
Abstract
Porphyromonas asaccharolytica and Porphyromonas uenonis are common inhabitants of the vaginal microbiome, but their presence has been linked to adverse health outcomes for women, including bacterial vaginosis and preterm birth. However, little is known about the pathogenesis mechanisms of these bacteria. The related oral opportunistic pathogen, Porphyromonas gingivalis, is comparatively well-studied and known to secrete numerous extracellular matrix-targeting proteases. Among these are the gingipain family of cysteine proteases that drive periodontal disease progression and hematogenic transmission to the placenta. In this study, we demonstrate that vaginal Porphyromonas species secrete broad-acting proteases capable of freely diffusing within the cervicovaginal niche. These proteases degrade collagens that are enriched within the cervix (type I) and chorioamniotic membranes (type IV), as well as fibrinogen, which inhibits clot formation. Bioinformatic queries confirmed the absence of gingipain orthologs and identified five serine, cysteine, and metalloprotease candidates in each species. Inhibition assays revealed that each species’ proteolytic activity can be partially attributed to a secreted metalloprotease with broad substrate specificity that is distantly related to the P. gingivalis endopeptidase PepO. This characterization of virulence activities in vaginal Porphyromonas species highlights their potential to alter the homeostasis of reproductive tissues and harm human pregnancy through clotting disruption, fetal membrane weakening, and premature cervical remodeling.
Similar content being viewed by others

Introduction
The vaginal microbiome of healthy reproductive-age women is typically characterized by low species diversity, with Lactobacillus dominating the vaginal and ectocervical niches of the lower genital tract1,2. A community shift towards high species diversity, with overgrowth of anaerobic bacteria, is associated with increased risk of bacterial vaginosis (BV)2,3, acquisition and transmission of sexually transmitted infections4,5,6, preterm birth7,8,9, and cervical cancer10,11,12. Intriguingly, some women harboring a diverse cervicovaginal microbiome are healthy and asymptomatic2, suggesting a need to untangle how specific species contribute to poor outcomes. Among BV-associated bacteria, Gram-negative anaerobic rods corresponding to Prevotella and black-pigmented Porphyromonas species are frequently detected in vaginal samples and significantly associated with the Bacteroides morphotype from Nugent scoring13. While Prevotella is an abundant, species-rich, and relatively well-studied vaginal clade, comparatively little is known about Porphyromonas species inhabiting the human vagina. No Porphyromonas species is currently thought to be specific to the human urogenital tract, but P. asaccharolytica, P. uenonis, P. bennonis, and P. somerae (in decreasing order of cervicovaginal microbiome citation frequency) exhibit a preference for these niches3,14,15. P. asaccharolytica and P. uenonis colonize the vagina in 15–50% of healthy women and although their prevalence and abundance increase with BV, they are typically considered low abundance taxa13,16,17,18,19,20. Recent studies show these species are predictors of spontaneous preterm labor9,21, pelvic inflammatory disease22,23, human papillomavirus (HPV) infections progressing to cervical neoplasia24,25, and uterine cancer26,27. Thus, an improved understanding of the functional capacity of vaginal Porphyromonas species is needed.
To date, the only Porphyromonas species that has been well-characterized is P. gingivalis, a low abundance species in the oral microbiome of both healthy patients and those with gingivitis28,29. As a keystone species driving oral (plaque) biofilm formation and periodontal disease progression30,31, P. gingivalis contributes to local tissue destruction directly and indirectly through the induction of inflammatory processes32,33. P. gingivalis can also disseminate via the bloodstream to distal infection sites such as the endocardium and joints34,35. During pregnancy, P. gingivalis has been isolated from the placenta and amniotic fluid of women who delivered preterm36,37,38, and in mouse infection models, P. gingivalis induces preterm labor via inflammatory activation of the chorioamniotic membranes34,39,40. Pathogenesis mechanisms contributing to these outcomes include a wide array of proteolytic activities carried out by numerous secreted proteases. Among these are the gingipain family of cysteine proteases that drive periodontal disease progression41, hematogenic transmission to the placenta29,40,42,43, and preterm labor induction in mice40. The gingipains degrade many extracellular matrix components, including collagen44,45, and amplify their effects by activating and upregulating host matrix metalloproteases (MMPs) that also degrade collagen46,47. Furthermore, gingipains can degrade immune factors including immunoglobulins44,48, complement components49,50, cytokines51,52, clotting factors53,54, and antimicrobial peptides55, giving rise to a favorable immune environment for P. gingivalis colonization.
Proteolytic activity has been previously detected in vaginal fluid from patients with BV56,57,58 and characterized in clinical isolates of BV-associated bacteria59,60. In fact, collagenase (gelatinase) and caseinase activity of P. asaccharolytica (formerly Bacteroides asaccharolyticus) was previously reported in screens of Bacteroides species detected in human infections61 and clinical isolates from reproductive tract infections60. However, further characterization of the enzymes responsible was not conducted, and proteolytic activity of P. uenonis has yet to be explored. Given their phylogenetic relatedness and epidemiological similarity to P. gingivalis—exhibiting high prevalence, low abundance, and association with the disease—we sought to determine whether vaginal Porphyromonas species possess the broad-acting proteolytic virulence activity of the periodontal pathogen. In this study, we show that P. asaccharolytica and P. uenonis are capable of degrading multiple extracellular matrix components in the female genital tract. Our study furthermore reveals differences between the species, suggesting preterm birth-associated P. asaccharolytica may secrete more enzymes that contribute to collagenase activity. Finally, we identify and functionally characterize a P. asaccharolytica virulence factor—a metalloprotease that is highly conserved in P. uenonis and distantly related to the PepO endopeptidase in P. gingivalis and other Porphyromonas species62,63. We demonstrate this protein exhibits broad-acting proteolytic capacity, which may make it a key microbial virulence factor in the pathogenesis of gynecological and reproductive health conditions.
Results
Vaginal Porphyromonas species degrade type I collagen, type IV collagen, and casein using secreted proteases
Collagenase activity of P. asaccharolytica and P. uenonis was evaluated using fluorescently quenched substrates (type I collagen or type IV collagen), where proteolytic digestion results in dequenching and measurable increases in fluorescence over time. Fluorometric collagenase assays confirmed that P. asaccharolytica and P. uenonis cell suspensions degrade type I collagen in a dose-dependent manner (Fig. 1a). Next, collagenase activity was measured in cell-free supernatants from P. asaccharolytica and P. uenonis, validating that both organisms secrete proteases capable of degrading type I and type IV collagen (Fig. 1b, c). Collagenase activity was further confirmed with gelatin zymography (Supplementary Fig. 2), where P. asaccharolytica supernatants produced three distinct high molecular weight zones of clearing (~85, 95, and 120 kDa), while P. uenonis supernatants generated four zones of clearing: three high molecular weight (~75, 90, and 110 kDa) and one low molecular weight (30 kDa). Due to its low-complexity tertiary structure, casein is regarded as a universal protease substrate that is highly susceptible to proteolytic degradation. Fluorometric assays revealed that supernatants from P. asaccharolytica and P. uenonis possess caseinase activity (Fig. 1d). This was further confirmed using agar-based casein degradation assays, where P. asaccharolytica, P. uenonis, and P. gingivalis cell suspensions all produced zones of clearing (Supplementary Fig. 3). Although P. asaccharolytica and P. uenonis can degrade similar substrates as P. gingivalis41, P. gingivalis showed substantially higher maximum fluorescence and area under the curve for collagen degradation (Supplementary Fig. 4 and Supplementary Tables 1, 2). The area under the curve for casein degradation by P. gingivalis was also much higher than for P. asaccharolytica or P. uenonis, while the maximum fluorescence and time to maximum fluorescence for casein degradation were comparable between all three Porphyromonas species (Supplementary Fig. 4 and Supplementary Table 2). To understand if proteolytic activity might contribute to pathogenesis in the female genital tract, we evaluated whether a common commensal vaginal microbe is also capable of degrading collagen and casein. No collagenase or caseinase activity was detected from Lactobacillus crispatus cell suspensions in the fluorometric collagenase or caseinase assays (Supplementary Fig. 5).

a Cell suspensions of P. asaccharolytica and P. uenonis at 107 or 108 CFU/reaction were incubated with fluorophore-conjugated type I collagen. Results are presented as mean ± standard error from three independent experiments performed in technical triplicate or quadruplicate. Collagen degradation was measured every 3 min by detecting the increase in fluorescence over a 2-h time course. b, c Cell-free supernatants of P. asaccharolytica and P. uenonis were incubated with fluorophore-conjugated b type I or c type IV collagen over an 18-h time course. Results are presented as mean ± standard error from seven independent experiments (type I collagen) and four independent experiments (type IV collagen) performed in technical triplicate. d Cell-free supernatants of P. asaccharolytica and P. uenonis were incubated with fluorescein (FITC)-conjugated casein over a 5-h time course. Results are presented as mean ± standard error from five independent experiments performed in technical triplicate. The mean fluorescence readings of the negative control were subtracted from experimental wells and relative fluorescence units (RFU) were plotted over time, with negative values adjusted to zero.
P. asaccharolytica and P. uenonis inhibit fibrin clot formation through fibrinogen degradation
Since gingipains are known to degrade fibrinogen and exacerbate gum bleeding41, we next investigated whether P. asaccharolytica and P. uenonis proteases can degrade fibrinogen and impair fibrin clot formation. To evaluate direct fibrinogen degradation, cell-free media controls and cell suspensions of P. asaccharolytica or P. uenonis were incubated in the presence and absence of human fibrinogen over a 24-h time course. Samples removed at defined intervals were separated by SDS-PAGE and stained with Coomassie Brilliant Blue. When P. asaccharolytica or P. uenonis were incubated with fibrinogen, complete degradation of the fibrinogen α and β chains was observed after 18 h, while the γ chain remained intact (Fig. 2a, b). To determine whether fibrinogen degradation translates to impaired fibrin clotting, thrombin-induced fibrin clot formation was measured after Porphyromonas cell suspensions were preincubated with citrated plasma. A significant delay in clot formation was observed with the highest dose of P. asaccharolytica or P. uenonis (9.0 × 109 CFU/reaction) compared to the no cell control (Fig. 2c, ****p < 0.0001) or a lower dose of P. asaccharolytica or P. uenonis (Fig. 2c; ****p < 0.0001). Turbidimetry at the experimental endpoint (30 min) allowed for quantitative evaluation of final clot size. In keeping with clotting times, fibrin clot size was significantly reduced in samples exposed to P. asaccharolytica or P. uenonis at 9.0 × 109 CFU/reaction compared with the no cell control (Fig. 2d ****p < 0.0001) or lower doses of bacteria (Fig. 2d, ***p = 0.0003 for P. asaccharolytica and **p = 0.003 for P. uenonis). These findings were further confirmed by visual assessment of clot formation at assay endpoints (Fig. 2e).

a, b SDS-PAGE of a P. asaccharolytica and b P. uenonis incubated with human fibrinogen (+Fg), saline (no Fg), or a cell-free media control (sBHI Media + Fg). Samples were assessed for fibrinogen degradation, indicated by the absence of bands corresponding to α, β, and γ fibrinogen chains in “Porphyromonas cells + Fg” treatments compared to “no Fg” or media controls. c Time from thrombin addition to fibrin clot formation. Citrated plasma was preincubated with P. asaccharolytica, P. uenonis, or no cell controls. Experiments were performed in technical duplicate and results are presented as mean ± standard error from five independent experiments. d Quantitative evaluation of clot size via well scan optical density measurements at the experimental endpoint. Experiments were performed in technical duplicate and control reactions without thrombin were used to blank the experimental reactions. Data were normalized to the average clot size of the no cell control within each experiment. Results are presented as mean ± standard error from five independent experiments. Significance was assessed by one-way ANOVA with Holm–Sidak’s multiple comparisons test (****p < 0.0001, ***p < 0.0003, **p = 0.003). e Endpoint qualitative evaluation of fibrin clots (>30 min) after clotting time assay with P. asaccharolytica (2.4 × 109 CFU/reaction), P. uenonis (3.0 × 109 CFU/reaction), or no cell control.
Identification of collagenase candidates in P. asaccharolytica and P. uenonis
Since our analyses confirmed previous reports that P. asaccharolytica and P. uenonis do not encode gingipain orthologs (Supplementary Note, Supplementary Fig. 1, and Supplementary Tables 3–5), we sought to identify other candidate proteins that may be responsible for the collagenolytic activity of vaginal Porphyromonas species. To identify putative collagenases amongst the 59 and 64 peptidases predicted for P. asaccharolytica and P. uenonis by the MEROPs database64, we cross-referenced a list of known and predicted microbial collagenases in the BRENDA enzyme information database65. This approach shortened the candidate peptidase list to 18 enzymes in P. asaccharolytica and 14 in P. uenonis. InterPro scans of each putative collagenase revealed proteins likely to be involved in cell wall synthesis or export machinery and narrowed the list to ten peptidases in P. asaccharolytica and nine peptidases in P. uenonis (Supplementary Fig. 1). Factoring in similarity to characterized collagenases, additional domains identified in InterPro, and the presence of secretion signals, a final short-list of the seven most promising candidate peptidases in each organism was generated (Tables 1, 2 and Supplementary Table 6). Intriguingly, each organism’s candidate collagenases could be organized into four groups: Ig-containing serine proteases (2x), C10 cysteine proteases (2x), M13 metalloproteases (1x), and U32 proteases (2x) (Tables 1, 2). Multiple sequence alignments of candidate collagenase pairs for a given type within each strain revealed low amino acid sequence identity (29–45%, Supplementary Table 7). Conversely, BLASTP searches between species allowed for the identification of orthologous protein pairs in P. asaccharolytica and P. uenonis, with amino acid identity ranging from 56 up to 97% (Supplementary Table 7). Notably, all within-genome candidate collagenases pairs had lower amino acid identity compared to the ortholog identified in the other Porphyromonas genome (Supplementary Table 7).
Serine proteases within the short-list all contained Ig-like folds (Tables 1, 2), which are also present in the binding domain of the well-characterized collagen-degrading metalloproteases from Clostridium histolyticum (ColG, ColH)66. The candidate cysteine proteases contained type 9 secretion system signal domains and a SpeB domain (Tables 1, 2), indicating sequence similarity with Streptococcus pyogenes streptopain, a cysteine protease capable of cleaving host components such as fibrinogen, immunoglobulins, and complement proteins67,68,69. The candidate metalloproteases from P. asaccharolytica (Poras_0079) and P. uenonis (Poru_00076) each contained a catalytic collagenase domain in addition to an M13 type metallopeptidase domain and a predicted N-terminal secretion signal (Tables 1,2). Finally, two U32 collagenases were detected in each vaginal Porphyromonas species indicating an orthologous relationship with the P. gingivalis U32 collagenase PrtC70,71. Of note, none of these putative U32 collagenases were found to possess secretion signals indicative of localization to the extracellular space (Tables 1, 2).
Vaginal Porphyromonas species encode metalloproteases targeting collagens, casein, and fibrinogen
To further narrow down candidate enzymes responsible for collagenolytic and fibrinogenolytic activities, inhibitors of the predicted serine, cysteine, and metalloproteases (Tables 1, 2) were incorporated into functional assays. Cell-free supernatants from P. asaccharolytica and P. uenonis were incubated with type I collagen in the presence of three doses of 1,10-phenanthroline, iodoacetamide, or aprotinin to inhibit metallo-, cysteine, and serine proteases, respectively (Fig. 3a–d and Supplementary Fig. 6). Treatment of P. asaccharolytica supernatants with 1,10-phenanthroline resulted in decreased collagenase activity, with a statistically significant reduction in max enzyme activity observed with the highest dose of inhibitor (Fig. 3a, c; 0.2 vs. 0.02 mM ***p = 0.0009; 0.2 vs. 0.002 mM ***p = 0.0005, and Supplementary Fig. 6). Iodoacetamide treatment revealed a trend toward a dose-dependent decrease in collagenase activity in individual experiments (Supplementary Fig. 7), but this trend was not observed when multiple experiments were combined (Fig. 3a, c and Supplementary Fig. 6). However, when P. asaccharolytica supernatants were treated with a combination of 1,10-phenanthroline and iodoacetamide, there was a further reduction in collagenase activity compared to 1,10-phenanthroline alone (Fig. 3a, c, **p = 0.0092). There was no statistically significant decrease in the max collagenase activity secreted by P. asaccharolytica with aprotinin (Fig. 3a, c and Supplementary Fig. 6). For P. uenonis, 1,10-phenanthroline also inhibited collagenase activity (Fig. 3b, d and Supplementary Fig. 6), with a significant reduction in max collagenase activity (Fig. 3d; 0.2 vs. 0.02 mM ***p = 0.0008; 0.2 vs. 0.002 mM **p = 0.0012). However, treatment with iodoacetamide or aprotinin did not reduce P. uenonis collagenase activity (Fig. 3b, d and Supplementary Fig. 6) and the combination treatment of 1,10-phenanthroline and iodoacetamide did not provide an additional reduction in collagenase activity or max fluorescence (Fig. 3b, d) as observed in P. asaccharolytica (Fig. 3a, c).

a, b Type I collagen degradation of a P. asaccharolytica and b P. uenonis cell-free supernatants in the presence of 0.2 mM 1,10-phenanthroline, 0.4 mM iodoacetamide, 0.01 mM aprotinin or 0.2 mM 1,10-phenanthroline + 0.4 mM iodoacetamide. Results are normalized to the no inhibitor control (set to 1) and presented as mean ± standard error from four independent experiments. c, d Maximum fluorescence of c P. asaccharolytica and d P. uenonis secreted collagenase activity, normalized to the no inhibitor control and presented as mean ± standard error from four independent experiments, three independent experiments (P. asaccharolytica +0.01 mM aprotinin) or two independent experiments (P. uenonis +0.01 mM aprotinin) performed in technical triplicate. Statistical significance was assessed by one-way ANOVA and Tukey’s post hoc comparison P. asaccharolytica: 1,10-phenanthroline 0.2 vs. 0.02 mM ***p = 0.0009; 1,10-phenanthroline 0.2 vs. 0.002 mM ***p = 0.0005; 1,10-phenanthroline 0.2 vs. 0.2 mM 1,10-phenanthroline +0.4 mM iodoacetamide **p = 0.0092. P. uenonis: 1,10-phenanthroline 0.2 vs. 0.02 mM ***p = 0.0008; 1,10-phenanthroline 0.2 vs. 0.002 mM **p = 0.0012. e, f Type IV collagen degradation by e P. asaccharolytica and f P. uenonis cell-free supernatants in the presence of 0.2 mM 1,10-phenanthroline, 0.4 mM iodoacetamide, 0.01 mM aprotinin, or 0.2 mM 1,10-phenanthroline + 0.4 mM iodoacetamide. Results are normalized to the no inhibitor control and presented as mean ± standard error from four independent experiments. g, h Maximum fluorescence of g P. asaccharolytica and h P. uenonis secreted collagenase activity in the presence of inhibitors. Results are presented as mean ± standard error from four independent experiments performed in technical triplicate. Statistical significance was assessed by one-way ANOVA and Tukey’s post hoc comparison P. asaccharolytica: 1,10-phenanthroline 0.2 mM vs. no inhibitor *p = 0.0352.; P. uenonis: 1,10-phenanthroline 0.2 mM vs. no inhibitor *p = 0.0398. The mean fluorescence readings of the negative control were subtracted from experimental wells and relative fluorescence units (RFU) was plotted over time, with negative values adjusted to zero.
Next, we evaluated whether the same protease classes degrade type IV collagen and casein. For both Porphyromonas species, the metalloprotease inhibitor 1,10-phenanthroline completely abrogated type IV collagenase activity from supernatants, while serine and cysteine protease inhibitors did not significantly reduce activity (Fig. 3e–h). P. asaccharolytica caseinase activity was similar to its type I collagenase activity (Fig. 4a, b) as proteolytic activity was reduced by treatment with 1,10-phenanthroline, and treatment with both 1,10-phenanthroline and iodoacetamide produced an additional downward shift in enzyme activity that resulted in a significant reduction in max fluorescence when compared to the no inhibitor control (Fig. 4a, c; p = 0.0413). In keeping with the type I collagen and type IV collagen degradation results, P. uenonis caseinase activity was significantly decreased by treatment with the metalloprotease inhibitor 1,10-phenanthroline (Fig. 4c, d; p = 0.018), while aprotinin and iodoacetamide treatment did not inhibit activity (Fig. 4b, d). Further to this, the 1,10-phenanthroline/iodoacetamide combination did not offer any additional reduction in protease activity (Fig. 4b, d).

a, b Cell-free supernatants of a P. asaccharolytica and b P. uenonis were incubated with fluorophore-conjugated casein in the presence of the metalloprotease inhibitor 1,10-phenanthroline (0.2 mM), the cysteine protease inhibitor iodoacetamide (0.4 mM), or the serine protease inhibitor aprotinin (0.01 mM). Casein degradation was measured every 10 min by detecting the increase in fluorescence over a 5-h time course. Results are presented as a ratio normalized to the no inhibitor control and presented as mean ± standard error from five independent experiments performed in technical triplicate. The mean fluorescence readings of the negative control were subtracted from experimental wells and relative fluorescence units (RFU) was plotted over time, with negative values adjusted to zero. c Maximum fluorescence of P. asaccharolytica supernatant caseinase activity in the presence of inhibitors. Results are presented as mean ± standard error from five independent experiments performed in technical triplicate. Statistical significance was assessed with a one-way ANOVA and Tukey’s post hoc comparison, where inhibitor combination vs. no inhibitor *p = 0.0413. d Maximum fluorescence of P. uenonis supernatant caseinase activity in the presence of inhibitors. Results are presented as mean ± standard error from five independent experiments performed in technical triplicate. Statistical significance was assessed with a one-way ANOVA and Tukey’s post hoc comparison, where 1,10-phenanthroline 0.2 mM vs. no inhibitor p = 0.0145, inhibitor combination vs. no inhibitor p = 0.0180.
Inhibitors were also incorporated into the fibrinogen degradation assay to determine whether fibrinogen is proteolyzed by the same enzyme classes as those observed in our collagen and casein experiments. Delayed fibrinogen degradation by P. asaccharolytica was observed in the presence of 1,10-phenanthroline, but not with aprotinin or iodoacetamide (Fig. 5a), suggesting that secreted metalloproteases from P. asaccharolytica contribute to fibrinogen degradation. Fibrinogen degradation patterns from P. asaccharolytica and P. uenonis supernatants (Fig. 5) differed from the results obtained with cell suspensions (Fig. 2a–b). For both species, the α chain was only partially degraded by the supernatants (Fig. 5). For P. uenonis, the β and γ chains remained intact for the 48-hour time course (Fig. 5b). Furthermore, P. uenonis partial degradation of the α chain was not impacted by the inhibitors included in this study (Fig. 5b).

a SDS-PAGE of P. asaccharolytica supernatants incubated with human fibrinogen in the presence of 1,10-phenanthroline (0.5 mM), iodoacetamide (1 mM), or aprotinin (0.01 mM) compared to no inhibitor and no fibrinogen controls. Samples collected over 48 h were assessed for fibrinogen degradation, indicated by the absence of bands corresponding to α, β, and γ fibrinogen chains as denoted within gel images. b SDS-PAGE of P. uenonis supernatants incubated with human fibrinogen in the presence of 1,10-phenanthroline (0.5 mM), iodoacetamide (1 mM), or aprotinin (0.01 mM) compared to no inhibitor and no fibrinogen controls. Samples collected over 48 h were assessed for fibrinogen degradation, indicated by the absence of bands corresponding to α, β, and γ fibrinogen chains.
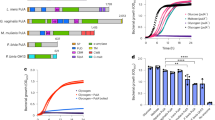
Finally, we sought to further characterize the Porphyromonas M13 metalloproteases identified in our bioinformatics inquiries (Tables 1, 2 and Fig. 6a). Genomic exploration of other Porphyromonas species detected in the urogenital tract and commonly isolated from other human body sites revealed that the M13 metalloproteases are ubiquitous (Fig. 6b). Notably, the M13 metalloprotease in P. gingivalis has been previously characterized as PepO, a secreted endopeptidase involved in host attachment/invasion and proteolytic activation of endothelin, a potent peptide that induces vasoconstriction62,63,72. To determine whether the vaginal Porphyromonas M13 metalloproteases have collagenase and caseinase activity, Poras_0079 (pepO), was cloned and expressed using the myTXTL® in vitro transcription/translation system. Expression of PepO was confirmed via SDS-PAGE with the appearance of a 76 kDa protein in PepO reactions but not in the RNA polymerase only control reactions (Fig. 7a). Fluorescent protease assays revealed that P. asaccharolytica PepO is capable of degrading casein and type I collagen, but not type IV collagen (Fig. 7b–d). Further, the metalloprotease inhibitor 1,10-phenanthroline fully abrogated the type I collagenase and caseinase activities of PepO (Fig. 7b, c).

a Domain structure of candidate host-degrading PepO metalloproteases from P. asaccharolytica and P. uenonis. Created with BioRender.com. b 16S rRNA gene phylogeny of Porphyromonas species identified in the urogenital tract presented as a Maximum Likelihood tree rooted to Prevotella buccalis (same order, different family). Red circles on each leaf indicate the percent amino acid identity of that species’ PepO ortholog compared to P. asaccharolytica PepO (Poras_0079 = 100%). The taxon previously identified as uncultivated Porphyromonas species type 1 was found to encompass two cultured, but unsequenced isolates (DNF00129 and C941, >99% 16S rRNA gene identity over 1120 nt). We identified a metagenome-assembled genome (MAG) in IMG/MER that encoded a 958 bp 16S rRNA gene fragment >99.5% identical to those from the cultured isolates DNF00129 and C941. Since this MAG represents the only genome sequence available for this species, we used it in our PepO queries, identifying an ortholog 42% identical to P. asaccharolytica PepO. As this taxon’s PepO ortholog was more distantly related to P. asaccharolytica PepO than the Prevotella buccalis PepO ortholog (47% identical), Porphyromonas species type 1 may represent a novel genus or family; we, therefore, labeled this taxon Porphyromonadaceae sp. type 1. Our phylogenetic analysis, along with inquiries through the Genome Taxonomy Database furthermore indicated that vaginal isolates KA00683 and KA00676 should be designated as belonging to the species P. pasteri, with each containing a PepO ortholog 58–59% identical to that from P. asaccharolytica.

a SDS-PAGE of control in vitro transcription/translation (myTXTL®) reaction (RNA polymerase only) and PepO myTXTL® reaction. b–d PepO myTXTL® reactions were incubated with b fluorescein (FITC)-casein or c fluorophore-conjugated type I collagen in the presence of 0.5 mM 1,10-phenanthroline. Casein degradation was measured by detecting the increase in fluorescence over a 24-h time course. Results are presented as mean ± standard error from three independent experiments. Collagen degradation was measured by detecting the increase in fluorescence over an 18-h time course. Results are presented as mean ± standard error from five independent experiments. d PepO myTXTL® reactions were incubated with fluorophore-conjugated type IV collagen over an 18-h time course and results are presented as mean ± standard deviation from one independent experiment. The mean fluorescence readings of the negative control were subtracted from experimental wells and relative fluorescence units (RFU) was plotted over time, with negative values adjusted to zero.
Discussion
It is well established that mucinase activity is elevated during BV73,74,75,76 and attributed to Gardnerella75,77 and Prevotella species75,78,79. In support of this, sialidase activity has been utilized as a diagnostic marker for BV78,80,81,82,83. Although most studies have focused on the degradation of mucin glycans, proteolytic activity in the vaginal fluid has also been linked with BV status56,57,58 and described among predominant BV-associated bacteria59,60. Isolates of Prevotella bivia from women with preterm premature rupture of membranes (PPROM) were shown to secrete proteases that degrade elastin, collagen, casein, and gelatin59. In another study screening bacterial strains from women with PPROM, preterm labor, or puerperal infection, protease activity was confirmed in phylogenetically diverse Gram-negative and Gram-positive organisms. This included the BV-associated bacteria Gardnerella vaginalis, Prevotella bivia (formerly Bacteroides bivius), and P. asaccharolytica (formerly Bacteroides asaccharolyticus)60. The authors found that P. bivia and P. asaccharolytica exhibited gelatin and casein degradation, while G. vaginalis exclusively degraded casein. Our findings confirm the collagen/casein degradation capacity of P. asaccharolytica and extend these activities to P. uenonis. Furthermore, we demonstrate that both vaginal Porphyromonas species also degrade type IV collagen, commonly found in reproductive tissues84,85, and describe the diverse protease types capable of coordinating these activities.
Our in vitro analyses show that the proteolytic activities of P. asaccharolytica and P. uenonis are dose-dependent across a range of 107–109 cells/mL. With concentrations of these microbes reported at 103–106 cells/mL in healthy women86,87, and up to 109 cells/mL in populations that include women with BV15, it is reasonable to expect that although vaginal Porphyromonas species are not high in relative abundance, they, like P. gingivalis, have the potential to cause disease in a protease- and community-dependent fashion88. To understand whether proteolytic activity is also observed among commensal bacteria that inhabit the vaginal niche, we investigated the proteolytic activity of Lactobacillus crispatus, demonstrating that L. crispatus is not capable of degrading type I collagen or casein. The absence of detectable proteolytic activity from a commensal Lactobacillus strain and the growing evidence for secreted proteolytic activity from BV-associated bacteria59,60, including vaginal Porphyromonas species, suggests that degradation of host proteins could be an important virulence trait of opportunistic pathogens in the female genital tract.
Our results demonstrate that vaginal Porphyromonas species are capable of directly degrading fibrinogen and impairing fibrin clot formation. Fibrinogen is detected in vaginal lavage fluid89,90 and is targeted by other reproductive pathogens91,92. Although the implications of altered fibrinogen levels in the female reproductive tract are not clear, impaired clotting functions could have severe consequences during labor and postpartum. Fibrinogen is known to increase substantially during pregnancy93,94, and decreased plasma fibrinogen levels have been associated with increased severity of postpartum hemorrhage95. Further to this, genetic fibrinogen abnormalities are significantly associated with miscarriage, placental abruption, and postpartum hemorrhage96,97,98,99.
Within the female genital tract, collagens are found within the vagina, cervix, uterus, and pelvic floor and their composition and content is significantly altered throughout pregnancy and during labor100. Vaginal and cervical tissue is primarily composed of fibrillar type I and III collagens, with type I collagen playing a critical role in tissue integrity101,102,103. Type IV collagen is typically found within basement membranes and is enriched within the placenta84. Both type I and type IV collagens are found within chorioamniotic membranes at the maternal–fetal interface85. During pregnancy, cervical collagens (type I, III) undergo a shift toward increased solubility and decreased abundance, contributing to cervical softening104,105,106, while increased collagenase activity is observed during cervical ripening to prepare for dilation and parturition107,108. Importantly, cervical remodeling during term and preterm labor occurs via the same mechanisms, with host matrix metalloproteinases (MMPs) coordinating cervical collagen degradation109. Premature preterm rupture of the membrane (PPROM) has been associated with infection110,111, increased host MMP collagenase activity112,113, and decreased collagen content114. Furthermore, microbial collagenases can reduce the tensile strength of chorioamniotic membranes ex vivo115, and collagenase activity has been detected in clinical isolates from PPROM patients59,60. Taken together, these findings imply that host and microbial modulation of collagen within the cervix and chorioamniotic membranes could play critical roles in preterm labor and PPROM. In the present study, our findings indicate that proteases secreted by P. asaccharolytica and P. uenonis are capable of degrading both type I and type IV collagens, uncovering a possible mechanism for how these microbes contribute to the initiation of preterm labor.
Although P. asaccharolytica and P. uenonis are phylogenetically related to the opportunistic periodontal pathogen P. gingivalis and our investigations demonstrated that P. asaccharolytica and P. uenonis possess gingipain-like activities, including degradation of type I and type IV collagen, casein, and fibrinogen, gingipain homologs are absent in P. asaccharolytica and P. uenonis. Our bioinformatics inquiries identified five candidate secreted collagenases in each vaginal Porphyromonas species. Interestingly, within-genome pairs of candidate collagenases had lower amino acid identity than interspecies pairs, suggesting that within-genome pairs are paralogs that resulted from a gene duplication event prior to speciation of P. asaccharolytica and P. uenonis116. By incorporating protease inhibitors into our functional assays, we determined that Porphyromonas secreted metalloproteases degrade collagens (type I, IV), casein, and fibrinogen. Although P. asaccharolytica and P. uenonis possess the same candidate collagenases, only P. asaccharolytica appears to secrete both metallo and cysteine proteases in the experimental conditions used in our study. Future investigations will need to address whether pH or redox state may affect the activity of the cysteine proteases. Additionally, due to the general proteolytic activity of the metallo and cysteine proteases, it is plausible that secreted proteases may degrade other proteins in the supernatants, including other proteases. Our results also indicate that P. uenonis may degrade fibrinogen using both secreted and cell surface-associated proteases, as P. uenonis cell suspensions degraded the α and β chains of fibrinogen, while cell-free supernatants partially degraded the α chain only. Interestingly, the secreted P. uenonis protease that contributes to partial degradation of the fibrinogen α chain is not impacted by the inhibitors included in this study, implying that the secreted fibrinogenolytic enzyme from P. uenonis is distinct from the secreted collagen and casein degrading enzymes.
Since 1,10-phenanthroline inhibited the secreted proteolytic activity of P. asaccharolytica and P. uenonis, the M13 metalloproteases (distant orthologs of P. gingivalis PepO [PgPepO]) were expected to confer the observed protease activity. Our results confirm that P. asaccharolytica PepO degrades type I collagen and casein, but not type IV collagen, suggesting there is an additional type IV collagen-degrading metalloprotease in Porphyromonas supernatants. Previous work characterizing P. gingivalis PepO revealed sequence conservation with the human endothelin converting enzyme 1 (ECE-1), which proteolytically processes inactive endothelin (big endothelin) into active endothelin. Activated endothelin peptides can induce vasoconstriction and cellular proliferation, alter vascular permeability and activate inflammatory cells117,118. PgPepO was confirmed to possess ECE-1 like activity, converting all three subtypes of big endothelin to active endothelin62. Numerous studies have also demonstrated that PgPepO plays a role in cellular invasion and intracellular survival of P. gingivalis63,72. However additional substrates for this endopeptidase and the functional consequences of bacterial endothelin activation have yet to be explored.
PepO orthologs have also been characterized in select Lactobacillus species: Lactobacillus lactis and Lactobacillus rhamnosus PepO can proteolyze casein, but these enzymes are either confirmed or predicted to localize to the cytoplasm119,120. PepO has also been explored in Streptococcus species, including Streptococcus pneumoniae and Streptococcus pyogenes (Group A Streptococci; GAS). In S. pneumoniae, PepO is detected on the bacterial cell surface and in culture supernatants. S. pneumoniae PepO binds to host cells and fibronectin, facilitating bacterial adhesion and invasion121. S. pneumoniae PepO also interacts with the host immune system, but there is conflicting evidence on whether this contributes to immune evasion or activation121,122,123,124. Surprisingly, none of these studies have shown any proteolytic activity of S. pneumoniae PepO. In GAS, PepO has been detected both in the cytoplasm and as a secreted protein125,126. Similar to S. pneumoniae, GAS PepO contributes to complement evasion126. GAS PepO also participates in regulating quorum sensing via direct degradation of peptide pheromones secreted by GAS125. Taken together, this body of work highlights the broad substrate specificity and diverse functionality of bacterial PepO. Our work characterizing P. asaccharolytica PepO demonstrates degradation of host extracellular matrix (type I collagen), adding another substrate to the proteolytic repertoire of PepO. The findings that PepO can degrade regulatory proteins secreted by host cells (P. gingivalis PepO: endothelin) and bacterial cells (GAS PepO: quorum sensing peptides) suggest that PepO enzymes could play a role in the dysregulation of proteolytic cascades. Further investigation is needed to better understand the substrates targeted by PepO proteins in complex mucosal body sites such as the female reproductive tract to reveal how this enzyme contributes to the pathogenesis of phylogenetically diverse bacteria.
Methods
Bacterial strains and growth conditions
Porphyromonas asaccharolytica CCUG 7834 (type strain, identical to DSM 20707, ATCC 25260, and JCM 6326), Porphyromonas uenonis CCUG 48615 (type strain, identical to DSM 23387, ATCC BAA-906, and JCM 13868), P. gingivalis W50 ATCC 53978 and Lactobacillus crispatus CCUG 42897 were cultured anaerobically on 1.5% brucella agar (BD Biosciences, Franklin Lakes, MD) supplemented with 5% defibrinated sheep’s blood (Dalynn Biologicals, Calgary, AB). For liquid cultivation, supplemented brain heart infusion (sBHI) was prepared by supplementing BHI (BD) with 2% gelatin (BD), 1% yeast extract (ThermoFisher Scientific, Burnaby, BC), 0.8% dextrose (BD) and 0.1% starch (ThermoFisher). Solid and liquid cultivation was conducted at 37 °C in an AS-580 anaerobic chamber (Anaerobe Systems, Morgan Hill, CA). Bacterial suspensions were prepared by harvesting cells from the solid medium after growth for 16–24 h (P. gingivalis, L. crispatus) or 36–48 h (P. asaccharolytica and P. uenonis) and resuspending cells in sBHI, sBHI (no gelatin), BHI (no supplements), or PBS. Optical density (600 nm, OD600) of bacterial suspensions was measured with a Genesys 300 visible spectrophotometer (ThermoFisher Burnaby, BC) and colony forming units per mL (CFU/mL) was calculated using empirically determined CFU/mL/OD600 for each strain. Serial dilution spot plating was used to verify the CFU/mL of the starting suspensions. Cell-free supernatants (SNs) were harvested during late-log to early stationary phase for each species (P. asaccharolytica OD600 1.3–1.8 [1.7 × 109–2.3 × 109 CFU/mL]; P. uenonis OD600 0.8–1.2 [2.1 × 109–3.2 × 109 CFU/mL]).
Collagenase/caseinase assays
Cell suspensions or cell-free supernatants were tested for collagenase activity with the EnzChek Gelatinase/Collagenase assay kit (Invitrogen, Carlsbad, CA) using fluorophore-labeled DQTM gelatin conjugate (predominantly type I collagen, Invitrogen) or a fluorophore-labeled DQTM type IV collagen conjugate from the human placenta (Invitrogen). Cell-free supernatants were harvested from liquid cultures by centrifuging at 10,000 × g for 10 min at room temperature before the supernatant was filter-sterilized (0.2 µm; Pall Laboratory, Mississauga, ON) and stored at −20 °C. Reactions were prepared by mixing 20 µL of the substrate at 0.25 mg/mL with 80 µL of reaction buffer and 100 µL of bacterial suspension, cell-free supernatant, or media in black optical bottom 96-well plates (Greiner Bio-One, Monroe, NC). Reactions testing general protease activity used fluorescein (FITC)-labeled casein as a substrate and were prepared by mixing 20 µL of the substrate at 50 µg/mL with 80 µL of Tris-buffered saline and 100 µL of cell-free supernatant. Plates were incubated in a Synergy H1 microplate reader (BioTek, Winooski, VT) at 37 °C in atmospheric conditions. Kinetic fluorescence reads were measured after 7 s shaking every 3–30 min at 485 nm excitation/527 nm emission. The mean fluorescence readings of the negative control (substrate in sBHI media) were subtracted from experimental wells and relative fluorescence units (RFU) were plotted over time, with negative values adjusted to zero.
Casein plate assay
Casein plates were prepared by autoclaving three solutions: 30 g/L instant skim milk power (Pacific Dairy), 19 g/L brain heart infusion (BHI, BD Biosciences), and 30 g/L agar (Fisher Scientific). The solutions were combined in equal volume and 10 mL was added to 100 × 15 mm petri dishes (Fisher Scientific) to solidify at room temperature. Bacterial suspensions were prepared in PBS and 5 µL was spotted onto casein agar plates. Zones of clearance were measured for each spot after incubating the plates at 37 °C under anaerobic conditions for 3 to 6 days.
Zymography
The total protein content of Porphyromonas cell-free supernatants was determined using a bicinchoninic acid microplate assay (BCA; Pierce, Rockford, IL). Cell-free supernatants were diluted to 8 mg/mL and 5 µL of the sample was combined with 5 µL of NovexTM Tris-Glycine SDS Sample Buffer (Invitrogen) to load 40 µg per well. Samples were separated on NovexTM 10% Zymogram Plus (Gelatin; Invitrogen) protein gels at a constant voltage of 125 V in NovexTM 1X Tris-Glycine Running Buffer (Invitrogen). After separation, gels were incubated in NovexTM 1X Renaturing Buffer for 30 min at room temperature with gentle agitation, followed by two consecutive incubations in NovexTM 1X Developing Buffer: room temperature for 30 min and 37 °C for 16 h. Gels were then stained in Coomassie brilliant blue R-250 solution (Fisher Scientific) and de-stained in 5% methanol/7.5% acetic acid in distilled water. The unprocessed scan of the zymogram gel is available in the Supplementary Information (Supplementary Fig. 8).
Clotting assays
Bacterial strains were harvested from a solid medium and suspended in 13 mM sodium citrate. Suspensions were centrifuged at 10,000 × g for 7 min at room temperature and resuspended in 13 mM sodium citrate. Duplicate cell suspensions, or cell-free controls, were incubated with 50 µL of sterile-filtered bovine plasma (Quad Five, Ryegate, MT) for 30 min at 37 °C under anaerobic conditions. After incubation, samples were blinded and centrifuged at 10,000 × g for 7 min at room temperature, and 50 µL of HEMOCLOT thrombin time reagent (Hyphen Biomed, Neuville-sur-Oise, France) was added to each reaction. The clotting time for each blinded sample was estimated using a stereo microscope to directly visualize clot formation, indicated by the presence of white precipitate, tendril formation, or increased viscosity of the samples. The clotting time for samples that did not form visible clots was recorded as 1800 s. To further evaluate final clot size, samples were transferred to a clear 96-well plate and OD (405 nm) was measured for the entire well using the well area scan feature of the Synergy H1 microplate reader (BioTek).
Fibrinogen degradation assay
Porphyromonas cell suspensions (107 CFU/reaction), sBHI media controls, or Porphyromonas cell-free supernatants (240 µg of total protein) were incubated with 120 µg of human fibrinogen (Sigma-Aldrich, St. Louis, MO). Reaction mixtures were incubated at 37 °C under anaerobic conditions or in an atmosphere of 5% CO2 for cell suspension or cell-free supernatants, respectively. Samples from each time point (cell suspensions: 0, 2, 18, 24 h; supernatants: 0, 2, 24, 48 h) were collected, mixed 1:1 with NovexTM 2X sample buffer (Invitrogen) with dithiothreitol (DTT, Fisher Scientific), heated at 95 °C for 10 min, and separated on NovexTM 10% Tris-Glycine polyacrylamide pre-cast gels (Invitrogen) at a constant voltage of 180 V. Gels were stained in 0.25% Coomassie brilliant blue R-250 solution (Fisher Scientific) and de-stained in 5% methanol/7.5% acetic acid in distilled water. Fibrinogen degradation was evaluated qualitatively by visualization of fibrinogen α chain (63.5 kDa), β chain (56 kDa), and γ chain (47 kDa) between experimental and control samples over the time-course. Gels are derived from the same experiments and were processed in parallel. Unprocessed scans of fibrinogen degradation protein gels are available in the Supplementary Information (Supplementary Figs. 9, 10).
Bioinformatic analyses
Gingipain ortholog queries
Gingipain amino acid sequences RgpA (PGN_1970; P28784), RgpB (PGN_1466; P95493), and Kgp (PGN_1728; B2RLK2) were obtained from P. gingivalis ATCC 33277 through UniProtKB. Each sequence was queried against all available P. asaccharolytica (DSM 20707; PR426713P-I) and P. uenonis (DSM 23387; 60-3) genomes using the “Selected Genomes” protein Basic Local Alignment Search Tool (BLAST; default settings) in the IMG/MER database to identify potential orthologs (Supplementary Fig. 1A). Each gingipain amino acid (AA) sequence was also submitted to the Pfam database and all Pfam IDs and names were recorded. Gingipain Pfam IDs were searched against all available P. asaccharolytica (DSM 20707; PR426713P-I) and P. uenonis (DSM 23387 [IMG Genome ID 2585427891 and 2528311143]; 60-3) strains using the advanced gene search function in the IMG/MER database (Supplementary Fig. 1A). All sequences returned by these IMG/MER BLAST and Pfam ID searches were used to query P. gingivalis ATCC 33277 in a reciprocal BLAST search with the National Center for Biotechnology Information (NCBI) protein BLAST tool (Supplementary Fig. 1A). Select hits were aligned with the P. gingivalis gingipains across their full length using the EMBL-EMI Clustal Omega alignment tool127.
Candidate collagenase queries
Peptidases from P. asaccharolytica CCUG 7834 and P. uenonis 60-3 were identified by searching for entries with MEROPS peptidase annotations in UniProt64,128. For the microbial peptidases from P. asaccharolytica and P. uenonis, the following enzyme information was exported from UniProtKB for each entry: gene ontology (biological process and molecular function), MEROPs, Pfam, PANTHER, PROSITE, SMART, SUPFAM. Next, a microbial collagenase enzyme number (EC3.4.24.3) was identified in BRENDA65 and searched against the UniProtKB database128, generating a list of 3417 entries corresponding to predicted and confirmed microbial collagenases. These microbial collagenase identifiers were cross-referenced against the exported peptidase information from P. asaccharolytica and P. uenonis to generate a short-list of 14–18 candidate collagenases in P. asaccharolytica and P. uenonis (Supplementary Fig. 1B). Short-list candidates were explored in UniProt and InterPro Scan128,129 to eliminate any proteins involved in cell wall synthesis or export machinery. Sequences were evaluated for the presence of secretion signals using SignalP v.5.0130 or InterPro129. Presence of the IDs: TIGR0483, IPR026444, or PF18962 from TIGR Fam, InterPro, and Pfam, respectively, within protein C-termini was indicative of secretion via the type IX secretion system. Since the MEROPs peptidase database only contained information for P. uenonis 60-3, IMG BLAST searches were used to identify the corresponding candidate collagenase in the experimental strain used in this study, P. uenonis CCUG 48615. Sequence identity and InterPro scans were evaluated for the top hit from each BLAST search in P. uenonis CCUG 48615 (Supplementary Table 6).
Phylogenetic analysis
16S rRNA gene sequences were collected using the Integrated Microbial Genomes & Microbiomes (IMG/MER)131 or National Center for Biotechnology Information (NCBI) databases for each Porphyromonas species reported in the human urogenital tract by Acuna-Amador in their comprehensive review14. Uncultured and recently cultured Porphyromonas species were also included15,132. Nomenclature choices were informed by the average nucleotide identity (ANI) tool within IMG/MER and the Genome Taxonomy Database (GTDB, Release 06-RS202, April 27, 2021)133. Whenever possible, type strain and near full-length sequences were selected. Multiple sequence alignment was performed with SINA aligner (v.1.2.11)134 using Silva’s Alignment, Classification and Tree Service (ACT)135. The phylogenetic tree was also computed through ACT using RAxML v.8.2.9136 with the Gamma model for likelihoods. The tree was edited using the interactive Tree of Life (iTOL137). The AA identity of PepO orthologs in each Porphyromonas species was queried through BLASTP searches in IMG/MER and NCBI using the P. asaccharolytica AA sequence as the query. With the exception of P. uenonis 60-3, the AA identity reported corresponds with a hit showing >98% query coverage.
Protease inhibition assays
The metalloprotease inhibitor, 1,10-Phenanthroline (Invitrogen), was prepared as a 2 M stock solution in ethanol. The cysteine protease inhibitor iodoacetamide (G-Biosciences, St. Louis, MO, or Sigma-Aldrich) was prepared as a 10 mM stock solution in HyPure H2O (Cytiva Life Sciences, Marlborough, MA). The serine protease inhibitor aprotinin (Roche, Mississauga, ON) was prepared as a 0.1 mM stock solution in HyPure H2O. Working solutions of protease inhibitors were diluted in EnzChek reaction buffer (Invitrogen) or TBS and incorporated into collagenase, caseinase, and fibrinogen degradation assays at the concentrations reported in the figure legends.
Expression construct cloning and in vitro transcription/translation
The Poras_0079 (PepO; IMG Gene ID 2504823953) DNA fragment encoding amino acid residues C21 to W682 was PCR amplified from a P. asaccharolytica DSM 20707 genomic DNA extraction (Qiagen DNeasy Blood & Tissue Kit, Germantown, MD) using the forward (5′-ATATCCATGGCTTGTAACAAGAAGCAGGAGAATC-3′) and reverse primers (5′-ATATCCCGGGCCAGACCACGACACGCTC-3′). The amplicon was cloned into the pTXTL-T7p14-aH plasmid (replacing alpha-hemolysin, Daicel Arbor Biosciences, Ann-Arbor, MI) using NcoI-HF and SmaI (New England Biolabs, Ipswitch, MA). The new plasmid construct (pSLP15) was transformed into Escherichia coli DH5α chemically competent cells and prepped using the QIAprep spin miniprep kit (Qiagen). Plasmid concentrations were determined using the Qubit dsDNA broad-range assay kit with a Qubit 3 fluorometer (Invitrogen). In vitro myTXTL® reactions were prepared by combining 5 nM of pSLP15, 1 nM of pTXTL-P70a-T7rnap (expressing the T7 RNA polymerase, Daicel Arbor Biosciences), and 9 µl of myTXTL® Sigma 70 Master Mix (Daciel Arbor Biosciences) to a final volume of 12 µL with HyClone HyPure water H2O (Cytiva); negative controls included 1 nM pTXTL-P70a-T7rnap and myTXTL® Sigma 70 Master Mix only. For each reaction, 10 µL was transferred to a PCR-clean polypropylene V bottom 96-well plate and covered with a silicone seal (Eppendorf, Mississauga, ON). Reactions were incubated at 29 °C for 16 h and final reactions stored at −20 °C. To assess proteolytic activity, control (RNAP only) and PepO reactions were incorporated into fluorescent collagenase and caseinase assays. For caseinase assays, myTXTL® reactions were diluted 60-fold in TBS and 10 µL was added to each well. For type I and type IV collagenase assays, 10 µL of undiluted myTXTL® reactions were added to each well. Caseinase and type I collagenase assays were also conducted in the presence of 0.5 mM 1,10-phenanthroline. Mean fluorescence readings of the negative control (substrate with RNAP myTXTL® reaction) were subtracted from the experimental wells and relative fluorescence units (RFU) were plotted over time, with negative values adjusted to zero. The unprocessed scan of the myTXTL® reaction protein gel is available in the Supplementary Information (Supplementary Fig. 11).
Bioinformatics and statistical analyses
Details of our bioinformatics analyses are provided in the Methods. Statistics were performed in GraphPad Prism (GraphPad, San Diego, CA) or Stata 15 (StataCorp, College Station, TX) and graphs were prepared in GraphPad. GraphPad was used to assess data variance and normality (Shapiro–Wilk test) and statistical significance was evaluated as described in figure legends. All schematic illustrations were created with BioRender.com.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors confirm that the data supporting the findings of this study are available within the paper and Supplementary files. Additional data are available from the corresponding author upon reasonable request. The unprocessed protein gel scans are available in the Supplemental Information (Supplemental Figs. 8–11). P. gingivalis ATCC 33277 gingipain protein sequences were accessed via UniProtKB using the following accession numbers: RgpA (PGN_1970; P28784), RgpB (PGN_1466; P95493), and Kgp (PGN_1728; B2RLK2). Gingipain BLASTP and Pfam searches were conducted against P. asaccharolytica and P. uenonis strains in IMG using the following IMG genome IDs: 649989985, 2504756018, 643886142, 2585427891, 2528311143. Candidate collagenases in P. asaccharolytica DSM 20707 and P. uenonis 60-3 were accessed via UniProtKB using the taxon identifiers 879243 and 596327, respectively. The protein sequence for the P. asaccharolytica PepO protease was retrieved through UniProtKB with the accession ID F4KL59. Cultured and uncultured vaginal Porphyromonas and Prevotella species’ 16S rRNA gene sequences were drawn from public databases as follows: Set I – (NCBI genome accession/locus tag): P. bennonis DSM 23058 (PRJNA169760/B088DRAFT_10000), P. asaccharolytica DSM 20707 (NC_015501/Poras_R0016), P. uenonis 60-3 (NZ_ACLR00000000/PORUE0001_1896), P. endodontalis ATCC 35406 (NZ_ACNN00000000/POREN0001_0839), P. somerae DSM 23386 (PRJNA165425/A3GKDRAFT_10000), P. pasteri KA00676 (LSDJ0100000/HMPREF3184_00469), P. buccalis ATCC 35310 (PRJNA40669/HMPREF_R0056); Set II – (IMG Gene ID): P. circumdentaria DSM 103022 (2828460842), P. crevioricanis ATCC 55563 (2587859826), P. macacae DSM 20710 (2517121994), P. gingivalis ATCC 33277 (2843222782), P. gulae DSM 15663 (2519001822), P. levii DSM 23370 (2558308191), P. gingivicanis JCM 15907 (2587113678), P. catoniae ATCC 51270 (2559434490), P. pasteri KA00683 (2860175207), P. uenonis DSM 23387 (2528767153), Porphyromonadaceae sp. type 1 HMP subject159005010-visit2-bin3300007272 4 (Ga0104871_10051020);Set III – (NCBI nucleotide accession): P. pasteri JCM 30531 (LC014934.1), Porphyromonadaceae sp. type 1 C941 (JF803519.1), Porphyromonadaceae sp. type 1 DNF00129 (KU726636.1).
References
van de Wijgert, J. H. et al. The vaginal microbiota: what have we learned after a decade of molecular characterization? PLoS ONE 9, e105998 (2014).
Ravel, J. et al. Vaginal microbiome of reproductive-age women. Proc. Natl Acad. Sci. USA 108, 4680–4687 (2011).
Onderdonk, A. B., Delaney, M. L. & Fichorova, R. N. The human microbiome during bacterial vaginosis. Clin. Microbiol. Rev. 29, 223–238 (2016).
Cohen, C. R. et al. Bacterial vaginosis associated with increased risk of female-to-male V-1 transmission: a prospective cohort analysis among African couples. PLoS Med 9, e1001251 (2012).
Brotman, R. M. et al. Bacterial vaginosis assessed by gram stain and diminished colonization resistance to incident gonococcal, chlamydial, and trichomonal genital infection. J. Infect. Dis. 202, 1907–1915 (2010).
Myer, L. et al. Bacterial vaginosis and susceptibility to HIV infection in South African women: a nested case-control study. J. Infect. Dis. 192, 1372–1380 (2005).
Hillier, S. L. et al. Association between bacterial vaginosis and preterm delivery of a low-birth-weight infant. The vaginal infections and prematurity study group. N. Engl. J. Med. 333, 1737–1742 (1995).
Fettweis, J. M. et al. The vaginal microbiome and preterm birth. Nat. Med. 25, 1012–1021 (2019).
Elovitz, M. A. et al. Cervicovaginal microbiota and local immune response modulate the risk of spontaneous preterm delivery. Nat. Commun. 10, 1305 (2019).
Norenhag, J. et al. The vaginal microbiota, human papillomavirus and cervical dysplasia: a systematic review and network meta-analysis. BJOG 127, 171–180 (2020).
Klein, C. et al. How the cervical microbiota contributes to cervical cancer risk in sub-Saharan Africa. Front. Cell Infect. Microbiol. 10, 23 (2020).
Laniewski, P. et al. Linking cervicovaginal immune signatures, HPV and microbiota composition in cervical carcinogenesis in non-Hispanic and Hispanic women. Sci. Rep. 8, 7593 (2018).
Srinivasan, S. et al. More than meets the eye: associations of vaginal bacteria with gram stain morphotypes using molecular phylogenetic analysis. PLoS ONE 8, e78633 (2013).
Acuna-Amador, L. & Barloy-Hubler, F. Porphyromonas spp. have an extensive host range in ill and healthy individuals and an unexpected environmental distribution: a systematic review and meta-analysis. Anaerobe 66, 102280 (2020).
McClelland, R. S. et al. Evaluation of the association between the concentrations of key vaginal bacteria and the increased risk of HIV acquisition in African women from five cohorts: a nested case-control study. Lancet Infect. Dis. 18, 554–564 (2018).
Hillier, S. L., Krohn, M. A., Rabe, L. K., Klebanoff, S. J. & Eschenbach, D. A. The normal vaginal flora, H2O2-producing lactobacilli, and bacterial vaginosis in pregnant women. Clin. Infect. Dis. 16, S273–S281 (1993).
Zozaya-Hinchliffe, M., Lillis, R., Martin, D. H. & Ferris, M. J. Quantitative PCR assessments of bacterial species in women with and without bacterial vaginosis. J. Clin. Microbiol. 48, 1812–1819 (2010).
Delaney, M. L. & Onderdonk, A. B. Nugent score related to vaginal culture in pregnant women. Obstet. Gynecol. 98, 79–84 (2001).
Albert, A. Y. et al. A study of the vaginal microbiome in healthy Canadian women utilizing cpn60-based molecular profiling reveals distinct Gardnerella subgroup community state types. PLoS ONE 10, e0135620 (2015).
Schellenberg, J. J. et al. Molecular definition of vaginal microbiota in East African commercial sex workers. Appl. Environ. Microbiol. 77, 4066–4074 (2011).
Holst, E., Goffeng, A. R. & Andersch, B. Bacterial vaginosis and vaginal microorganisms in idiopathic premature labor and association with pregnancy outcome. J. Clin. Microbiol. 32, 176–186 (1994).
Petrina, M. A. B., Cosentino, L. A., Wiesenfeld, H. C., Darville, T. & Hillier, S. L. Susceptibility of endometrial isolates recovered from women with clinical pelvic inflammatory disease or histological endometritis to antimicrobial agents. Anaerobe 56, 61–65 (2019).
Haggerty, C. L., Hillier, S. L., Bass, D. C. & Ness, R. B. Bacterial vaginosis and anaerobic bacteria are associated with endometritis. Clin. Infect. Dis. 39, 990–995 (2004).
Chao, X. et al. Research of the potential biomarkers in vaginal microbiome for persistent high-risk human papillomavirus infection. Ann. Transl. Med. 8, 100 (2020).
Chen, Y. et al. Human papillomavirus infection and cervical intraepithelial neoplasia progression are associated with increased vaginal microbiome diversity in a Chinese cohort. BMC Infect. Dis. 20, 629 (2020).
Walther-Antonio, M. R. et al. Potential contribution of the uterine microbiome in the development of endometrial cancer. Genome Med 8, 122 (2016).
Summanen, P. H. et al. Porphyromonas somerae sp. nov., a pathogen isolated from humans and distinct from porphyromonas levii. J. Clin. Microbiol. 43, 4455–4459 (2005).
Eloe-Fadrosh, E. A. & Rasko, D. A. The human microbiome: from symbiosis to pathogenesis. Annu Rev. Med. 64, 145–163 (2013).
Yang, H. W., Huang, Y. F. & Chou, M. Y. Occurrence of Porphyromonas gingivalis and Tannerella forsythensis in periodontally diseased and healthy subjects. J. Periodontol. 75, 1077–1083 (2004).
How, K. Y., Song, K. P. & Chan, K. G. Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front. Microbiol. 7, 53 (2016).
Hajishengallis, G., Darveau, R. P. & Curtis, M. A. The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 10, 717–725 (2012).
Katz, J. et al. Hydrolysis of epithelial junctional proteins by Porphyromonas gingivalis gingipains. Infect. Immun. 70, 2512–2518 (2002).
Takeuchi, H. et al. Porphyromonas gingivalis induces penetration of lipopolysaccharide and peptidoglycan through the gingival epithelium via degradation of junctional adhesion molecule 1. PLoS Pathog. 15, e1008124 (2019).
Ao, M. et al. Dental infection of Porphyromonas gingivalis induces preterm birth in mice. PLoS ONE 10, e0137249 (2015).
Mougeot, J. C. et al. Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. J. Oral. Microbiol. 9, 1281562 (2017).
Katz, J., Chegini, N., Shiverick, K. T. & Lamont, R. J. Localization of P. gingivalis in preterm delivery placenta. J. Dent. Res. 88, 575–578 (2009).
McCuaig, R. et al. Periodontal pathogens in the placenta and membranes in term and preterm birth. Placenta 68, 40–43 (2018).
Puertas, A. et al. Association of periodontitis with preterm birth and low birth weight: a comprehensive review. J. Matern. Fetal Neonatal Med. 31, 597–602 (2018).
Konishi, H. et al. Fetal membrane inflammation induces preterm birth via toll-like receptor 2 in mice with chronic gingivitis. Reprod. Sci. 26, 869–878 (2019).
Takii, R., Kadowaki, T., Tsukuba, T. & Yamamoto, K. Inhibition of gingipains prevents Porphyromonas gingivalis-induced preterm birth and fetal death in pregnant mice. Eur. J. Pharm. 824, 48–56 (2018).
Imamura, T., Travis, J. & Potempa, J. The biphasic virulence activities of gingipains: activation and inactivation of host proteins. Curr. Protein Pept. Sci. 4, 443–450 (2003).
Mysak, J. et al. Porphyromonas gingivalis: major periodontopathic pathogen overview. J. Immunol. Res. 2014, 476068 (2014).
Rafiei, M. et al. Study of Porphyromonas gingivalis in periodontal diseases: a systematic review and meta-analysis. Med. J. Islam Repub. Iran. 31, 62 (2017).
Bedi, G. S. & Williams, T. Purification and characterization of a collagen-degrading protease from Porphyromonas gingivalis. J. Biol. Chem. 269, 599–606 (1994).
Tada, H. et al. Proteolysis of ICAM-1 on human oral epithelial cells by gingipains. J. Dent. Res. 82, 796–801 (2003).
Zhou, J. & Windsor, L. J. Porphyromonas gingivalis affects host collagen degradation by affecting expression, activation, and inhibition of matrix metalloproteinases. J. Periodontal Res. 41, 47–54 (2006).
Andrian, E., Mostefaoui, Y., Rouabhia, M. & Grenier, D. Regulation of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinases by Porphyromonas gingivalis in an engineered human oral mucosa model. J. Cell Physiol. 211, 56–62 (2007).
Kadowaki, T., Yoneda, M., Okamoto, K., Maeda, K. & Yamamoto, K. Purification and characterization of a novel arginine-specific cysteine proteinase (argingipain) involved in the pathogenesis of periodontal disease from the culture supernatant of Porphyromonas gingivalis. J. Biol. Chem. 269, 21371–21378 (1994).
Schenkein, H. A., Fletcher, H. M., Bodnar, M. & Macrina, F. L. Increased opsonization of a prtH-defective mutant of Porphyromonas gingivalis W83 is caused by reduced degradation of complement-derived opsonins. J. Immunol. 154, 5331–5337 (1995).
Jagels, M. A. et al. Cleavage of the human C5A receptor by proteinases derived from Porphyromonas gingivalis: cleavage of leukocyte C5a receptor. Adv. Exp. Med. Biol. 389, 155–164 (1996).
Calkins, C. C., Platt, K., Potempa, J. & Travis, J. Inactivation of tumor necrosis factor-alpha by proteinases (gingipains) from the periodontal pathogen, Porphyromonas gingivalis. Implications of immune evasion. J. Biol. Chem. 273, 6611–6614 (1998).
Darveau, R. P., Belton, C. M., Reife, R. A. & Lamont, R. J. Local chemokine paralysis, a novel pathogenic mechanism for Porphyromonas gingivalis. Infect. Immun. 66, 1660–1665 (1998).
Scott, C. F., Whitaker, E. J., Hammond, B. F. & Colman, R. W. Purification and characterization of a potent 70-kDa thiol lysyl-proteinase (Lys-gingivain) from Porphyromonas gingivalis that cleaves kininogens and fibrinogen. J. Biol. Chem. 268, 7935–7942 (1993).
Grenier, D. Inactivation of human serum bactericidal activity by a trypsinlike protease isolated from Porphyromonas gingivalis. Infect. Immun. 60, 1854–1857 (1992).
Carlisle, M. D., Srikantha, R. N. & Brogden, K. A. Degradation of human alpha- and beta-defensins by culture supernatants of Porphyromonas gingivalis strain 381. J. Innate Immun. 1, 118–122 (2009).
Cauci, S., Monte, R., Driussi, S., Lanzafame, P. & Quadrifoglio, F. Impairment of the mucosal immune system: IgA and IgM cleavage detected in vaginal washings of a subgroup of patients with bacterial vaginosis. J. Infect. Dis. 178, 1698–1706 (1998).
Lewis, W. G. et al. Hydrolysis of secreted sialoglycoprotein immunoglobulin A (IgA) in ex vivo and biochemical models of bacterial vaginosis. J. Biol. Chem. 287, 2079–2089 (2012).
Borgdorff, H. et al. Cervicovaginal microbiome dysbiosis is associated with proteome changes related to alterations of the cervicovaginal mucosal barrier. Mucosal Immunol. 9, 621–633 (2016).
Doust, R. & Mobarez, A. M. Collagenase activity in Prevotella bivius isolated from patients with premature rupture of membranes. Med. J. Islamic Repub. Iran. 18, 61–66 (2004).
McGregor, J. A., Lawellin, D., Franco-Buff, A., Todd, J. K. & Makowski, E. L. Protease production by microorganisms associated with reproductive tract infection. Am. J. Obstet. Gynecol. 154, 109–114 (1986).
Steffen, E. K. & Hentges, D. J. Hydrolytic enzymes of anaerobic bacteria isolated from human infections. J. Clin. Microbiol. 14, 153–156 (1981).
Awano, S. et al. Sequencing, expression and biochemical characterization of the Porphyromonas gingivalis pepO gene encoding a protein homologous to human endothelin-converting enzyme. FEBS Lett. 460, 139–144 (1999).
Ansai, T., Yu, W., Urnowey, S., Barik, S. & Takehara, T. Construction of a pepO gene-deficient mutant of Porphyromonas gingivalis: potential role of endopeptidase O in the invasion of host cells. Oral. Microbiol. Immunol. 18, 398–400 (2003).
Rawlings, N. D. et al. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res 46, D624–D632 (2018).
Schomburg, I., Chang, A. & Schomburg, D. BRENDA, enzyme data and metabolic information. Nucleic Acids Res 30, 47–49 (2002).
Bauer, R. et al. Structures of three polycystic kidney disease-like domains from Clostridium histolyticum collagenases ColG and ColH. Acta Crystallogr. D. Biol. Crystallogr. 71, 565–577 (2015).
Elliott, S. D. A proteolytic enzyme produced by group a Streptococci with special reference to its effect on the type-specific M antigen. J. Exp. Med. 81, 573–592 (1945).
Matsuka, Y. V., Pillai, S., Gubba, S., Musser, J. M. & Olmsted, S. B. Fibrinogen cleavage by the Streptococcus pyogenes extracellular cysteine protease and generation of antibodies that inhibit enzyme proteolytic activity. Infect. Immun. 67, 4326–4333 (1999).
Nelson, D. C., Garbe, J. & Collin, M. Cysteine proteinase SpeB from Streptococcus pyogenes - a potent modifier of immunologically important host and bacterial proteins. Biol. Chem. 392, 1077–1088 (2011).
Takahashi, N., Kato, T. & Kuramitsu, H. K. Isolation and preliminary characterization of the Porphyromonas gingivalis prtC gene expressing collagenase activity. FEMS Microbiol. Lett. 68, 135–138 (1991).
Kato, T., Takahashi, N. & Kuramitsu, H. K. Sequence analysis and characterization of the Porphyromonas gingivalis prtC gene, which expresses a novel collagenase activity. J. Bacteriol. 174, 3889–3895 (1992).
Park, Y., Yilmaz, O., Jung, I. Y. & Lamont, R. J. Identification of Porphyromonas gingivalis genes specifically expressed in human gingival epithelial cells by using differential display reverse transcription-PCR. Infect. Immun. 72, 3752–3758 (2004).
Wiggins, R., Hicks, S. J., Soothill, P. W., Millar, M. R. & Corfield, A. P. Mucinases and sialidases: their role in the pathogenesis of sexually transmitted infections in the female genital tract. Sex. Transm. Infect. 77, 402–408 (2001).
Olmsted, S. S., Meyn, L. A., Rohan, L. C. & Hillier, S. L. Glycosidase and proteinase activity of anaerobic gram-negative bacteria isolated from women with bacterial vaginosis. Sex. Transm. Dis. 30, 257–261 (2003).
Briselden, A. M., Moncla, B. J., Stevens, C. E. & Hillier, S. L. Sialidases (neuraminidases) in bacterial vaginosis and bacterial vaginosis-associated microflora. J. Clin. Microbiol. 30, 663–666 (1992).
Howe, L. et al. Mucinase and sialidase activity of the vaginal microflora: implications for the pathogenesis of preterm labour. Int J. STD AIDS 10, 442–447 (1999).
Lewis, W. G., Robinson, L. S., Gilbert, N. M., Perry, J. C. & Lewis, A. L. Degradation, foraging, and depletion of mucus sialoglycans by the vagina-adapted Actinobacterium Gardnerella vaginalis. J. Biol. Chem. 288, 12067–12079 (2013).
Wiggins, R. et al. Use of 5-bromo-4-chloro-3-indolyl-alpha-D-N-acetylneuraminic acid in a novel spot test to identify sialidase activity in vaginal swabs from women with bacterial vaginosis. J. Clin. Microbiol. 38, 3096–3097 (2000).
Moncla, B. J., Braham, P. & Hillier, S. L. Sialidase (neuraminidase) activity among gram-negative anaerobic and capnophilic bacteria. J. Clin. Microbiol. 28, 422–425 (1990).
Zhang, X. et al. [Relationship between vaginal sialidase bacteria vaginosis and chorioammionitis]. Zhonghua Fu Chan Ke Za Zhi 37, 588–590 (2002).
Smayevsky, J., Canigia, L. F., Lanza, A. & Bianchini, H. Vaginal microflora associated with bacterial vaginosis in nonpregnant women: reliability of sialidase detection. Infect. Dis. Obstet. Gynecol. 9, 17–22 (2001).
Sumeksri, P., Koprasert, C. & Panichkul, S. BVBLUE test for diagnosis of bacterial vaginosis in pregnant women attending antenatal care at Phramongkutklao Hospital. J. Med Assoc. Thai 88, S7–S13 (2005).
Myziuk, L., Romanowski, B. & Johnson, S. C. BVBlue test for diagnosis of bacterial vaginosis. J. Clin. Microbiol. 41, 1925–1928 (2003).
Oefner, C. M. et al. Collagen type IV at the fetal-maternal interface. Placenta 36, 59–68 (2015).
Strauss, J. F. 3rd Extracellular matrix dynamics and fetal membrane rupture. Reprod. Sci. 20, 140–153 (2013).
Beamer, M. A. et al. Bacterial species colonizing the vagina of healthy women are not associated with race. Anaerobe 45, 40–43 (2017).
Fredricks, D. N. et al. Extra-vaginal bacterial colonization and risk for incident bacterial vaginosis in a population of women who have sex with men. J. Infect. Dis. https://doi.org/10.1093/infdis/jiaa233 (2020).
Hajishengallis, G. et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10, 497–506 (2011).
Tang, L. J. et al. Proteomic analysis of human cervical-vaginal fluids. J. Proteome Res. 6, 2874–2883 (2007).
Dasari, S. et al. Comprehensive proteomic analysis of human cervical-vaginal fluid. J. Proteome Res. 6, 1258–1268 (2007).
Wang, N. Y. et al. Group B streptococcal serine-rich repeat proteins promote interaction with fibrinogen and vaginal colonization. J. Infect. Dis. 210, 982–991 (2014).
Harris, T. O., Shelver, D. W., Bohnsack, J. F. & Rubens, C. E. A novel streptococcal surface protease promotes virulence, resistance to opsonophagocytosis, and cleavage of human fibrinogen. J. Clin. Invest. 111, 61–70 (2003).
Katz, D. & Beilin, Y. Disorders of coagulation in pregnancy. Br. J. Anaesth. 115, ii75–ii88 (2015).
Kadir, R., Chi, C. & Bolton-Maggs, P. Pregnancy and rare bleeding disorders. Haemophilia 15, 990–1005 (2009).
Cortet, M. et al. Association between fibrinogen level and severity of postpartum haemorrhage: secondary analysis of a prospective trial. Br. J. Anaesth. 108, 984–989 (2012).
Ness, P. M., Budzynski, A. Z., Olexa, S. A. & Rodvien, R. Congenital hypofibrinogenemia and recurrent placental abruption. Obstet. Gynecol. 61, 519–523 (1983).
Evron, S., Anteby, S. O., Brzezinsky, A., Samueloff, A. & Eldor, A. Congenital afibrinogenemia and recurrent early abortion: a case report. Eur. J. Obstet. Gynecol. Reprod. Biol. 19, 307–311 (1985).
Goodwin, T. M. Congenital hypofibrinogenemia in pregnancy. Obstet. Gynecol. Surv. 44, 157–161 (1989).
Kobayashi, T., Kanayama, N., Tokunaga, N., Asahina, T. & Terao, T. Prenatal and peripartum management of congenital afibrinogenaemia. Br. J. Haematol. 109, 364–366 (2000).
Woessner, J. F. & Brewer, T. H. Formation and breakdown of collagen and elastin in the human uterus during pregnancy and post-partum involution. Biochem. J. 89, 75–82 (1963).
Montoya, T. I., Maldonado, P. A., Acevedo, J. F. & Word, R. A. Effect of vaginal or systemic estrogen on dynamics of collagen assembly in the rat vaginal wall. Biol. Reprod. 92, 43 (2015).
Kerkhof, M. H., Hendriks, L. & Brolmann, H. A. Changes in connective tissue in patients with pelvic organ prolapse-a review of the current literature. Int. Urogynecol. J. Pelvic Floor Dysfunct. 20, 461–474 (2009).
Minamoto, T., Arai, K., Hirakawa, S. & Nagai, Y. Immunohistochemical studies on collagen types in the uterine cervix in pregnant and nonpregnant states. Am. J. Obstet. Gynecol. 156, 138–144 (1987).
Myers, K., Socrate, S., Tzeranis, D. & House, M. Changes in the biochemical constituents and morphologic appearance of the human cervical stroma during pregnancy. Eur. J. Obstet. Gynecol. Reprod. Biol. 144, S82–S89 (2009).
Myers, K. M., Paskaleva, A. P., House, M. & Socrate, S. Mechanical and biochemical properties of human cervical tissue. Acta Biomater. 4, 104–116 (2008).
Uldbjerg, N., Ekman, G., Malmstrom, A., Olsson, K. & Ulmsten, U. Ripening of the human uterine cervix related to changes in collagen, glycosaminoglycans, and collagenolytic activity. Am. J. Obstet. Gynecol. 147, 662–666 (1983).
Osmers, R. et al. Collagenase activity in the cervix of non-pregnant and pregnant women. Arch. Gynecol. Obstet. 248, 75–80 (1990).
Rath, W., Adelmann-Grill, B. C., Pieper, U. & Kuhn, W. Collagen degradation in the pregnant human cervix at term and after prostaglandin-induced cervical ripening. Arch. Gynecol. 240, 177–184 (1987).
Gonzalez, J. M., Dong, Z., Romero, R. & Girardi, G. Cervical remodeling/ripening at term and preterm delivery: the same mechanism initiated by different mediators and different effector cells. PLoS ONE 6, e26877 (2011).
Parry, S. & Strauss, J. F. 3rd Premature rupture of the fetal membranes. N. Engl. J. Med 338, 663–670 (1998).
Jones, H. E. et al. Differing prevalence and diversity of bacterial species in fetal membranes from very preterm and term labor. PLoS ONE 4, e8205 (2009).
Wang, H. et al. Genetic and epigenetic mechanisms combine to control MMP1 expression and its association with preterm premature rupture of membranes. Hum. Mol. Genet. 17, 1087–1096 (2008).
McLaren, J., Taylor, D. J. & Bell, S. C. Increased concentration of pro-matrix metalloproteinase 9 in term fetal membranes overlying the cervix before labor: implications for membrane remodeling and rupture. Am. J. Obstet. Gynecol. 182, 409–416 (2000).
Skinner, S. J., Campos, G. A. & Liggins, G. C. Collagen content of human amniotic membranes: effect of gestation length and premature rupture. Obstet. Gynecol. 57, 487–489 (1981).
McGregor, J. A. et al. Bacterial protease-induced reduction of chorioamniotic membrane strength and elasticity. Obstet. Gynecol. 69, 167–174 (1987).
Zallot, R., Harrison, K. J., Kolaczkowski, B. & de Crecy-Lagard, V. Functional annotations of paralogs: a blessing and a curse. Life (2016).
Filep, J. G. Endothelin peptides: biological actions and pathophysiological significance in the lung. Life Sci. 52, 119–133 (1993).
Noveral, J. P., Rosenberg, S. M., Anbar, R. A., Pawlowski, N. A. & Grunstein, M. M. Role of endothelin-1 in regulating proliferation of cultured rabbit airway smooth muscle cells. Am. J. Physiol. 263, L317–L324 (1992).
Pritchard, G. G., Freebairn, A. D. & Coolbear, T. Purification and characterization of an endopeptidase from Lactococcus lactis subsp. cremoris SK11. Microbiol 140(Pt 4), 923–930 (1994).
Christensson, C. et al. Cloning and expression of an oligopeptidase, PepO, with novel specificity from Lactobacillus rhamnosus HN001 (DR20). Appl. Environ. Microbiol. 68, 254–262 (2002).
Agarwal, V. et al. Streptococcus pneumoniae endopeptidase O (PepO) is a multifunctional plasminogen- and fibronectin-binding protein, facilitating evasion of innate immunity and invasion of host cells. J. Biol. Chem. 288, 6849–6863 (2013).
Agarwal, V. et al. Binding of Streptococcus pneumoniae endopeptidase O (PepO) to complement component C1q modulates the complement attack and promotes host cell adherence. J. Biol. Chem. 289, 15833–15844 (2014).
Shu, Z. et al. Streptococcus pneumoniae PepO promotes host anti-infection defense via autophagy in a Toll-like receptor 2/4 dependent manner. Virulence 11, 270–282 (2020).
Zhang, H. et al. Streptococcus pneumoniae endopeptidase O (PepO) elicits a strong innate immune response in mice via TLR2 and TLR4 signaling pathways. Front. Cell Infect. Microbiol. 6, 23 (2016).
Wilkening, R. V., Chang, J. C. & Federle, M. J. PepO, a CovRS-controlled endopeptidase, disrupts Streptococcus pyogenes quorum sensing. Mol. Microbiol. 99, 71–87 (2016).
Honda-Ogawa, M. et al. Streptococcus pyogenes endopeptidase O contributes to evasion from complement-mediated bacteriolysis via binding to human complement factor C1q. J. Biol. Chem. 292, 4244–4254 (2017).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47, W636–W641 (2019).
The UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, (2021).
Blum, M. et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 49, D344–D354 (2021).
Almagro Armenteros, J. J. et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423 (2019).
Chen, I. A. et al. The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res 49, D751–D763 (2021).
Srinivasan, S. et al. More Easily Cultivated Than Identified: Classical Isolation With Molecular Identification of Vaginal Bacteria. J. Infect. Dis. 214(Suppl 1), S21–S28 (2016).
Parks, D. H. et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 38, 1079–1086 (2020).
Pruesse, E., Peplies, J. & Glockner, F. O. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49(W1), W293–W296 (2021).
Acknowledgements
The authors would like to thank Dr. Antoine Dufour and Dr. Morley Hollenberg for their technical assistance and advice on this work. This work was supported by the Canadian Institutes of Health Research (L.K.S., FRN 161348), the National Institutes of Health (L.K.S., STI-CRC Developmental Research Program Award associated with U19AI113173), the Canadian Foundation for Innovation John R. Evans Leaders Fund (L.K.S, 36603), Alberta Innovates (K.V.L. and A.D.), the Government of Alberta, the University of Calgary’s Calvin, Phoebe and Joan Snyder Institute for Chronic Diseases, the International Microbiome Centre, the O’Brien Centre for the Bachelor of Health Sciences (V.C.H.B.), and the Program for Undergraduate Research Experience (E.K.).
Author information
Authors and Affiliations
Contributions
L.K.S. conceived the hypothesis and experimental design in collaboration with K.V.L. and V.C.H.B. K.V.L. and V.C.H.B. carried out bacterial collagenase assays, K.V.L., V.C.H.B., and E.K. conducted clotting inhibition assays, K.V.L., A.D., and L.K.S. conducted bioinformatic analyses and S.K. performed the cloning of PepO protein. K.V.L. conducted supernatant and PepO collagenase and caseinase assays and fibrinogen degradation assays. K.V.L. and L.K.S. performed statistical analyses, prepared figures, and wrote the manuscript. All authors read, revised, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lithgow, K.V., Buchholz, V.C.H., Ku, E. et al. Protease activities of vaginal Porphyromonas species disrupt coagulation and extracellular matrix in the cervicovaginal niche. npj Biofilms Microbiomes 8, 8 (2022). https://doi.org/10.1038/s41522-022-00270-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-022-00270-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
