
Abstract
An understanding of connections between gut microbiome and liver has provided important insights into the pathophysiology of liver diseases. Since gut microbial dysbiosis increases gut permeability, the metabolites biosynthesized by them can reach the liver through portal circulation and affect hepatic immunity and inflammation. The immune cells activated by these metabolites can also reach liver through lymphatic circulation. Liver influences immunity and metabolism in multiple organs in the body, including gut. It releases bile acids and other metabolites into biliary tract from where they enter the systemic circulation. In this review, the bidirectional communication between the gut and the liver and the molecular cross talk between the host and the microbiome has been discussed. This review also provides details into the intricate level of communication and the role of microbiome in Gut-Liver-Brain, Gut-Liver-Kidney, Gut-Liver-Lung, and Gut-Liver-Heart axes. These observations indicate a complex network of interactions between host organs influenced by gut microbiome.
Similar content being viewed by others
Introduction
The role of gut microbial community (microbiome) in maintaining host health has gained a lot of attention in the recent past1. Scientific studies indicate links between dysbiosis or disturbance in microbiome and diseases that not only affect the gut, but also organs like brain, liver, lung, kidneys, etc. The pathophysiology of diseases effecting distal organs has often been associated with gastrointestinal discomfort or disorders. The crosstalk between the gut microbiome and distal organs is being increasingly recognized and host-microbiome interactions are being delineated piece by piece2,3. The increasing socio-economic burden of various diseases associated with changes in gut microbiome suggest the importance of understanding the molecular events facilitating such interactions.
Diseases affecting distal organ like liver (e.g., non-alcoholic/alcoholic fatty liver, non-alcoholic/alcoholic steatohepatitis, liver fibrosis/cirrhosis) are often seen to be associated with dysbiosis in gut microbiome4,5,6. Increase in fat cells in liver leads to a state known as fatty liver disease. Fatty liver may occur due to excessive intake of alcohol termed as ‘alcoholic fatty liver’ or may also be observed in individuals with no or negligible intake of alcohol termed as non-alcoholic fatty liver7. In both cases the observed pathological spectra may range from simple hepatic steatosis, steatohepatitis to liver cirrhosis7. In cases where an accumulation of fat leads to inflammation and damage, an advanced form of non-alcoholic fatty liver disease (NAFLD) called as Non-alcoholic steatohepatitis (NASH) is observed. Similarly, alcoholic steatohepatitis is the inflammatory state of Alcoholic liver disease (ALD)8. Since, these liver associated diseases are linked with dysbiosis in gut microbiome, it is important to understand the mechanisms involved in the cross-talk between gut and liver.
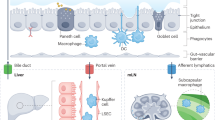
Certain mechanisms govern the tight bidirectional communication between the gut and liver (Fig. 1). For example, metabolic machinery of the host and resident gut microbiome metabolize several exogenous dietary and environmental components as well as endogenous substrates like amino acids and bile acid. The products generated during this process are carried to liver by portal vein, thereby influencing hepatic physiology9. Similarly, the immune cells activated by several dietary compounds as well as metabolites from gut microbiome can enter lymphatic system and modulate immune responses in distal organ like the liver9. On the other hand, the liver communicates with the gut through the release of bile acids and other metabolites into biliary tract of the systemic circulation9. The release of bile salts by liver also helps to control unrestricted gut microbial growth9.

Translocation of metabolites and pathogen associated molecular patterns (PAMPs) biosynthesized by microbes in the gut through the portal circulation to liver where they exert multiple effects on liver health condition.
Liver dysfunction influences immunity and metabolism of not only the gut, but also other organs. For example, brain malfunction due to Hepatic encephalopathy (HE) as well as kidney disorders are often observed in people with liver ailments10. Hepatic encephalopathy (HE) leads to an impaired brain function often observed in patients with advanced liver diseases. The factors like decreased metabolism of ammonia associated with liver failure have often been associated with occurrence of HE11. Similarly, cardiovascular diseases effecting heart and blood vessels (CVD) are seen to be associated with fatty liver and other liver disorders12. Thus, it is important to understand the mechanisms involved in the interaction of Gut–Liver axis with distal organs. The beneficial effects of certain probiotics and Fecal Microbial Transfer (FMT) in liver diseases as well as ailments (like HE and CVDs) which are linked to Gut-Liver interaction with other organs further highlight the importance of the close-knit interaction of gut microbiome with host physiology.
Gut microbiome and liver diseases
‘Non-alcoholic Fatty Liver Disease’ (NAFLD) and ‘Non-alcoholic Steatohepatitis’ (NASH) are liver ailments that have been associated with dysbiosis in gut microbiome and ‘Small Intestinal Bacterial Overgrowth’ (SIBO)13,14. Similar symptoms have also been observed in individuals suffering from ‘Alcoholic Liver Disease’ (ALD) due to excessive abuse of alcohol. The non- progressive form of these diseases (e.g. NAFLD) often involves fat accumulation in liver or steatosis, while the progressive form (e.g. NASH) is diagnosed by liver injury and inflammation (steatohepatitis)9. Analysis of stool samples of 57 patients showed lower levels of Prevotella and higher Bacteroides as well as Ruminococcus are seen in the gut of patients with NASH at stage 2 fibrosis or higher as compared to those in control subjects with fibrosis stage 115. This indicates that gut microbiome changes are associated with severity of disease (fibrosis stage 1 vs. stage 2 in this case). Whole genome sequencing of gut microbial community obtained using stool samples indicates higher abundances of Escherichia coli and Bacteroides vulgatus in the gut of NAFLD patients in early (72 patients) as well as advanced stages of fibrosis (14 patients)16. Similarly, pediatric subjects suffering with NASH are seen to have higher occurrence of genus Escherichia as compared to obese non-NASH subjects9. Multiple studies on gut samples (stool as well as biopsy) of human as well as animal systems have indicated that while patients with ALD have an increased number of bacteria belonging to the family Enterobacteriaceae in their gut, they have lower abundances of genera Lactobacillus and Bacteroidetes17,18. Alterations in gut microbial community have also been observed in cirrhosis patients. Interestingly, analysis of stool samples of 95 liver cirrhosis patients and 47 healthy controls indicated an invasion of oral microbes like Streptococcus and Veillonella into the small intestine is observed in cirrhosis patients19,20. Bacterial genera like Veillonella, Megasphaera, Dialister, Atopobium, and Prevotella are also found in higher abundances in biopsies of distal duodenum from 30 cirrhosis patients as compared to 28 healthy controls used in the study21.
In addition to ascertaining the role of gut microbiome in liver diseases certain studies have indicated potential use of probiotics as a therapy for chronic liver diseases. The outcomes of these therapeutic strategies differ in terms of their efficacy and long-term impacts as well as effects on host-microbiome balance have yet to be elucidated. some of these studies have been mentioned in Supplementary Table 1. While most of these studies indicate a decrease in pro-inflammatory markers like cytokines, Lipopolysaccharides (LPS) etc. as well as improvement in liver lesions, few studies also indicate that no significant change is observed with the intake of probiotics in liver disease (Supplementary Table 1). The efficacy of a synbiotic (combination of a prebiotic (fructo-oligosaccharides and a probiotic Bifidobacterium animalis subsp. lactis BB-12) administered for 10–14 months on the gut microbiome, liver fat and fibrosis was studied as a part of a placebo controlled study called Insyte. The study recruited 55 NAFLD patients with symbiotic administration while 49 were administered a placebo. The results indicated that although a gut microbiome change was observed, no significant changes in liver pathophysiology could be observed22. Thus, long-term randomized controlled trials with larger number of participants are needed to clearly understand the efficacy of probiotics/prebiotics or symbiotics in amelioration of liver disease.
Gut–Liver axis
Mechanisms of communication
Intestinal barrier integrity
The intestinal barrier, comprising of tightly bound cells, ensures selective transfer of nutrients and restricts the movement of pathogenic organisms from the gut lumen into the host system23. The gut microbiome influences gut barrier integrity by either maintaining immune signaling mechanisms or by producing metabolites like short chain fatty acids (SCFAs)23. Thus, disturbances in any of these factors can lead to an increase in gut permeability. For example, dysbiosis in gut microbiome in cases of inflammatory diseases or due to intake of high fat diet, alcohol and antibiotics can bring about loss of gut barrier integrity24,25.
A compromised gut barrier integrity is likely to lead to translocation of microorganisms and microbes- derived molecules into the portal system26. Under such condition, these microbes as well as their biosynthesized metabolites can translocate to the liver from where they can be carried through the portal system to distal organs, thereby causing their inflammation and injury (Fig. 1)26. Certain metabolites formed in the intestine may also directly interact with host factors in order to bring about exacerbation of liver disease27,28.
Transfer of microbes and microbes-derived metabolites through portal circulation
The intestinal dysbiosis is accompanied by loss of gut barrier integrity and transfer of pathogen associated molecular patterns (PAMPS) to the portal circulation29 (Fig. 1). This leads to induction of pattern recognition receptors (PRR) like TOLL-like receptors and NOD-like receptors in liver cells, which results in activation of pro-inflammatory signaling cascades, which in turn bring about local inflammatory responses29. Toll-like receptors are one category of PRRs which are suppressed in healthy liver conditions30 (Fig. 1). The delivery of pathogens or/and the molecules biosynthesized by them to the liver leads to activation of toll-like receptor (TLR) signaling. This leads to an increase in the production of cytokines like Tumor Necrosis Factor α (TNF α) and Interleukin-1β, both of which are known to act on bacteria and viruses. An elevated TLR signaling and expression of these cytokines due to prolonged stimulation can worsen hepatic injury in several liver diseases31. For example, NASH is known to affect the levels of TLR2 (lipopolysaccharide), TLR4 (peptidoglycan), TLR5 (flagellin), and TLR9 (bacterial DNA), all of which are activated by microbial antigens, thereby leading to inflammatory signaling cascades31.
Systemic levels of LPS, a component of Gram-negative bacteria, are higher in cases of liver diseases like NAFLD and NASH. Injecting LPS in mice model for NAFLD enhances liver injury as well as elevates the expression of pro-inflammatory cytokines32,33. Wild type mice fed with high fat diet develop steatohepatitis with an increased TLR4 expression and proinflammatory cytokines32,33. Further, TLR4 mutants are resistant to LPS induced release of pro-inflammatory cytokines, thus confirming role of TLR4 signaling in NAFLD and NASH30. Presence of bacterial DNA (which is higher in NASH patients) leads to elevated expression of TLR9 in NASH models30. Experiments with TLR9-deficient models fed with choline-deficient amino acid-defined (CDAA) diet show lesser inflammation, steatosis or fibrosis as compared to those in wild type model30. TLR9 signaling affects expression of inflammasome in macrophages, thereby resulting in formation of proinflammatory IL-1β and enhancement of the progression of hepatic injury in NASH30.
TLR2 interacts with such Gram-positive bacterial cell wall components like lipoteichoic acids and peptidoglycan. Based on the experimental observations on mice models, insulin resistance induced by high fat diet can be prevented by inhibition of TLR2 signaling34. Further, TLR2-deficient mice are seen to be resistant to ‘Choline Deficient Amino Acid’ (CDAA) induced steatohepatitis and have reduced expression of pro-inflammatory cytokines35. In contrast, TLR2-deficient mice on ‘Methionine-Choline Deficient’ (MCD) diet display similar or severe steatohepatitis as compared to wild type mice30. Although, MCD diet may lead to features of steatohepatitis, it helps in increasing the insulin sensitivity and promotes weight reduction. On the other hand, high fat and CDAA diets lead to weight gain and insulin resistance36.
TLR5 binds to bacterial flagellin and plays a protective role for the intestine. TLR5-knockouts develop not only obesity and steatosis, but also display an imbalance in the gut microbiome30. Further, transfer of gut microbial communities from TLR5 knockout mice to WT germ-free mice gives rise to metabolic syndrome30. Thus, an interplay of gut microbiome and TLR5 probably contribute towards metabolic syndrome pathophysiology.
Metabolites transferred from gut through systemic and portal circulation
Several metabolites biosynthesised in the gut exert multiple effects in liver (depicted in Fig. 2).

Metabolites biosynthesized by the microbiome and their impact on onset of liver disease.
Trimethylamine and trimethylamine oxide
Choline, a dietary macronutrient, is involved in multiple physiological processes in liver, which include phospholipid biosynthesis (phosphatidyl choline and other membrane lipids), cholesterol metabolism and enterohepatic circulation of bile and lipids9. Deficiency of choline in the diet leads to impairment in liver and brain function as well as metabolic processes and muscle movement. Free choline is absorbed by small intestine which then either gets integrated into the membrane or is transferred to liver where it is likely to get converted to betaine, lecithin, etc37. Lesser availability of choline leads to accumulation of triglycerides (due to lower formation of phosphatidyl choline by the host) in liver, a factor which has been associated with NASH and also in the manifestation of NAFLD38. While a choline-deficient diet induces steatohepatitis, excess choline in diet (exceeding the absorptive capacity of host) moves to the large intestine to get assimilated to Trimethylamine (TMA) by gut microbes (Supplementary File 1–1.1)39. Another route for biosynthesis of TMA involves degradation of carnitine obtained from dietary sources like red meat and dairy products (Supplementary File 1–1.1). The TMA thus formed is transferred to liver through portal circulation and gets converted to Trimethylamine Oxide (TMAO), a component which has been implicated in multiple cardiometabolic disorders, hepatic diseases, etc39.
Short-chain fatty acids
Metabolites like short chain fatty acids (SCFAs) primarily include butyrate, propionate and acetate and are formed in the large intestine as a result of dietary assimilation of polysaccharides, resistant starch, fiber, etc40. The SCFAs work as nutrient and energy source for intestinal epithelium and act as precursors for lipogenesis and gluconeogenesis41. The butyrate level in the gut helps in maintaining the intestinal integrity as well as permeability42. A decrease in butyrate is observed in several liver ailments and alcohol influenced liver injuries43.
SCFAs bind and activate G-protein coupled receptors (GPCRs) GPR41 and GPR4344. This activation influences peptide-YY secretion as well as causes inhibition of gut motility, thereby increasing the nutrient utilization and yielding of energy. The signaling across GPR41 and GPR43 leads to secretion of GLP1 which in turn reduces the food intake as well as emptying of gastric tract44. Further, GPCR signaling also affects regulation of fatty acid oxidation and insulin sensitivity by hepatocytes. Apart from this, GPR43 activation also leads to inhibition of lipolysis and reduced plasma fatty acids44.
In addition to GPCR-based signaling, SCFAs can reach the liver through the portal circulation and can have either beneficial or deleterious effects on the liver. For example, increased acetate can be channeled to fatty acid biosynthesis pathway, thereby leading to triglyceride accumulation which has often been correlated to liver ailments41,45. Similarly, propionate which acts as a precursor for gluconeogenesis has also been associated to NAFLD41,46. On the other hand, butyrate may utilize multiple mechanisms to reduce the pathophysiology associated with liver diseases. For instance, butyrate can activate ‘AMP activated Protein Kinase’ (AMPK), which in turn reduces inflammation and influences glucose as well as lipid metabolism. AMPK further suppresses lipogenic genes41. AMPK expression in liver (regulated by butyrate) reduces insulin resistance and obesity. Butyrate can also function as inhibitors of ‘Histone deacetylases’ (HDACs) which can prevent development of liver diseases like NASH and NAFLD at epigenetic level41.
Administration of SCFAs has beneficial effects like reduction in hepatic steatosis and insulin resistance47. On the contrary, while enrichment of formate and acetate are found in adult subjects at advanced stages of NAFLD, butyrate and propionate are seen to be higher in mild NAFLD16. These differences in overall functioning of SCFAs in liver diseases may be affected by factors like diet and environment.
Ethanol and acetaldehyde
Ethanol is absorbed mostly in stomach and small intestine via diffusion by gastrointestinal mucosa48. Majority of ethanol in large intestine is obtained from systemic circulation. Some of the gut microbes can convert ethanol to acetaldehyde and to lesser extent acetate using alcohol metabolizing enzymes such as alcohol dehydrogenase49. Liver also expresses enzymes for ethanol metabolism in response to systemic ethanol content50.
Interestingly, while certain small amounts of ethanol are observed in the bloodstream of subjects who do not consume alcohols, pediatric subjects with NASH are seen to possess higher serum ethanol levels as compared to obese children without NASH51. These levels of ethanol could be contributed by metabolism by the gut microbiome52. Consumption of ethanol is likely to add to the pathophysiology of liver diseases (NASH, NAFLD, etc.) since it may cause not only an increase in intestinal permeability, but also may assist in production of inflammatory cytokines25. Endogenous ethanol can increase availability of acetate, a precursor of triglyceride formation through mechanisms involving inhibition of TCA cycle53. Ethanol oxidation by CYP2E1 can lead to production of free radicals which is likely to elevate inflammation54. Apart from this, ethanol can be metabolized to acetaldehyde which may either disrupt the tight junctions in the intestinal epithelium55 or may have oxidant-dependent cytotoxic and metabolic effect on intestinal goblet like cells56.
Bile aids
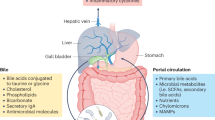
Oxidation of cholesterol to form primary bile acids, cholic acid, and chenodeoxycholic acid takes place in the hepatocytes through a multi-step process57. These bile acids are further conjugated to glycine or taurine which function as fat emulsifiers in the duodenum for solubilizing fats57. The released bile acids enter canaliculi through an export pump and move to the gallbladder where they get stored58. The bile acids are released into the duodenum upon consumption of food as a response to increase in production of cholecystokinin57. The intestinal microbiome converts these primary bile acids to secondary bile acids such as deoxycholic, lithocholic, and ursodeoxycholic acids59.
Chenodeoxycholic acid (CDCA) activates FXR signaling, which in turn helps in not only regulating glucose levels and metabolism (increase insulin sensitivity, glycogen synthesis and inhibit gluconeogenic genes), but also influences cholesterol transport, inhibits lipogenesis and enhances fatty acid oxidation60. Bile acids also lead to reduction in expression of lipogenic genes as well as help in reducing triglyceride levels by activating FXR and the pathway involving small heterodimer partner (SHP) and the sterol regulatory element-binding protein 1 (SREBP-1)61. FXR also increases the proliferator-activated receptor alpha (PPARalpha) expression which in turn exerts anti-inflammatory effects and regulates lipid as well as glucose metabolism62.
While most (95%) of the bile acids are reabsorbed in the distal part of ileum and transported back to liver through portal vein, the remaining gets deconjugated by gut microbes and excreted out in the feces57. A small fraction of reabsorbed bile acids is likely to escape uptake into liver and reach the peripheral tissues through systemic circulation. The changes observed in bile acids in liver disease have been detailed in Supplementary File 1–1.2.
Immune surveillance by liver affects distal organs
Liver plays an important role in immune regulation as well as immunomodulation and possesses almost 80% of all tissue-based macrophages63. It affects the innate immunity in other organs and is responsible for secretion of inflammation mediators like serum IL-6 and the acute phase protein CRP64,65 in. Thus, it is important to understand the role of gut microbiome, the biosynthesized metabolites in liver and the overall effects on distal organs.
Gut–Liver–Brain axis
Hepatic encephalopathy (HE), linking brain function with liver diseases, involves a vast range of neurological and psychiatric abnormalities, ranging from subclinical alterations to coma11. HE has often been observed as one of the major complications in individuals with hepatic insufficiency which includes diseases like liver cirrhosis and fibrosis10. HE is observed in almost 30–45% of patients with liver cirrhosis and 24–53% of patients with transjugular intrahepatic portosystemic shunt (TIPS)66. Having seen the link between the gut and liver, it is important to view this connection with respect to brain pathophysiology as observed in HE, i.e. Gut–Liver–Brain axis
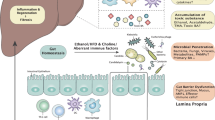
Gut microbial products like ammonia and oxindole, obtained after metabolism of amino acids, are deleterious for brain12 (Fig. 3). Oxindole functions as a sedative by acting as a ligand for voltage operated sodium channels in the brain. Ammonia functions by influencing neurotransmission, pH, membrane potential, astrocyte swelling, etc. Liver diseases like cirrhosis are often associated with insufficiency in detoxification of ammonia and indole derivatives by the liver10,11. The reduction in clearance of ammonia from portal vein in cirrhotic patients is accompanied by higher ammonia uptake by brain astrocytes which has been associated to neurological symptoms (Fig. 3). Ammonia is primarily formed in gastrointestinal tract by the action of glutaminase or urease enzymes as well as metabolism of other nitrogen-rich compounds67. This ammonia gets into portal circulation and reaches the liver where it is further detoxified by urea cycle. Individuals with portosystemic shunts or liver failure often have compromised liver detoxification abilities which lead to excessive accumulation of nitrogen wastes in systemic circulation (Fig. 3). Excess ammonia is likely to cross blood brain barrier and be absorbed into astrocytes where it possibly gets converted to glutamine12. Glutamine thus formed may cause oxidative or osmotic stress and astrocyte swelling, further manifesting as cerebral edema and increased GABAergic activity (Fig. 3)68.

Impairment of liver urea cycle in liver disease condition leads to increase in uremic toxins and ammonia which reach the brain and affect neurotransmitter signaling and astrocyte swelling.
Systemic inflammation and sepsis have also been considered as factors which are involved in exacerbation of HE69. HE patients often show higher occurrence of inflammatory cytokines like IL-6, IL-18 and TNFα11. The presence of systemic and local inflammation has been shown to augment the effect of hyperammonemia in HE. Proinflammatory cytokines can be produced in brain, thereby giving rise to neuroinflammation70. Systemic inflammation can occur in case of liver cirrhosis due to multiple factors, one of them being increase in intestinal permeability which can lead to translocation of bacteria and their products into systemic circulation68. These bacteria along with their PAMPs help in activating the immune response with release of pro-inflammatory cytokines. The loss in gut barrier integrity in case of liver failure can happen due to factors like reduction in formation of tight junction proteins, reduction in SCFA levels, dysbiosis in gut, endotoxemia, etc.71. Systemic inflammation and hyperammonemia may lead to activation of resident macrophages in central nervous system called microglial cells72. This may result in formation of brain derived proinflammatory cytokines and result in neuronal death.
Gut–Liver–kidney axis
Hepatic failure is often linked to kidney dysfunction or chronic kidney disease (CKD)73. A reduction in the estimated glomerular filtration rate (eGFR) of <60 mL/min for more than 3 months is considered as the diagnosis of CKD in cirrhosis. A study in 2019 showed that 46.8% of hospitalized patients with cirrhosis were diagnosed with CKD74. Similarly, there is a correlation between loss of kidney function and dyslipidemia75. NAFLD leads to lipid accumulation, often involved in aggravating insulin resistance, inflammation, hypertension and obesity, which in turn may influence kidney dysfunction76. Increase in biosynthesis of pro-inflammatory, pro-thrombotic factors in NAFLD may contribute towards renal damage77. Further, changes in the expression of hepatic lipase lead to high triglyceride levels. Levels of Apolipoprotein B-100 containing lipoproteins biosynthesized in liver also show abnormalities in CKD patients78. Systemic inflammation during CKD can cause multiple effects and contribute to NAFLD.
Gut microbiome plays an important role in the connection between liver and kidneys (‘Gut–Liver–Kidney axis’) depicted in Fig. 4a. Dysbiosis in gut microbiome or high level of proteins in diet lead to high protein fermentation in the gut, thereby giving rise to formation of ammonia, indole, p-cresol, etc.79 (Fig. 4a). While indole is formed by fermentation of tryptophan by intestinal bacteria, p-cresol is formed by decarboxylation of 4-hydroxyphenylacetic acid which is a product of tyrosine degradation by host enzyme80,81. These products are absorbed by intestinal mucosa and taken to the liver where they are further modified by host sulfotransferases or glucoronotransferases to give rise to indoxyl-sulfate, indoxyl glucuronate, p-cresyl-sulfate, and p-cresyl-glucuronate, all of which are uremic toxins82. These toxins move into systemic circulation and are cleared from the system by renal filtration. Such toxins also affect the progression of renal ailments and are observed to be elevated in patients with CKD and end stage renal disease (ESRD)82 (Fig. 4a). The uremic toxins are expected to act as agonists of aryl hydrocarbon receptor (AhR) and influence release of pro-inflammatory cytokines as well as increase inflammation and oxidative stress83. The observed alterations in expression of genes like hepatic cytochrome P450 (CYP) and drug transporter function are expected since these genes have AhR sites on their promoters84. This leads to changes in drug metabolism in hepatocytes (Fig. 4a). In patients with CKD and advanced liver disease or cirrhosis, the activity of enzymes responsible for modification in liver (sulfotransferases) is lowered and could contribute towards reduction in the uremic toxin formation82 (Fig. 4b). Thus, the amount of uremic toxins in the body in case of kidney damage is also influenced by the liver condition. Further, uremic toxins could have regulatory effects in liver (Fig. 4b).

a Gut–Liver–kidney axis without liver damage: Oxindole and cresol produced by gut microbiome are converted to uremic toxins in liver. The uremic toxins reach kidneys through portal circulation. b Gut–Liver–Kidney axis with liver damage: Oxindole and cresol produced in the gut are not converted to uremic toxins in liver.
TMA produced by gut microbiome from choline metabolism is converted to TMAO in liver by flavin-like monooxygenases. The TMAO is carried to kidneys by systemic circulation and cleared by glomerular filtration85. TMA and TMAO levels are increased in individuals with renal diseases, CKD, ‘End Stage Renal Disease’ (ESRD), fibrosis, etc86. Further, a higher presence of TMAO is associated with liver ailments like NAFLD, NASH, liver cirrhosis, etc87. TMAO helps in suppression of bile acid mediated farnesoid X receptor signaling in liver, which in turn leads to aggravation in liver steatosis87. This indicates that change in TMAO levels during kidney dysfunction may also influence the physiology of liver.
Gut–Liver–Lung axis
An increase in innate immunity concomitant with an increase in inflammatory markers like C-reactive protein (CRP) has been associated with deterioration in lung function and exacerbation of diseases like Chronic Obstructive Pulmonary Disease (COPD)88. COPD refers to a lung disease that causes airflow blockage to the lungs and leads to breathing-related problems. The prevalence of steatosis, NASH and fibrosis in COPD patients have been reported to be 41.4%, 36.9% and 61.3%, respectively89. The higher innate immune response as well as pro-inflammatory markers like IL-6 have been correlated to pathophysiology of lung ailments88. The liver works as a site of immunomodulation with the mevalonate pathway playing a major role. Statins which inhibits the mevalonate pathway in liver is observed to reduce the lung damage90. Lovastatin is seen to reduce deleterious effects of macrophage activation in mouse models. Atorvastatin is shown to reduce not only lung inflammatory cells by 30–60%, but also expression of pro-inflammatory genes including reduction in CRP and IL-691.
In addition to mevalonate pathway, liver plays a crucial role in building up an innate immune response in terms of recruitment of macrophages and the neutrophils at the site of lung injury. Studies on mouse model indicate its role in increasing release of IL-6 and acute phase proteins by alveolar macrophages92. These proteins are likely to generate chronic inflammation leading to activation of innate immunity in circulation as well as in lungs (termed as innate immune hyper-responsiveness), especially in cases of injury as seen in diseases causing lung damage. Thus, the “Liver–Lung axis” links the innate immune responsiveness and lung injury with the innate immune response regulated by the liver and the mevalonate pathway88.
To assess possible link between diet and respiratory diseases, outcomes based on a study on ~120,000 subjects indicate significant reduction in occurrence of COPD with intake of high-fiber diet comprising of whole food grains93. Similarly, intake of whole grains shows substantial improvements in FEV1 (Forced Expiratory Volume in 1 s) of smokers (200 ml change across diet quartiles) as compared to non-smokers (50 ml change across dietary quartiles)94. Evidences indicate the effect of dietary fibers on immunomodulation of innate immune response. Comparison of effects of dietary fiber intake on specific causes of death suggest dietary fiber’s role in reduction in mortality in individuals suffering from respiratory ailments95. Similarly, 50–60% reduction in mortality due to intake of high-fiber diet comprising of whole grains (HR = 0.47 for men and 0.40 for women) has been observed in a European study on >450,000 individuals96. A study on a cohort of 35,339 Swedish women showed that a long-term intake of dietary fiber could be associated with a 30% lower risk of COPD97. Epidemiological and clinical studies also suggest role of high-fiber diets in reducing systemic inflammation and leading to decrease markers of inflammation like CRP and IL-698.
Looking into the ‘Gut–Liver–Lung axis’, one of the possible ways by which dietary fiber provides benefit is by stimulating the growth of beneficial bacteria in gut which help in biosynthesis of SCFAs through fermentation3. Some SCFAs get absorbed and enter portal circulation, thereby affecting organs like liver99. SCFAs function by modulating innate immune activation. High-fiber diet is seen to reduce pulmonary inflammation in murine models. SCFAs influence the migration of neutrophils and macrophages via GPCR activation which helps in reducing pulmonary inflammatory response44. Furthermore, SCFAs inhibit the HMG-CoA reductase which catalyzes the rate limiting step of mevalonate pathway41. This inhibition enables reduction in inflammatory markers and lowering of the innate immune response. HMG-CoA reductase inhibition in the liver ‘dampens’ the innate immune response and lowers serum levels of IL-6 through the IL-6 trans-signaling pathway88. This leads to a downstream inhibitory effect on the pro-inflammatory transcription factors NF-ĸB and signal transducer and activator of transcription88.
Gut–Liver–Heart axis
Liver diseases like NAFLD, NASH, and cirrhosis, which have been associated with changes in gut microbiome, also correlate with the occurrence cardiovascular (CV) ailments comprising disorders of heart and blood vessels (‘Gut–Liver–Heart axis’)100. Non-alcoholic fatty liver disease (NAFLD) is associated with a higher risk of cardiovascular disease (CVD) which includes coronary heart disease (CHD), heart failure, stroke, and arrhythmia101. A follow up study on 285 individuals with biopsy proven NAFLD and no incidence of CVD showed an occurrence of a cardiovascular event in 9.1% of these advanced stage individuals within 5 years of follow up102. Exposure to lipopolysaccharide and its binding to TLR4 lead to an inflammatory immune response with release of pro-inflammatory cytokines103. This process promotes LDL oxidation, formation of atherosclerotic plaques and thrombogenesis104.
Dietary intake which are higher in choline, betaine or carnitine (e.g. red meat) leads to formation of TMA by gut bacteria, which further gets converted to TMAO in liver39,87,105. The increase in TMAO levels is associated to liver as well as CV events86. Thus, TMAO can be considered as marker for a deteriorating liver condition or Cardiovascular health86. TMAO concentrations have been related to atherosclerosis which is one of the major causes of CVDs as well as ‘Major adverse cardiovascular events’ (MACE) including myocardial infarction, stroke, etc.86. Higher TMAO levels and expression of pro-inflammatory cytokines (TNF-α and IL-1β) are observed to be accompanied with cardiac dysfunction in mouse models106. The inhibition of choline TMA lyase enzyme by chemicals like 3,3-dimethyl-1-butanol (DMB) can prevent increase in TMA levels as well as other outcomes107.
There exists a link between endothelial dysfunction and TMAO levels. TMAO treatment carried out on human monocytic THP-1 cells and human umbilical vein endothelial cells (HUVEC) reveal an increase in monocyte adhesion which lead to increased expression of VCAM-1108. Additionally, lipid metabolism is also regulated by TMAO which alters cholesterol and sterol metabolism106. Catabolism of cholesterol involves bile acid synthesizing enzyme Cyp7a1 catalyzing the rate limiting step. TMAO lowers the expression of this enzyme which has been associated with atherosclerosis109. Supplementation of choline, carnitine or TMAO may decrease reverse cholesterol transport. Further, TMAO influences the increase in expression of CD36 and SR-A1 (scavenger receptors) which leads to lipid accumulation and foam cell formation106,110. These effects are induced by oxidative modification of LDL in presence of TMAO. The inhibition of MAPK by inhibitors leads to a reduction in expression of CD36 as well as foam cell formation, indicating action of MAPK/JNK pathway in atherosclerosis induced by TMAO111.
Some inconsistencies regarding link of plasma TMAO levels and CVDs still exist. For example, short- and long-term higher plasma TMAO levels are observed in people after bariatric surgery. The result is unexpected as high TMAO concentrations increase the CVD risk while the aim of bariatric surgery is to reduce CVD risk39. However, recording their diet and gut microbiome could have thrown some light on whether TMAO levels were found to be higher in subjects as a result of a surgery-induced change in gut microbiome or due to a greater ingestion of carnitine (a TMA precursor) which is often promoted as a weight loss inducing supplement.
There are also certain contrasting observations regarding role of TMAO in CVDs. Although, TMAO is shown to correlate with inflammatory markers and endothelial dysfunction, some studies indicate such associations only in case of HIV and type-II diabetes39. Few studies also indicate no significant correlation of TMAO levels with inflammatory marker CRP. Protective role of TMAO in CVDs have also been reported. After carnitine supplementation, improvement is seen in some CVDs despite an increase in TMAO and TMA112. Food items like marine fish contain high levels of TMAO which are observed in circulation after dietary intake. Despite that, studies on mice show that supplementation of fish oil along with a high fat diet alleviate damage caused by TMAO including increased glucose tolerance and inflammation of adipose tissue113.
In summary, the Gut–Liver axis refers to bidirectional communication between gut, its microbiome and the liver. The metabolites produced by gut microbiome are connected with liver through systemic circulation, portal circulation and the bile duct. While the metabolites produced in the gut influence immunity, metabolism and bile acid production, the bile acids produced in liver in turn regulate the gut microbial composition as well as gut epithelial barrier integrity. Therefore, a dysbiosis in gut microbiome not only leads to a change in the bile acid pool within the host, but also often been observed in liver related pathophysiologies like NAFLD, NASH, ALD, etc. Further, since some gut bacteria are capable of metabolizing bile acid, the bile acid pool determines and influences the composition of gut microbiome. The shifting level of bile acids impacts the intestinal integrity and metabolism by affecting FXR signaling. Exposure of liver immune cells to metabolites like TMAO produced by gut bacteria can increase liver inflammation. Further, the liver regulates the innate immunity as well as metabolism of various toxins and metabolites in other organs. In other words, a deterioration in liver condition can also impact the metabolism signaling and immunity in other important host organs. Hence, the Gut–Liver axis can be extended to distal organs like Gut–Liver–Brain, Gut–Liver–Kidney, Gut–Liver-Heart and Gut-Liver-Lung axes.
Findings from the Gut-Liver-X (X being Brain or Kidney or Heart or Lung) axes indicate potential of utilizing gut microbiome as diagnostic and therapeutic strategy for early detection and management of not only liver diseases, but also diseases effecting other organs (e.g., chronic kidney disease, hepatic encephalopathy, cardiovascular ailments, respiratory obstructions, etc.). Identifying microbiome signatures which can be indicative of different health conditions is an active area of research. An understanding of Gut–Liver axis and interactions with distal organs can further help in identifying probiotic and fecal transplant strategies as preventive therapeutic regimes for liver ailments. Although certain studies have indicated potential use of probiotics as a therapy for chronic liver diseases, long-term impacts as well as effects on host-microbiome balance have yet to be elucidated (Supplementary Table 1). Clinical trials with standardized dosage of probiotics and extended duration of administration along with regular follow-ups are necessary to confirm the efficacy of the probiotics in manipulating the Gut–Liver axis as well as understanding their impacts on other organs like brain, kidney, lung and heart.
Data availability
All required data has been provided within the manuscript.
References
Schmidt, T. S. B., Raes, J. & Bork, P. The human gut microbiome: from association to modulation. Cell 172, 1198–1215 (2018).
Kho, Z. Y. & Lal, S. K. The human gut microbiome – a potential controller of wellness and disease. Front. Microbiol. 9, 1835 (2018).
Anand, S. & Mande, S. S. Diet, microbiota and gut-lung connection. Front. Microbiol. 9, 2147 (2018).
Bajaj, J. S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 16, 235–246 (2019).
Kirpich, I. A., Marsano, L. S. & McClain, C. J. Gut-liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 48, 923–930 (2015).
Milosevic, I. et al. Gut-liver axis, gut microbiota, and its modulation in the management of liver diseases: a review of the literature. Int. J. Mol. Sci. 20, 395 (2019).
Toshikuni, N., Tsutsumi, M. & Arisawa, T. Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J. Gastroenterol. 20, 8393–8406 (2014).
Gao, B. & Tsukamoto, H. Inflammation in alcoholic and nonalcoholic fatty liver disease: friend or foe? Gastroenterology 150, 1704–1709 (2016).
Tripathi, A. et al. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 15, 397–411 (2018).
vom Dahl, S., Kircheis, G. & Haussinger, D. Hepatic encephalopathy as a complication of liver disease. World J. Gastroenterol. 7, 152–156 (2001).
Ferenci, P. Hepatic encephalopathy. Gastroenterol. Rep. 5, 138–147 (2017).
Mancini, A., Campagna, F., Amodio, P. & Tuohy, K. M. Gut: liver: brain axis: the microbial challenge in the hepatic encephalopathy. Food Funct. 9, 1373–1388 (2018).
Augustyn, M., Grys, I. & Kukla, M. Small intestinal bacterial overgrowth and nonalcoholic fatty liver disease. Clin. Exp. Hepatol. 5, 1–10 (2019).
Fitriakusumah, Y. et al. The role of Small Intestinal Bacterial Overgrowth (SIBO) in Non-alcoholic Fatty Liver Disease (NAFLD) patients evaluated using Controlled Attenuation Parameter (CAP) Transient Elastography (TE): a tertiary referral center experience. BMC Gastroenterol. 19, 43 (2019).
Boursier, J. et al. The severity of NAFLD is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–775 (2016).
Loomba, R. et al. Gut microbiome based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 25, 1054–1062.e5 (2017).
Hartmann, P., Seebauer, C. T. & Schnabl, B. Alcoholic liver disease: the gut microbiome and liver cross talk. Alcohol. Clin. Exp. Res. 39, 763–775 (2015).
Szabo, G. Gut-liver axis in alcoholic liver disease. Gastroenterology 148, 30–36 (2015).
Ling, Z., Chen, D., Liu, Y., Yang, F. & Li, L. Disorganized gut microbiome contributed to liver cirrhosis progression: a meta-omics-based study. Front. Microbiol. 9, 3166 (2018).
Liu, G., Zhao, Q. & Wei, H. Characteristics of intestinal bacteria with fatty liver diseases and cirrhosis. Ann. Hepatol. 18, 796–803 (2019).
Chen, Y. et al. Dysbiosis of small intestinal microbiota in liver cirrhosis and its association with etiology. Sci. Rep. 6, 34055 (2016).
Scorletti, E. et al. Synbiotics alter fecal microbiomes, but not liver fat or fibrosis, in a randomized trial of patients with nonalcoholic fatty liver disease. Gastroenterology 158, 1597–1610.e7 (2020).
Suzuki, T. Regulation of the intestinal barrier by nutrients: the role of tight junctions. Anim. Sci. J. 91, e13357 (2020).
Rohr, M. W., Narasimhulu, C. A., Rudeski-Rohr, T. A. & Parthasarathy, S. Negative effects of a high-fat diet on intestinal permeability: a review. Adv. Nutr. 11, 77–91 (2020).
Bishehsari, F. et al. Alcohol and gut-derived inflammation. Alcohol Res. Curr. Rev. 38, 163–171 (2017).
Seki, E. & Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: crosstalk between the liver and gut. J. Physiol. 590, 447–458 (2012).
Filliol, A. et al. RIPK1 protects hepatocytes from Kupffer cells-mediated TNF-induced apoptosis in mouse models of PAMP-induced hepatitis. J. Hepatol. 66, 1205–1213 (2017).
Wu, X., Wang, Y., Wang, S., Xu, R. & Lv, X. Purinergic P2X7 receptor mediates acetaldehyde-induced hepatic stellate cells activation via PKC-dependent GSK3β pathway. Int. Immunopharmacol. 43, 164–171 (2017).
Arab, J. P., Martin-Mateos, R. M. & Shah, V. H. Gut-liver axis, cirrhosis and portal hypertension: the chicken and the egg. Hepatol. Int. 12, 24–33 (2018).
Miura, K. & Ohnishi, H. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J. Gastroenterol. 20, 7381–7391 (2014).
Kiziltas, S. Toll-like receptors in pathophysiology of liver diseases. World J. Hepatol. 8, 1354–1369 (2016).
Fukunishi, S. et al. Lipopolysaccharides accelerate hepatic steatosis in the development of nonalcoholic fatty liver disease in Zucker rats. J. Clin. Biochem. Nutr. 54, 39–44 (2014).
Nakanishi, K. et al. Exogenous administration of low-dose lipopolysaccharide potentiates liver fibrosis in a choline-deficient l-amino-acid-defined diet-induced murine steatohepatitis model. Int. J. Mol. Sci. 20, 2724 (2019).
Himes, R. W. & Smith, C. W. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB J. 24, 731–739 (2010).
Miura, K. et al. Toll-like receptor 2 and palmitic acid cooperatively contribute to the development of nonalcoholic steatohepatitis through inflammasome activation in mice. Hepatology 57, 577–589 (2013).
Van Herck, M. A., Vonghia, L. & Francque, S. M. Animal models of nonalcoholic fatty liver disease—a starter’s guide. Nutrients 9, 1072 (2017).
Wiedeman, A. M. et al. Dietary choline intake: current state of knowledge across the life cycle. Nutrients 10, 1513 (2018).
Lombardi, B., Ugazio, G. & Raick, A. Choline-deficiency fatty liver: relation of plasma phospholipids to liver triglycerides. Am. J. Physiol. -Leg. Content 210, 31–36 (1966).
Janeiro, M. H., Ramírez, M. J., Milagro, F. I., Martínez, J. A. & Solas, M. Implication of trimethylamine N-oxide (TMAO) in disease: potential biomarker or new therapeutic target. Nutrients 10, 1398 (2018).
Topping, D. L. & Clifton, P. M. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 81, 1031–1064 (2001).
den Besten, G. et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340 (2013).
Silva, Y. P., Bernardi, A. & Frozza, R. L. The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. 11, 25 (2020).
Juanola, O. et al. Circulating levels of butyrate are inversely related to portal hypertension, endotoxemia, and systemic inflammation in patients with cirrhosis. FASEB J. 33, 11595–11605 (2019).
Priyadarshini, M., Kotlo, K. U., Dudeja, P. K. & Layden, B. T. Role of short chain fatty acid receptors in intestinal physiology and pathophysiology. Compr. Physiol. 8, 1091–1115 (2018).
Aragonès, G., González-García, S., Aguilar, C., Richart, C. & Auguet, T. Gut Microbiota-Derived Mediators as Potential Markers in Nonalcoholic Fatty Liver Disease. BioMed Research International 2019, e8507583 (2019).
Jasirwan, C. O. M., Lesmana, C. R. A., Hasan, I., Sulaiman, A. S. & Gani, R. A. The role of gut microbiota in non-alcoholic fatty liver disease: pathways of mechanisms. Biosci. Microbiota Food Health 38, 81–88 (2019).
Deng, M. et al. SCFAs alleviated steatosis and inflammation in mice with NASH induced by MCD. J. Endocrinol. 245, 425–437 (2020).
Bode, C. & Bode, J. C. Alcohol’s role in gastrointestinal tract disorders. Alcohol Health Res. World 21, 76–83 (1997).
Salaspuro, M. Microbial metabolism of ethanol and acetaldehyde and clinical consequences. Addict. Biol. 2, 35–46 (1997).
Zakhari, S. Overview: How is alcohol metabolized by the body?. Alcohol Res. Health 29, 245–254 (2006).
Clemente, M. G., Mandato, C., Poeta, M. & Vajro, P. Pediatric non-alcoholic fatty liver disease: recent solutions, unresolved issues, and future research directions. World J. Gastroenterol. 22, 8078–8093 (2016).
Purohit, V. et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol 42, 349–361 (2008).
Grunnet, N. & Kondrup, J. The effect of ethanol on the beta-oxidation of fatty acids. Alcohol. Clin. Exp. Res. 10, 64S–68S (1986).
French, S. W. The importance of CYP2E1 in the pathogenesis of alcoholic liver disease and drug toxicity and the role of the proteasome. Subcell. Biochem. 67, 145–164 (2013).
Elamin, E. et al. Effects of ethanol and acetaldehyde on tight junction integrity: in vitro study in a three dimensional intestinal epithelial cell culture model. PLoS ONE 7, e35008 (2012).
Elamin, E., Masclee, A., Troost, F., Dekker, J. & Jonkers, D. Cytotoxicity and metabolic stress induced by acetaldehyde in human intestinal LS174T goblet-like cells. Am. J. Physiol. Gastrointest. Liver Physiol. 307, G286–G294 (2014).
Chiang, J. Y. L. Bile acid metabolism and signaling. Compr. Physiol. 3, 1191–1212 (2013).
Boyer, J. L. Bile formation and secretion. Compr. Physiol. 3, 1035–1078 (2013).
Molinero, N., Ruiz, L., Sánchez, B., Margolles, A. & Delgado, S. Intestinal bacteria interplay with bile and cholesterol metabolism: implications on host physiology. Front. Physiol. 10, 185 (2019).
Prawitt, J., Caron, S. & Staels, B. Bile acid metabolism and the pathogenesis of type 2 diabetes. Curr. Diab. Rep. 11, 160–166 (2011).
Watanabe, M. et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Invest. 113, 1408–1418 (2004).
Pineda Torra, I. et al. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol. Endocrinol. 17, 259–272 (2003).
Bogdanos, D. P., Gao, B. & Gershwin, M. E. Liver immunology. Compr. Physiol. 3, 567–598 (2013).
Norris, C. A. et al. Synthesis of IL-6 by hepatocytes is a normal response to common hepatic stimuli. PLoS ONE 9, e96053 (2014).
Nehring, S. M., Goyal, A. & Patel, B. C. C Reactive Protein. In StatPearls (StatPearls Publishing, 2022).
Mandiga, P., Foris, L. A. & Bollu, P. C. Hepatic Encephalopathy. In StatPearls (StatPearls Publishing, 2022).
Liu, J., Lkhagva, E., Chung, H.-J., Kim, H.-J. & Hong, S.-T. The pharmabiotic approach to treat hyperammonemia. Nutrients 10, 140 (2018).
Campion, D. et al. Dietary approach and gut microbiota modulation for chronic hepatic encephalopathy in cirrhosis. World J. Hepatol. 11, 489–512 (2019).
Tranah, T. H., Vijay, G. K. M., Ryan, J. M. & Shawcross, D. L. Systemic inflammation and ammonia in hepatic encephalopathy. Metab. Brain Dis. 28, 1–5 (2013).
Jayakumar, A. R., Rama Rao, K. V. & Norenberg, M. D. Neuroinflammation in hepatic encephalopathy: mechanistic aspects. J. Clin. Exp. Hepatol. 5, S21–S28 (2015).
Nicoletti, A. et al. Intestinal permeability in the pathogenesis of liver damage: from non-alcoholic fatty liver disease to liver transplantation. World J. Gastroenterol. 25, 4814–4834 (2019).
Zemtsova, I. et al. Microglia activation in hepatic encephalopathy in rats and humans. Hepatology 54, 204–215 (2011).
Bucsics, T. & Krones, E. Renal dysfunction in cirrhosis: acute kidney injury and the hepatorenal syndrome. Gastroenterol. Rep. 5, 127–137 (2017).
Kumar, R., Priyadarshi, R. N. & Anand, U. Chronic renal dysfunction in cirrhosis: a new frontier in hepatology. World J. Gastroenterol. 27, 990–1005 (2021).
Mikolasevic, I., Žutelija, M., Mavrinac, V. & Orlic, L. Dyslipidemia in patients with chronic kidney disease: etiology and management. Int. J. Nephrol. Renovasc. Dis. 10, 35–45 (2017).
Marcuccilli, M. & Chonchol, M. NAFLD and chronic kidney disease. Int. J. Mol. Sci. 17, 562 (2016).
Targher, G. & Byrne, C. D. Non-alcoholic fatty liver disease: an emerging driving force in chronic kidney disease. Nat. Rev. Nephrol. 13, 297–310 (2017).
Kwon, S. et al. Apolipoprotein B is a risk factor for end-stage renal disease. Clin. Kidney J. https://doi.org/10.1093/ckj/sfz186 (2021).
Geypens, B. et al. Influence of dietary protein supplements on the formation of bacterial metabolites in the colon. Gut 41, 70–76 (1997).
Roager, H. M. & Licht, T. R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 9, 3294 (2018).
Saito, Y., Sato, T., Nomoto, K. & Tsuji, H. Identification of phenol- and p-cresol-producing intestinal bacteria by using media supplemented with tyrosine and its metabolites. FEMS Microbiol. Ecol. 94, fiy125 (2018).
Lin, C.-J. et al. The role of liver in determining serum colon-derived uremic solutes. PLoS ONE 10, e0134590 (2015).
Brito, J. S. de et al. Aryl hydrocarbon receptor and uremic toxins from the gut microbiota in chronic kidney disease patients: is there a relationship between them? Biochemistry https://doi.org/10.1021/acs.biochem.8b01305 (2019).
Santana Machado, T., Cerini, C. & Burtey, S. Emerging roles of aryl hydrocarbon receptors in the altered clearance of drugs during chronic kidney disease. Toxins 11, 209 (2019).
Fennema, D., Phillips, I. R. & Shephard, E. A. Trimethylamine and trimethylamine N-oxide, a flavin-containing monooxygenase 3 (FMO3)-mediated host-microbiome metabolic axis implicated in health and disease. Drug Metab. Dispos. 44, 1839–1850 (2016).
Tang, W. H. W. et al. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 116, 448–455 (2015).
Tan, X. et al. Trimethylamine N-oxide aggravates liver steatosis through modulation of bile acid metabolism and inhibition of farnesoid x receptor signaling in nonalcoholic fatty liver disease. Mol. Nutr. Food Res. 63, e1900257 (2019).
Young, R. P., Hopkins, R. J. & Marsland, B. The gut-liver-lung axis. modulation of the innate immune response and its possible role in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 54, 161–169 (2016).
Viglino, D. et al. Nonalcoholic fatty liver disease in chronic obstructive pulmonary disease. Eur. Respir. J. 49, 1601923 (2017).
Young, R. P. & Hopkins, R. J. The mevalonate pathway and innate immune hyper-responsiveness in the pathogenesis of COPD and lung cancer: potential for chemoprevention. Curr. Mol. Pharmacol. 10, 46–59 (2017).
Hothersall, E., McSharry, C. & Thomson, N. C. Potential therapeutic role for statins in respiratory disease. Thorax 61, 729–734 (2006).
Hilliard, K. L. et al. The lung-liver axis: a requirement for maximal innate immunity and hepatoprotection during pneumonia. Am. J. Respir. Cell Mol. Biol. 53, 378–390 (2015).
Varraso, R. et al. Alternate Healthy Eating Index 2010 and risk of chronic obstructive pulmonary disease among US women and men: prospective study. BMJ 350, h286 (2015).
Lee, P. N. & Fry, J. S. Systematic review of the evidence relating FEV1 decline to giving up smoking. BMC Med. 8, 84 (2010).
Scoditti, E., Massaro, M., Garbarino, S. & Toraldo, D. M. Role of diet in chronic obstructive pulmonary disease prevention and treatment. Nutrients 11, 1357 (2019).
Chuang, S.-C. et al. Fiber intake and total and cause-specific mortality in the European Prospective Investigation into Cancer and Nutrition cohort. Am. J. Clin. Nutr. 96, 164–174 (2012).
Szmidt, M. K., Kaluza, J., Harris, H. R., Linden, A. & Wolk, A. Long-term dietary fiber intake and risk of chronic obstructive pulmonary disease: a prospective cohort study of women. Eur. J. Nutr. https://doi.org/10.1007/s00394-019-02038-w (2019).
North, C. J., Venter, C. S. & Jerling, J. C. The effects of dietary fibre on C-reactive protein, an inflammation marker predicting cardiovascular disease. Eur. J. Clin. Nutr. 63, 921–933 (2009).
Cummings, J. H., Pomare, E. W., Branch, W. J., Naylor, C. P. & Macfarlane, G. T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28, 1221–1227 (1987).
Ismaiel, A. & Dumitraşcu, D. L. Cardiovascular risk in fatty liver disease: the liver-heart axis—literature review. Front. Med. 6, 202 (2019).
Przybyszewski, E. M., Targher, G., Roden, M. & Corey, K. E. Nonalcoholic fatty liver disease and cardiovascular disease. Clin. Liver Dis. 17, 19–22 (2021).
Henson, J. B. et al. Advanced fibrosis is associated with incident cardiovascular disease in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 51, 728–736 (2020).
Soares, J.-B., Pimentel-Nunes, P., Roncon-Albuquerque, R. & Leite-Moreira, A. The role of lipopolysaccharide/toll-like receptor 4 signaling in chronic liver diseases. Hepatol. Int. 4, 659–672 (2010).
Lin, J., Kakkar, V. & Lu, X. Essential roles of toll-like receptors in atherosclerosis. Curr. Med. Chem. 23, 431–454 (2016).
Zhu, Y. et al. Carnitine metabolism to trimethylamine by an unusual Rieske-type oxygenase from human microbiota. Proc. Natl Acad. Sci. USA 111, 4268–4273 (2014).
Yang, S. et al. Gut microbiota-dependent marker TMAO in promoting cardiovascular disease: inflammation mechanism, clinical prognostic, and potential as a therapeutic target. Front. Pharmacol. 10, 1360 (2019).
Wang, Z. et al. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell 163, 1585–1595 (2015).
Ma, G. et al. Trimethylamine N-oxide in atherogenesis: impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 37, BSR20160244 (2017).
Canyelles, M. et al. Trimethylamine N-oxide: a link among diet, gut microbiota, gene regulation of liver and intestine cholesterol homeostasis and HDL function. Int. J. Mol. Sci. 19, 3228 (2018).
Wang, Z. et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63 (2011).
Geng, J. et al. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed. Pharmacother. Biomed. Pharmacother. 97, 941–947 (2018).
Fukami, K. et al. Oral L-carnitine supplementation increases trimethylamine-N-oxide but reduces markers of vascular injury in hemodialysis patients. J. Cardiovasc. Pharmacol. 65, 289–295 (2015).
Gao, X. et al. Fish oil ameliorates trimethylamine N-oxide-exacerbated glucose intolerance in high-fat diet-fed mice. Food Funct. 6, 1117–1125 (2015).
Acknowledgements
We thank TCS research for their constant support. We also acknowledge pixabay for some of the copyright free images used in this manuscript.
Author information
Authors and Affiliations
Contributions
S.A. and S.M. designed the review and drafted the manuscript. Both authors approved the final version of the manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
S.M. and S.A. are employed by company Tata Consultancy Services Ltd., (TCS) and are part of TCS Research. Both authors declare no competing interests.
Consent for publication
No individual’s data was utilized in the preparation of the manuscript.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Anand, S., Mande, S.S. Host-microbiome interactions: Gut-Liver axis and its connection with other organs. npj Biofilms Microbiomes 8, 89 (2022). https://doi.org/10.1038/s41522-022-00352-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-022-00352-6
This article is cited by
-
Investigating the causal role of the gut microbiota in esophageal cancer and its subtypes: a two-sample Mendelian randomization study
BMC Cancer (2024)
-
Colonization with ubiquitous protist Blastocystis ST1 ameliorates DSS-induced colitis and promotes beneficial microbiota and immune outcomes
npj Biofilms and Microbiomes (2023)
-
Titanium dioxide nanoparticles: revealing the mechanisms underlying hepatotoxicity and effects in the gut microbiota
Archives of Toxicology (2023)
-
Non-alcoholic fatty liver disease and gut microbial dysbiosis- underlying mechanisms and gut microbiota mediated treatment strategies
Reviews in Endocrine and Metabolic Disorders (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
