effective (graduates). The VC arm was open for enrollment to HER2-negative (HER2−) patients, and VC was eligible for graduation in three signatures: HER2-negative, HR-positive HER2-negative (HR+HER2−), and TN.

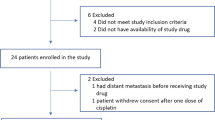
a I-SPY 2 TRIAL design schematic. Only patients with HER2− disease were eligible for randomization to the VC arm. b Consort diagram for the VC arm and HER2− concurrent controls, showing data availability for biomarker analysis
VC successfully graduated in the TN signature;23 but not all TN patients in the VC arm achieved a pCR, and many outside this subset responded. There is a need for additional robust biomarkers that predict VC sensitivity. Just as the I-SPY 2 standing platform trial is an efficient mechanism for screening promising agents for Phase 3 testing, the biomarker component of I-SPY 2, where samples are profiled for gene expression, protein levels and mutation status, provides a centralized resource for investigators to propose and test additional mechanism-of-action-based biomarkers using a rigorous, pre-defined analysis methodology in the context of the established clinically relevant biomarkers already incorporated within the trial. Previous studies have proposed homologous DNA repair deficiency (HRD) biomarkers, such as BRCA1/2 mutation,24, 25 BRCA1 promoter methylation,26,27,28 ‘BRCAness’ expression and CGH signatures,27, 29 Fanconi F and D methylation or loss,30 p53 and PTEN loss of function mutations or expression level,31,32,33 cell line based HRD/PARPi signatures,34 telomeric allelic imbalance,35 loss of heterozygocity HRD scoring,36 and HRD ‘genomic scarring’,37 among others,27, 38, 39 as possible markers of sensitivity to PARP inhibition. Through the I-SPY 2 Data access and Publication Committee process, five investigator-submitted qualifying biomarker concept proposals related to DNA damage repair deficiency were approved and individually evaluated in baseline (pre-treatment) biopsies as specific biomarkers of VC response.
The five proposed mechanism-of-action-based biomarkers were: (1) BRCA1/BRCA2 germline mutation; (2) a 7-gene DNA-repair deficiency expression signature (PARPi-7) that predicts breast cancer cell line sensitivity to the PARP inhibitor olaparib; 34 (3) a 77-gene BRCA1ness expression signature;40, 41 (4) the CIN70 chromosomal instability expression signature;42, 43 and (5) PARP1 protein levels, the target of veliparib. In addition, we also consider MP1/2 status, because the restricted enrollment of the VC arm to HER2− patients precluded the MP2 signature from being eligible for graduation. In this report, we present our evaluation of the predictive performance of each of these six biomarkers in the VC arm relative to the control arm, analyzed in isolation and in the context of the graduating TN subset. In addition, we compare these biomarkers to determine whether they are identifying the same patients, and perform an exploratory analysis that combines the most successful candidates into a composite VC sensitivity score.
Results
Patient characteristics summary
A total of 72 patients were randomized to the VC arm, and treated with veliparib and carboplatin in addition to standard taxane/anthracycline chemotherapy (VC+T−>AC). There were 44 concurrently randomized HER2− controls treated with standard chemotherapy (T−>AC).23 Patients who received non-protocol therapy, left their treating institution, or withdrew consent prior to surgery were considered non-pCR as per protocol. Patient and baseline clinical characteristics, such as age, ethnicity, HR status, tumor size and nodal status, were similar between the experimental and control arms.23 All 116 patients have Agilent 44 K expression arrays available. 98% of patients (n = 114) were evaluated for BRCA germline mutation status; and 89% (n = 103) have available RPPA data (Fig. 1b).
BRCA1/2 germline mutation status
Of the 114 patients evaluated for BRCA1/2 germline mutation status, 15 were found to carry a deleterious or suspected deleterious BRCA1/2 mutation, with the majority (n = 11) occurring within the TN subtype. In the VC arm, BRCA mutation carriers were more likely to achieve a pCR compared to wild-type patients (75 vs. 29%) (Fig. S1). This translates to an odds ratio of 7.25 between the BRCA1/2 germline mutation carriers and wildtype patients (Fisher Exact test p-value = 0.006, Table 1). The small number of mutation carriers in the control arm (n = 3) precluded further evaluation.
The PARPi-7 and BRCA1ness signatures and MP1/2 classification specifically predict VC response
We evaluated as proposed three dichotomous expression based signatures—the BRCA1ness signature, the CIN70 signature and the MP1/2 risk classification—and one continuous signature (PARPi-7) as qualifying biomarkers of VC response using our predefined qualifying biomarker evaluation (QBE) process (see Fig. 2).

Biomarker analysis approach. Qualifying biomarker candidates are evaluated as specific predictors of response to VC using a predefined 3-step Qualifying Biomarker Evaluation (QBE) methodology, as shown in this flow diagram
The PARPi-7 and BRCA1ness signatures and the MP1/2 risk classification associated with response in the VC arm (p < 0.05), but not the control arm (Table 1). Using logistic regression, for each of these three signatures, the biomarker x treatment interaction term reflecting the relative biomarker performance between arms had a likelihood ratio (LR) test p value of <0.05, which remains significant (LR p < 0.05) upon adjusting for receptor subtype (Table 1).
Neither the CIN70 signature nor the total and cleaved PARP protein levels succeeded as specific biomarkers of response to VC (Table 1 and Fig. S2).
For potential clinical utility and ease of comparison to the other successful signatures, we dichotomize the patients using the PARPi-7 signature at the published threshold (0.0372)34 [see Fig. S3] and use the dichotomous version of this signature henceforth. Figure 3a shows the biomarker signature prevalence and composition in the VC and control arms. pCR rates by receptor subtype, treatment arm, and biomarker are shown in Fig. S4. As shown in the mosaic plots in Fig. 3b–d, PARPi-7 High, BRCA1ness and MP2 patients all had a higher pCR rate in the VC arm (Fisher’s exact test p < 0.05), but not the control arm (p > 0.05), when compared to biomarker-negative patients.

Qualifying biomarker performance. a Ordered heatmap showing the prevalence of all dichotomized biomarkers evaluated in this study, stratified by HR status. b–d Mosaic plots showing patient response stratified by treatment arm and b PARPi-7, c BRCA1ness (this figure panel is also jointly published in ref. [41]), and d MP1/2 signatures, respectively. e–g Bayesian estimated pCR probability distributions by treatment arm, for e PARPi7-high, f BRCA1ness, and g MP2 patients
We used Bayesian modeling, similar to the I-SPY 2 primary efficacy analysis, to estimate the pCR probabilities to VC and standard chemotherapy in biomarker predicted-sensitive patient subsets (PARPi-7 High, BRCA-like, and MP2). As shown in Fig. 3e–g, estimated pCR probabilities in the VC arm range from 50–61% vs. 18–27% in the control arm, in these subsets. We also computed the predictive probability of VC demonstrating superiority to control in a 1:1 randomized neoadjuvant phase 3 trial of 300 biomarker predicted-sensitive patients. These probabilities are >90% (95% for PARPi-7 High, 96% for BRCA1ness and 94% for MP2 patients). If we exclude the 15 BRCA1/2 germline mutation carriers from the analysis and consider only BRCA1/2 wild-type patients, these probabilities are 75% for PARPi-7 High, 81% for BRCA1ness and 87% for MP2 patients.
Expanding the ‘predicted-responder’ group beyond TN using qualifying biomarkers
We evaluated the PARPi-7, BRCA1ness and MP1/2 signatures in relation to the graduating TN signature, by adding biomarker-positive HR+/HER2− patients to the TN group and evaluating whether the treatment effect of VC remains significant in this expanded population of ‘predicted sensitive’ patients. For all three signatures, the VC treatment effect in the expanded population remains similar to that observed within the graduating TN signature (see Table S2 for details); and the predicted probability of success in phase 3 in these expanded groups are comparable to the graduating signature (Fig. S5). For instance, if the PARPi-7 is used to expand the population by adding PARPi-7 High HR+HER2− patients to the TN group, the probability of VC demonstrating superiority to control in phase 3 is 90%, while increasing the prevalence of ‘predicted sensitive’ patients by 23% of HR+/HER2− patients in the trial. Similar results were observed when the BRCA1ness and MP1/2 signatures were considered (Fig. S5).
Comparing and combining qualifying biomarkers in the context of HR and HER2 status to improve response prediction
Since PARPi-7, BRCA1ness, and MP1/2 all appear to succeed as specific predictors of VC combination therapy in I-SPY 2, we wondered whether these signatures identify the same patients, and if not, whether individual biomarkers could be combined to better identify likely responders within the TN and HR+/HER2− subsets. As shown in Fig. 3a, the concordance between biomarker pairs over all patients in the trial ranges from 66–78%, indicating that these biomarkers are not identifying exactly the same patients.
In our exploratory analysis that was not pre-specified, we combined pairs of biomarkers using a simple voting scheme. As shown in Fig. 4a, if patients are classified as resistant by both biomarkers, or resistant by one biomarker, they are classified as ‘resistant’. Patients are classified as ‘sensitive’ only if both biomarkers are in the ‘sensitive’ state.

Combining VC-sensitivity markers in TN patients. a Simple voting scheme for combining biomarkers to refine sensitivity prediction. b Venn diagram showing overlap between VC-sensitivity biomarkers, including the graduating TN signature. c–f Bayesian estimated pCR probability distributions by treatment arm, for c unselected triple negative [TN], d TN/MP2, e TN/PARPi7-high, and f TN/BRCA1ness patients, respectively. g, h Bayesian estimated pCR probability distributions by treatment arm, for g predicted sensitive (TN/MP2/PARPi7-high) and h predicted resistant (TN/(MP1 or PARPi7-low)) triple negative patients. i Pie chart showing relative proportion of predicted sensitive vs. resistant TN patients
Biomarker combinations within the TN subtype
Though 95% (57/60) of TN patients are PARPi7-high or MP2 or BRCA1ness, only 38% would be classified as sensitive by all three biomarkers (Fig. 4b).
Figure 4c shows the pCR probability distributions of TN patients in the VC and control arms, with estimated pCR rates of 51% in the VC arm and 26% in the control arm. In TN patients predicted VC-sensitive by a single additional biomarker, there is a greater separation in the pCR probability distributions between arms, with estimated pCR rates to VC ranging from 56% to 70% (Fig. 4d–f). If we combine the two biomarkers yielding the highest estimated pCR rate in the VC arm (PARPi-7 and MP2) to select TN patients who are TN/MP2/PARPi7-high (Fig. 4g), the separation of the estimated pCR rates between arms is greater still (75% in VC vs. 23% in control), with a predictive probability of success in phase 3 of 99% (Table S3). This group constitutes a sizeable 42% of TN patients in the trial (Fig. 4h). In contrast, the pCR probability distributions of TN patients with one or more markers in the resistant state (TN/PARPi7-low or TN/MP1) are nearly overlapping (Fig. 4i).
If we exclude the BRCA1/2 germline mutation carriers from the analysis, the separation of estimated pCR rates between arms is still large in the TN/MP2/PARPi7-high group (63% in VC vs. 23% in control), with a predictive probability of success in phase 3 of 95% (Fig. S6).
Biomarker combinations within the HR+/HER2− subtype
Only 34% (19/56) of HR+/HER2− patients are positive for one or more VC sensitivity biomarkers. 23% (13/56) are PARPi7-high, 20% (11/56) are MP2, and 14% (8/56) are BRCA1ness (Fig. 5a).

Combining VC-sensitivity markers in HR+HER2− patients. a Venn diagram showing overlap between VC-sensitivity biomarkers in the HR+HER2− subset. b–g Bayesian estimated pCR probability distributions by treatment arm, for b unselected HR+HER2−, c HR+HER2−/MP2, d HR+HER2−/PARPi7-high, and e HR+HER2−/BRCA1ness patients, respectively, and for HR+HER2− patients who are f predicted sensitive (HR+HER2−/MP2/PARPi7-high) and g predicted resistant (HR+HER2−/(MP1 or PARPi7-low)) by our simple voting scheme. h Pie chart showing relative proportion of predicted sensitive vs. resistant HR+HER2− patients
HR+/HER2− patients had low estimated pCR rates in both arms with nearly overlapping probability distributions (estimated pCR: 15% in VC, 19% in control; Fig. 5b). In HR+HER2− patients predicted VC sensitive by a single additional biomarker, there is a greater separation in the pCR probability distributions between arms with estimated pCR rates to VC of 33% in the HR+HER2−/MP2 and HR+HER2−/PARPi7-High subsets (Fig. 5c–e). If we combine MP1/2 and PARPi-7 per above to select HR+HER2− patients who are HR+HER2-/MP2/PARPi7-high (Fig. 5f), the separation of the estimated pCR rates between arms increases further (41% in VC vs. 15% in control), with a predictive probability of success in phase 3 of 82% (Fig. 5f; Table S3). In contrast, HR+/HER2− patients negative for at least one VC sensitivity marker (HR+HER2−/PARPi7-low, or HR+HER2-/MP1) have nearly overlapping pCR probability distributions with estimated pCR rates of 13% in VC vs. 19% in controls (Fig. 5g).
Although our results demonstrate that ‘double biomarker-positive’ in both TN and HR+HER2− patients may be more sensitive to VC than patients with fewer markers in the ‘sensitive’ state, the prevalence of MP2/PARPi7-high patients in the TN subset is relatively high (42%; Fig. 4i), whereas HR+/HER2− patients with these markers are relatively rare (11%; Fig. 5h).
Discussion
The I-SPY 2 TRIAL design presents both opportunities and challenges for evidence-based biomarker testing. On one hand, the biomarker-rich nature of the trial, in which pre-treatment tumor biopsies are assayed on Agilent 44 K expression and RPPA protein/phospho-protein platforms, provides an excellent resource to investigate the molecular correlates of response and resistance. As well, collection of serial biopsies and blood samples over the course of therapy generates an invaluable repository for additional molecular profiling.
On the other hand, as the I-SPY 2 TRIAL was designed to efficiently evaluate multiple novel regimens compared to a shared control arm for future Phase 3 testing, the sample sizes available for our biomarker studies are small. As well, VC is given as a combination therapy within the I-SPY 2 trial. While assessing the relative performance of a biomarker in the experimental vs. control arms enables us to identify specific predictors of response to VC, it is impossible to assess whether the successful biomarkers described in our report are predictive of response to the individual agents within the combination. In addition, I-SPY 2 uses adaptive randomization within HR/HER2/MP defined subtypes to enable efficient matching of novel regimens with their most responsive clinically relevant signatures. This may result in the unbalanced prevalence of biomarker-positive subsets in the experimental and control arms if the biomarker of interest is correlated with a HR/HER2/MP subtype that is preferentially enriched or depleted in the experimental arm by the randomization engine. As well, there are multiple genes in each signature measured on several platforms, creating the problem of multiplicity, which is compounded by the evaluation of multiple proposals. Altogether, these challenges limit our ability to draw definitive conclusions. Thus, our statistics are descriptive rather than inferential; and our findings require further validation in future trials.
Four of the five evidence-based, mechanism-of-action related (qualifying) biomarkers included in this report are markers of DNA repair deficiency because of the large body of in vitro and in vivo evidence that DNA repair deficient cancers selectively respond to DNA damaging agents potentiated by PARP inhibition. Many studies support BRCA1/2 germline mutation status as the ‘gold standard’ biomarker for predicting sensitivity to PARP inhibitors as monotherapy. In I-SPY 2, BRCA1/2 mutation carriers were indeed more likely to respond to VC than BRCA1/2 wildtype patients, but the mutation prevalence was too low for a comparison to response in the control arm.
Of the three DNA repair based expression signatures we evaluated, two (PARPi7 and BRCA1ness) were associated with response in the VC arm, but not in the control arm, and had a significant biomarker x treatment interaction that retained significance upon adjusting for HR status. These two successful qualifying biomarker signatures were derived in very different ways. The PARPi-7 signature was the result of an effort to build a predictor of response to a different PARP inhibitor, olaparib, using a breast cancer cell line panel and a candidate gene set restricted to DNA repair genes in one of the six major repair pathways. The BRCA1ness signature, on the other hand, was developed as a classifier to identify cancers exhibiting a BRCA1-mutated copy number profile. Indeed, 80% (12/15) of BRCA+ patients were classified as BRCA1ness, along with many non-mutation carriers. Interestingly, the concordance between these signatures is only moderate, and while both BRCA1ness and PARPi7 have predictive signal, the nature of this signal differs. In the TN subset, the differential response in the VC and control arms in BRCA1ness cancers mostly manifests as a depletion of responders in the control arm rather than an increase in responders in the VC arm relative to unselected TNs. Whereas the difference in estimated pCR rates in VC vs. control for PARPi7-high patients is driven by a higher response rate in VC.
Neither the PARP1 protein levels nor the dichotomized CIN70 signature specifically predict response to VC. The lack of association observed between PARP1 protein levels and differential response has been seen elsewhere in PARP inhibitor trials, perhaps due to saturation properties of the antibody or to a disconnect between DNA repair deficiency and PARP1 levels as measured. For CIN70, the lack of association with response may be due to the small size of the population or a non-ideal quantile cutoff for dichotomizing the I-SPY 2 patients, as it associates with response as a continuous variable.
We did not test non-expression based HRD biomarkers found to associate with response to platinum-based DNA damaging agents, including the HRD-LOH/HRD-LST score or Myriad’s HRD test quantifying the level of ‘DNA scarring’ on tumor genomes.36, 37 As these measures are in a sense closer to a direct read-out of DNA repair deficiency, it may be interesting to examine their correlation to the expression-based biomarkers we evaluated and to compare their predictive performance in I-SPY 2. But tissue availability is limited; and careful consideration is needed in determining the optimal use of samples within the I-SPY 2 tissue repository.
Our analysis suggests that had HER2−/MP2 or TN/MP2 signatures been eligible for graduation of VC from the I-SPY 2 trial, they would have graduated along with the TN signature (with >85% predictive probability of success in phase 3). Though not explicitly developed as a DNA repair deficiency biomarker, preliminary analysis of pathway differences between MP1 and MP2 class tumors suggest that MP2 cancers have higher expression of cell cycle genes and DNA repair pathways other than homologous repair.44 This may explain why MP1/2 and PARPi-7 are not highly concordant, as the latter largely reflects HRD.
The biomarker component of I-SPY 2 must function in the context of I-SPY 2’s innovative adaptive trial design. VC graduated with a >85% probability of success in a future phase 3 trial, conducted not in the general breast cancer population, but in the TN subset. Thus, TN (HR−/HER2−) status is already a successful biomarker for VC, and we must try to understand how the novel mechanism-of-action based biomarkers we are testing add value to the graduating TN signature. To this end, we imagined two different purposes for a biomarker.
One purpose of a biomarker concerns clinical decision making for individual patients, also known as personalized oncology. For this application, the objective is to use all the available information to predict the likelihood of an individual patient’s response to a set of treatment options. For this type of analysis we combine successful biomarkers, such as the qualifying biomarkers and the graduating signature to refine the sensitivity prediction. For instance, we combine TN status with the PARPi-7 signature, to show that TN patients who are also PARPi7-high have a 70% probability of achieving pCR in the VC arm compared to 30% in the control arm. We also show how a simple voting method can be used to combine information from multiple sensitivity biomarkers (PARPi7 and MP2) to improve VC response prediction to 75% pCR probability. Moreover, MP2/PARPi7-high remains a strong predictor of response to VC relative to control in TN patients who are BRCA1/2 wild-type. This information would be useful for a patient selecting a treatment protocol, but may not be appropriate for patient selection in a trial because the improved performance of the biomarker comes at the expense of excluding a large segment of the TN population.
Another purpose is to use the novel biomarker together with the graduating group to improve patient selection in a future phase 3 trial of VC. In this setting, an ideal biomarker is one which identifies the maximum number of patients likely to have a sufficiently improved response to VC (relative to control) so as to succeed in a subsequent phase 3 trial conducted in this ‘biomarker-positive’ group. Maximizing the size of this biomarker-positive subset is desirable because phase 3 testing and thus approval of VC will likely be limited to this group. When we added single biomarker positive HR+HER2− patients to the graduating TN group, the Bayesian probabilities of success in phase 3 of this expanded biomarker-positive population is similar to the graduating TN signature, while increasing prevalence by adding 14–23%. Using the voting method, only 11% of HR+HER2− patients would be added to the predicted responder group. This example illustrates a generic trade-off between biomarker performance and prevalence (or specificity and sensitivity), with ‘fit for purpose’ implications for how these biomarkers are best used combinatorially.
Our future plans include evaluating whether the biomarkers predictive of VC response in the I-SPY 2 TRIAL can also predict sensitivity to other PARP inhibitors combined with DNA damaging agents or even carboplatin alone. In addition, we plan to use these biomarkers as part of a SMART-trial design to adapt treatment within patients who are not responding to their initial therapy.
Methods
In the I-SPY 2 TRIAL (NCT01042379), HER2− patients were randomized to receive standard chemotherapy (paclitaxel followed by doxorubicin/cyclophosphamide; T−>AC) or the oral PARP inhibitor veliparib in combination with carboplatin and chemotherapy (VC+T−>AC).23 Pre-treatment samples from patients in the veliparib/carboplatin (VC) and concurrent control arms of the I-SPY 2 trial23 were profiled using Agilent 44 K expression arrays (Agendia, Inc) and reverse phase protein arrays (RPPA). Details of the sample preparation and data processing are provided in the Supplemental Methods. In addition, BRCA1/2 mutation status was assessed (Myriad Genetics Laboratories).
We evaluated six biomarkers as specific predictors of VC response using a pre-specified QBE methodology. Briefly, we follow a three-step methodology, first evaluating the relative performance of the biomarker between arms, followed by assessing biomarker performance in the context of the graduating (TN) signature. Finally, we perform Bayesian analysis to estimate pCR rates in the arms and the predictive probability of VC showing superiority to control in biomarker defined subsets. The six biomarkers evaluated are: (1) BRCA germline mutation; (2) a 7-gene DNA-repair deficiency expression signature (PARPi-7)34; (3) a 77-gene BRCA1ness expression signature;40, 41 (4) the CIN70 chromosomal instability expression signature;42, 43 (5) PARP1 protein levels; and (6) MP1/2 status. Details of the definition and scoring of each biomarker and our evaluation methodology are available in the Supplemental Methods. Each individual biomarker was evaluated separately; and these analyses did not account for multiplicities outside of the study.
In an exploratory analysis, we evaluate the concordance between successful biomarkers using the Kappa statistic. We use a simple voting method to combine the two most successful VC-sensitivity biomarkers and use Bayesian modeling to estimate the pCR rates and predictive probability of phase 3 trial success of biomarker-positive TN and HR+HER2− patients.
I-SPY 2 TRIAL investigators
I-SPY 2 TRIAL Investigators, by site/affiliation: Avera Cancer Institute: Leyland-Jones B; British Colombia Cancer Agency: Chia S, Serpanchy R, Yu C; Emory University: McMillan S, Mosley R, Nguyen K, Wood EC, Zelnak A; Georgetown University: Dillis C, Donnelly R, Harrington T, Isaacs C, Kallakury B, Liu M, Lynce F, Oppong B, Pohlmann P, Tousimis E, Warren R, Willey S, Wong JE, Zeck J; Loyola University Chicago Medical Center: Albain K, Bartolotta MB, Bova D, Brooks C, Busby B, Czaplicki K, Duan X, Gamez R, Ganesh K, Gaynor E, Godellas C, Grace-Louthen C, Kuritza T, Lo S, Nagamine A, Perez C, Robinson P, Rosi D, Vaince F, Ward K; Innova Fairfax Hospital: Choquette K, Edmiston K, Gallimore H, McGovern J, Mokarem K, Pajaniappan M, Rassulova S, Scott K, Sherwood K, Wright J; Mayo Clinic, Arizona: Anderson KS, Gray RJ, Myers SJ, Northfelt DE, Pockaj BA, Roedig J, Wasif N; Mayo Clinic, Rochester: Arens AM, Boughey JC, Brandt KR, Carroll JL, Chen B, Connors AL, Degnim AC, Farley DR, Greenlee SM, Haddad TC, Hieken TJ, Hobday TJ, Jakub JW, Liberte LL, Liu MC, Loprinzi CL, Menard LR, Moe MM, Moynihan TJ, O’Sullivan C, Olson EA, Peethambaram PP, Ruddy KJ, Russell BA, Rynearson AL, Smith DR, Visscher DW, Windish AJ; H. Lee Moffitt Cancer Center and Research Institute: Cox K, Dawson K, Newton O, Ramirez W; Oregon Health and Science University: Bengtson H, Bucher J, Chui S, Gilbert-Ghormley B, Hampton R, Kemmer KA, Kurdyla D, Nauman D, Spear J, Wilson A; Swedish Cancer Institute: Beatty D, Dawson P, Ellis ER, Fer M, Hanson J, Goetz MP, Haddad TC, Iriarte D, Kaplan HG, Porter B, Rinn K, Thomas H, Thornton S, Tickman R, Varghis N; University of Alabama at Birmingham: Caterinichia V, Delos Santos J, Falkson C, Forero A, Krontiras H, Vaklavas C, Wei S; University of Arizona: Bauland A, Inclan L, Lewallen D, Powell A, Roney C, Schmidt K, Viscusi RK, Wright H; University of California, San Diego: Blair S, Boles S, Bykowski J, Datnow B, Densley L, Eghtedari M, Genna V, Hasteh F, Helsten T, Kormanik P, Ojeda-Fournier H, Onyeacholem I, Parker B, Podsada K, Schwab R, Wallace A, Yashar C; University of California, San Francisco: Alvarado MD, Au A, Balassanian R, Benz C, Buxton M, Chen YY, Chien J, D’Andrea C, Davis SE, Esserman L, Ewing C, Goga A, Hirst GL, Hwang M, Hylton N, Joe B, Lyandres J, Kadafour M, Krings G, Melisko M, Moasser M, Munter P, Ngo Z, Park J, Price E, Rugo H, van’t Veer L, Wong J, Yau C; University of Chicago: Abe H, Jaskowiak NT, Nanda R, Olopade F, Schacht DV; University of Colorado, Denver: Borges V, Colvin T, Diamond J, Elias AD, Finlayson C, Fisher C, Hardesty L, Kabos P, Kounalakis N, Mayordomo J, McSpadden T, Murphy C, Rabinovitch R, Sams S, Shagisultanova E; University of Kansas: Baccaray S, Khan Q; University of Minnesota: Beckwith H, Blaes A, Emory T, Haddad TC, Hui J, Klein M, Kuehn-Hajder J, Nelson M, Potter D, Tuttle T, Yee D, Zera R; University of Pennsylvania: Bayne L, Bradbury A, Clark A, DeMichele A, Domchek S, Fisher C, Fox K, Frazee D, Lackaye M, Matro J, McDonald E, Rosen M, Shah P, Tchou J, Volpe M; University of Texas, MD Anderson Cancer Center: Alvarez R, Barcenas C, Berry DA, Booser D, Brewster A, Brown P, Gonzalez-Angulo A, Ibrahim N, Karuturi M, Koenig K, Moulder S, Murray J, Murthy R, Pusztai L, Saigal B, Symmans WF, Tripathy D, Theriault R, Ueno N, Valero V; University of Southern California: Brown M, Carranza M, Flores Y, Lang J, Luna A, Perez N, Tripathy D, Watkins K; University of Texas Southwestern Medical Center: Armstrong S, Boyd C, Chen L, Clark V, Frankel A, Euhus DM, Froehlich T, Goudreau S, Haley B, Harker-Murray A, Klemow D, Leitch AM, Leon R, Li H, Morgan T, Qureshi N, Rao R, Reeves M, Rivers A, Sadeghi N, Seiler S, Staves B, Tagoe V, Thomas G, Tripathy D, Unni N, Weyandt S, Wooldridge R, Zuckerman J; University of Washington: Korde L, Griffin M, Butler B, Cundy A, Rubinstein L, Hixson C
References
Iglehart, J. D. & Silver, D. P. Synthetic lethality—a new direction in cancer-drug development. N. Engl. J. Med. 361, 189–191 (2009).
Ashworth, A., Lord, C. J. & Reis-Filho, J. S. Genetic interactions in cancer progression and treatment. Cell 145, 30–38 (2011).
Ciccia, A. & Elledge, S. J. The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204 (2010).
Sancar, A., Lindsey-Boltz, L. A., Unsal-Kacmaz, K. & Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 (2004).
Buisson, R. et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat. Struct. Mol. Biol. 17, 1247–1254 (2010).
McCabe, N. et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 66, 8109–8115 (2006).
Williamson, C. T. et al. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol. Cancer Ther. 9, 347–357 (2010).
Ferraris, D. et al. Design and synthesis of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. Part 4: biological evaluation of imidazobenzodiazepines as potent PARP-1 inhibitors for treatment of ischemic injuries. Bioorg. Med. Chem. 11, 3695–3707 (2003).
Lee, Y. R. et al. New approaches of PARP-1 inhibitors in human lung cancer cells and cancer stem-like cells by some selected anthraquinone-derived small molecules. PLoS One 8, e56284 (2013).
Vinayak, S. & Ford, J. M. PARP inhibitors for the treatment and prevention of breast cancer. Curr. Breast Cancer Rep. 2, 190–197 (2010).
Plummer, R. Poly(ADP-ribose) polymerase inhibition: a new direction for BRCA and triple-negative breast cancer? Breast Cancer Res. 13, 218 (2011).
Scott, C. L., Swisher, E. M. & Kaufmann, S. H. Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J. Clin. Oncol. 33, 1397–1406 (2015).
Samol, J. et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest. New Drugs 30, 1493–1500 (2012).
Audeh, M. W. et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet 376, 245–251 (2010).
Sandhu, S. K. et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 14, 882–892 (2013).
Benafif, S. & Hall, M. An update on PARP inhibitors for the treatment of cancer. Onco Targets Ther. 8, 519–528 (2015).
Dent, R. A. et al. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 15, R88 (2013).
Calabrese, C. R. et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J. Natl. Cancer Inst. 96, 56–67 (2004).
Guha, M. PARP inhibitors stumble in breast cancer. Nat. Biotechnol. 29, 373–374 (2011).
Kaufman, B. et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 33, 244–250 (2015).
Tutt, A. et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet 376, 235–244 (2010).
Gelmon, K. A. et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 12, 852–861 (2011).
Rugo, H. S. et al. Adaptive randomization of veliparib-carboplatin treatment in breast cancer. N. Engl. J. Med. 375, 23–34 (2016).
Bryant, H. E. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005).
Rottenberg, S. et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 105, 17079–17084 (2008).
Esteller, M. et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 92, 564–569 (2000).
Turner, N., Tutt, A. & Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 4, 814–819 (2004).
Jacinto, F. V. & Esteller, M. Mutator pathways unleashed by epigenetic silencing in human cancer. Mutagenesis 22, 247–253 (2007).
Oonk, A. M. et al. Clinical correlates of ‘BRCAness’ in triple-negative breast cancer of patients receiving adjuvant chemotherapy. Ann. Oncol. 23, 2301–2305 (2012).
Do, K. & Chen, A. P. Molecular pathways: targeting PARP in cancer treatment. Clin. Cancer Res. 19, 977–984 (2013).
Saal, L. H. et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat. Genet. 40, 102–107 (2008).
Chuang, H. C., Kapuriya, N., Kulp, S. K., Chen, C. S. & Shapiro, C. L. Differential anti-proliferative activities of poly(ADP-ribose) polymerase (PARP) inhibitors in triple-negative breast cancer cells. Breast Cancer Res. Treat. 134, 649–659 (2012).
Hay, T. et al. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res. 69, 3850–3855 (2009).
Daemen, A. et al. Cross-platform pathway-based analysis identifies markers of response to the PARP inhibitor olaparib. Breast Cancer Res. Treat. (2012).
Birkbak, N. J. et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2, 366–375 (2012).
Abkevich, V. et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 107, 1776–1782 (2012).
Watkins, J. A., Irshad, S., Grigoriadis, A. & Tutt, A. N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 16, 211 (2014).
Popova, T. et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 72, 5454–5462 (2012).
Turner, N. C. & Ashworth, A. Biomarkers of PARP inhibitor sensitivity. Breast. Cancer Res. Treat. 127, 283–286 (2011).
Glas, A. et al. 532 Evaluation of a BRCAness signature as a predictive biomarker of response to veliparib/carboplatin plus standard neoadjuvant therapy in high-risk breast cancer: results from the I-SPY 2 trial. Eur. J. Cancer 50. doi:10.1016/S0959-8049(14)70658-6 (2014).
Severson, T. M. et al. The BRCA1ness signature is associated significantly with response to PARP inhibitor treatment versus control in the I-SPY 2 randomized neoadjuvant setting. Breast Cancer Res. doi:10.1186/s13058-017-0861-2 (2017).
Carter, S. L., Eklund, A. C., Kohane, I. S., Harris, L. N. & Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 38, 1043–1048 (2006).
Birkbak, N. J. et al. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 71, 3447–3452 (2011).
Wolf, D. M. et al. In Proceedings: AACR 107th Annual Meeting 2016. doi:10.1158/1538-7445.AM2016-859 (2016).
Acknowledgements
We thank the patients, patient advocates and investigators who participated in the I-SPY 2 study. We are grateful for support from our study sponsors: the Foundation for the National Institutes of Health (2010 to 2012) and Quantum Leap (2013 to present). We are thankful for support from the Safeway Foundation, Bill Bowes Foundation, Quintiles Transnational Corporation, Johnson & Johnson, Genentech, Amgen, Eli Lilly, Pfizer, Eisai Company, the San Francisco Foundation, Give Breast Cancer the Boot, the Breast Cancer Research Foundation, Side Out Foundation, Harlan Family, the Avon Foundation for Women, Alexandria Real Estate Equities, National Cancer Institute Specialized Program of Research Excellence in Breast Cancer, American College of Radiology Imaging Network, Cancer and Leukemia Group B, National Cancer Institute Center for Bioinformatics, the NCI via a Big Data To Knowledge (BD2K) grant, and Bruce and Martha Atwater.
Author information
Authors and Affiliations
Contributions
D.M.W. and C.Y. contributed equally to this study. D.M.W, C.Y., and L.V., designed the study and wrote the manuscript. D.M.W. and C.Y. performed the qualifying biomarker analysis, and scored samples for PARPi7 and CIN70 signatures. A.S. contributed Bayesian simulation code; A.G. managed expression profiling and MP scoring/classification; C.P. and J.W. contributed protein biomarkers; T.M.S and S.L. contributed the BRCA1ness signature; L.B. supervised sample preparation in the I-SPY lab; G.H. managed concept proposals/approval; M.B., A.D., N.H., F.S., and M.P. served on the Executive Operations Committee for I-SPY 2 to oversee trial operations. All I-SPY 2 TRIAL Investigators treated patients and enrolled them in the trial. H.R. and O.O. chaperoned VC; L.E. and D.B. are trial P.I.’s, and contributed to study design and oversight.
Corresponding authors
Ethics declarations
Competing interests
L.V. is a co-inventor of the MammaPrint signature and a co-founder of Agendia, Inc. The remaining authors declare that they have no competing financial interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Clinical Trial Registration
I-SPY 2 Trial is registered in the www.ClinicalTrials.gov as NCT01042379.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wolf, D.M., Yau, C., Sanil, A. et al. DNA repair deficiency biomarkers and the 70-gene ultra-high risk signature as predictors of veliparib/carboplatin response in the I-SPY 2 breast cancer trial. npj Breast Cancer 3, 31 (2017). https://doi.org/10.1038/s41523-017-0025-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-017-0025-7
This article is cited by
-
Gene expression alterations predict the pathological complete response in triple-negative breast cancer exploratory analysis of the NACATRINE trial
Scientific Reports (2023)
-
Outcomes and clinicopathologic characteristics associated with disseminated tumor cells in bone marrow after neoadjuvant chemotherapy in high-risk early stage breast cancer: the I-SPY SURMOUNT study
Breast Cancer Research and Treatment (2023)
-
Predictive biomarkers for molecularly targeted therapies and immunotherapies in breast cancer
Archives of Pharmacal Research (2022)
-
Safety and efficacy of HSP90 inhibitor ganetespib for neoadjuvant treatment of stage II/III breast cancer
npj Breast Cancer (2022)
-
RAB5A expression is a predictive biomarker for trastuzumab emtansine in breast cancer
Nature Communications (2021)
