
Abstract
Cationic antimicrobial peptides are multifunctional molecules that have a high potential as therapeutic agents. We have identified a histone H1-derived peptide from the Komodo dragon (Varanus komodoensis), called VK25. Using this peptide as inspiration, we designed a synthetic peptide called DRGN-1. We evaluated the antimicrobial and anti-biofilm activity of both peptides against Pseudomonas aeruginosa and Staphylococcus aureus. DRGN-1, more than VK25, exhibited potent antimicrobial and anti-biofilm activity, and permeabilized bacterial membranes. Wound healing was significantly enhanced by DRGN-1 in both uninfected and mixed biofilm (Pseudomonas aeruginosa and Staphylococcus aureus)-infected murine wounds. In a scratch wound closure assay used to elucidate the wound healing mechanism, the peptide promoted the migration of HEKa keratinocyte cells, which was inhibited by mitomycin C (proliferation inhibitor) and AG1478 (epidermal growth factor receptor inhibitor). DRGN-1 also activated the EGFR-STAT1/3 pathway. Thus, DRGN-1 is a candidate for use as a topical wound treatment. Wound infections are a major concern; made increasingly complicated by the emerging, rapid spread of bacterial resistance. The novel synthetic peptide DRGN-1 (inspired by a peptide identified from Komodo dragon) exhibits pathogen-directed and host-directed activities in promoting the clearance and healing of polymicrobial (Pseudomonas aeruginosa & Staphylococcus aureus) biofilm infected wounds. The effectiveness of this peptide cannot be attributed solely to its ability to act upon the bacteria and disrupt the biofilm, but also reflects the peptide’s ability to promsote keratinocyte migration. When applied in a murine model, infected wounds treated with DRGN-1 healed significantly faster than did untreated wounds, or wounds treated with other peptides. The host-directed mechanism of action was determined to be via the EGFR-STAT1/3 pathway. The pathogen-directed mechanism of action was determined to be via anti-biofilm activity and antibacterial activity through membrane permeabilization. This novel peptide may have potential as a future therapeutic for treating infected wounds.
Similar content being viewed by others


Introduction
The increasing prevalence of multidrug-resistant (MDR) pathogens demands new antibiotics. Due to their potent and broad antimicrobial activity, antimicrobial peptides (AMPs), and in particular cationic antimicrobial peptides (CAMPs), are a possible alternative to conventional antibiotics. Although the mode of action of AMPs is not fully understood, it is believed that their major targets are the cytoplasmic membrane and intracellular molecules.1 In addition, AMPs can have both antimicrobial and antibiofilm activities, and that antimicrobial and antibiofilm activity may be independent mechanisms of the same peptide.1,2,3,4,5 It is also believed that it is difficult for bacteria to develop resistance against CAMPs that kill quickly through their actions on the cytoplasmic membrane or that act through complex mechanisms inside the bacterial cell.5 Antimicrobial peptides have been found to have a broad-spectrum activity, with active peptides identified against both gram-positive and gram-negative bacteria, membrane-bound viruses, and fungi. Antimicrobial peptides can also have significant immunomodulatory activity directed towards the host, which is likely to be an additional critical aspect of their function in vivo.6,7,8 Some peptides have been identified as having a role in promoting wound-healing.9 For these reasons, AMPs are potentially useful in combating MDR bacteria and may be useful as potential treatments for infected wounds.
Animals are a promising source of CAMPs because of their evolved defenses to bacteria. The American alligator and other crocodilians are evolutionarily ancient animals, whose plasma and leukocyte extracts have been previously shown to have antimicrobial activity.10,11,12 Discovering and identifying CAMPs from blood, however, is labor-intensive and expensive. We developed a new bioprospecting-particle and proteomics approach to CAMP discovery.13 This platform includes the ability to harvest efficiently from small sample volumes (e.g., 100 µl), advanced middle-down mass spectrometry techniques, and the identification of CAMPs through de novo peptide sequencing.
In the present work, we applied this process to the discovery of novel AMPs from plasma obtained from another reptile, the Komodo dragon (Varanus komodoensis), a large species of lizard found on the Indonesian island of Komodo. The saliva of wild Komodo dragons is thought to contain many different strains of bacteria, some of which are known to cause sepsis.14, 15 However, this bacteria-laden saliva never sickens the Komodo dragon. Investigators therefore hypothesized that proteins in the dragon's saliva or blood might provide immunity. Using our AMP discovery bioprospector platform,13 we identified a Komodo dragon peptide from Komodo plasma that we named VK25 (Fig. S1). Inspired by this VK25 peptide, we designed and created a new short synthetic peptide, which is a rearrangement of the two amino acids at the N-terminal of the VK25 peptide, and which we named DRGN-1. DRGN-1 exhibited promising antimicrobial and anti-biofilm properties. Moreover, the DRGN-1 peptide significantly promoted wound healing in vitro and in vivo, in both uninfected and mixed biofilm infected wounds.
Results
Antimicrobial activity of DRGN-1
To investigate whether DRGN-1 could exert antimicrobial effects, we tested the activity of CAMPs against the gram-negative Pseudomonas aeruginosa, Francisella novicida and Burkholderia thailandensis and the gram-positive Staphylococcus aureus. It should be noted that VK25 is a histone H1-derived peptide from Komodo, and histone peptides are generally weakly active or inactive under minimal inhibitory concentration (MIC) conditions, but may still have activity under low-salt, phosphate buffer conditions (EC50).16, 17 The results showed that DRGN-1 demonstrated the antimicrobial activity in an EC50 assay against the gram-negative bacterium P. aeruginosa (but not F. novicida or B. thailandensis) as well as the gram-positive bacterium S. aureus within the range of 0.77–7.1 µg/ml (0.50–4.62 µM). This was compared to VK25, which was significantly less active with anti-Pseudomonas activity in the range of 25–30 µg/ml and anti-Staphylococcus activity of more than 100 µg/ml (Table 1). These peptides do not show antimicrobial activity in full broth under MIC conditions, as is known for many other peptides, including the human cathelicidin LL-37.17, 18
Anti-biofilm activity of DRGN-1
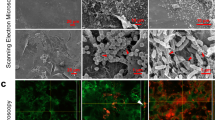
Next, the ability of DRGN-1 to prevent the formation of biofilm after 18 h was examined by crystal violet staining and confocal microscopy (Fig. 1). When cultured in a polystyrene tube under shaking conditions, P. aeruginosa and S. aureus form biofilms along the sidewall of the culture tube, which was strongly inhibited by DRGN-1 in a concentration-dependent manner (Fig. 1a, b). For confocal imaging, DRGN-1 (final concentration, 25 μg/ml) was incubated with bacteria overnight in the wells of a chamber slide. P. aeruginosa Green-fluorescent protein (GFP)+ and S. aureus Red-fluorescent protein (RFP)+ were used for these studies.19 The cells were incubated for 24 h in tryptic soy broth (TSB) medium in the presence of DRGN-1. The next day, the wells were rinsed and the development of a biofilm was compared with a control well (without DRGN-1). Biofilms of both P. aeruginosa and S. aureus were significantly reduced by DRGN-1. In mixed-culture biofilms grown for 24 h without treatment, P. aeruginosa significantly outgrows S. aureus,19 thus the appropriate starting ratio was established as a 99:1 ratio S. aureus to P. aeruginosa. Treatment with DRGN-1 had a significant inhibitory effect on the mixed bacterial cultures in contrast with untreated control.

Anti-biofilm activity of DRGN-1. a DRGN-1 anti-biofilm activity against S. aureus using three interdependent methods. The activity was analyzed by crystal violet staining of biofilm in culture tube at 24 h. b DRGN-1 anti-biofilm activity against P. aeruginosa. Pictures (right inlet in a and b) are a representative biofilm stained with crystal violet. P values were calculated using a two-tailed t test (assuming unequal variances) comparing test strains to untreated control (*P < 0.05; **P < 0.01; ***P < 0.001). c Biofilm evaluation by confocal microscopy. Representative images of biofilms of P. aeruginosa PAO1-pTDK-GFP expressing GFP, S. aureus SH1000 pAH9-RFP expressing RFP or mixed cultures exposed to PBS (control) or DRGN-1 (25 µg/ml) for 24 h. DRGN-1-exposed biofilms are thinner and more sparse than untreated controls. Scale bar: 20 µm
Ability of DRGN-1 to affect P. aeruginosa-infected HEKa keratinocytes
To gain insight into the ability of DRGN-1 to exert activity against internalized P. aeruginosa, HEKa keratinocytes were infected by P. aeruginosa ATCC 9027, and subsequently treated with DRGN-1 or VK25. As highlighted in Fig. 2, the killing of infected keratinocytes was pronounced 2 h after their exposure to the bacteria, presumably due to P. aeruginosa-induced apoptotic cell death, which was strongly prevented by exogenous treatment with DRGN-1 (Fig. 2a–d). Exogenous application of DRGN-1, but not VK25, significantly reduced the number of internalized bacteria with the peptide causing 64% reduction in survival of the internalized bacteria at the concentration used (30 µg/ml) after 2 h (Fig. 2e). LL-37 treatment of infected HEKa cells also caused a significant reduction in internalized P. aeruginosa cells. Both of these reductions were statistically significant compared to untreated or VK-25-treated cells. We conclude that DRGN-1 treatment of P. aeruginosa-infected HEKa keratinocytes leads to increased killing of those bacteria, whether directly by the peptide (direct antibacterial effect) or by a peptide-induced response of the keratinocyte cells (host-directed effect).

Treatment of cells with peptide reduces intracellular P. aeruginosa bacteria. a Light microscopy images of uninfected HEKa keratinocytes. b–d About 100,000 cells were infected with P. aeruginosa ATCC 9027 (1 × 107 CFU/ml) for 2 h, washed with PBS, treated with gentamicin to remove extracellular bacteria for 1 h, and b not-treated, or c treated with DRGN-1 (30 µg/ml), or (D) VK25 (30 µg/ml) for 2 h at 37 °C. e Bacterial survival within infected HEKa keratinocytes upon peptide treatment at 2 h post treatment. 100% represents the number of bacteria found inside of untreated HEKa cells. f Cytotoxicity testing: the peptides were tested for their cytotoxic effect against HEKa cells. The number of metabolically active cells remaining was determined by the MTT assay and is expressed as percentage with respect to the non-peptide-treated control cells. P values were calculated using a two-tailed t test (assuming unequal variances) comparing test strains to untreated control (*P < 0.05; **P < 0.01)
The cytotoxic effect of DRGN-1 and VK25 on HEKa cells was studied using an MTT (3-(4, 5-dimethylthiazolyl-2)- 2,5-diphenyltetrazolium bromide) based assay. As shown in Fig. 2f, both DRGN-1 and VK25 were devoid of toxic effects against these mammalian cells at a concentration range of 12.5–100 µg/ml. However, the control CAMP, LL-37, caused a significant decrease in HEKa cell viability at concentrations ranging from 50–100 µg/ml (Fig. 2f). DRGN-1 also did not show any toxicity against erythrocytes in a hemolysis assay at a high concentration (Fig. S2).
Effects of DRGN-1 on membrane permeability
Measuring the uptake of the fluorescent probe, ethidium bromide (EtBr), allowed us to assess the permeabilization of P. aeruginosa ATCC 9027 by these peptides. EtBr will only fluoresce if it is able to permeate both the inner and outer membranes through pores, and intercalate into the bacterial nucleic acid. The uptake of EtBr was significantly higher in the presence of DRGN-1 or LL-37 than in the presence of VK25 or the untreated control (Fig. 3a), suggesting significant disruption of the bacterial cytoplasmic membrane.

Membrane permeability, and membrane potential. a Fluorescent dye uptake for EtBr by the P. aeruginosa ATCC 9027 in the presence of peptides (20 µg/ml). P values were calculated using a two-tailed t test (assuming unequal variances) comparing with untreated control at 20 min (**P < 0.01). b Membrane depolarization was monitored by the change in the intensity of fluorescence emission of the membrane-potential-sensitive dye DiSC3(5) (excitation wavelength at 622 nm, emission wavelength at 670 nm) on addition of 20 µg/ml peptides or 0.1% Triton X-100 for a positive control
The effect of CAMPs on the membrane potential was measured by monitoring the membrane potential using the fluorescent dye 3,3'-dipropylthiadicarbocyanine iodide (DiSC3(5)) (Fig. 3b).20 DiSC3(5) dye is taken up by bacterial cell membranes in proportion to the electrical potential gradient across the bacterial cytoplasmic membrane. The dye concentrates in the cytoplasmic membrane, where it is able to self-quench its own fluorescence. Any compound that can permeabilize the cytoplasmic membrane and thus depolarize the gradient, such as an antimicrobial peptide, will lead to the release of DiSC3(5) and thus leads to an increase in fluorescence.21 The peptides were tested against P. aeruginosa ATCC 9027 at a concentration of 20 µg/ml, which was expected to cause a large and immediate increase in fluorescence (indicative of loss of membrane potential). DRGN-1 showed ~10% depolarization relative to Triton X-100 (which served as the “100%” positive control), while VK25 did not have any effect. These results suggest that in addition to membrane disruption, DRGN-1, much more than VK25, permeabilizes the membranes and slightly depolarizes the membrane potential.
Structure analysis of DRGN-1 by circular dichroism (CD) spectroscopy
The reversal of two N-terminal amino acids (ser–pro) in a histone H1-derived peptide of V. komodoensis caused drastic changes in antimicrobial activity, anti-biofilm activity, membrane permeability, and DNA binding. Therefore, we sought to determine the secondary structure of the peptides in the membrane-mimicking media, sodium dodecyl sulfate (SDS) and trifluoroethanol (TFE) (Fig. 4) using CD. Cathelicidin AMPs (such as LL-37) maintain a random or disordered structure until associated with a bacterial membrane or detergent micelle.22 The detergent SDS is able to form micelles that have a negatively charged surface,23 which mimics the bacterial membrane and forces the CAMP into a more ordered conformation such as a helix.24, 25 TFE is used in CD to promote a helical structure26 and stabilize secondary structure.27

CD spectra of peptides in the presence of SDS or TFE. a, b CD spectra of 100 μg/ml concentration of peptides in the presence of the indicated concentrations of sodium phosphate buffer (PB), SDS or TFE. c Helical wheel plot of DRGN-1 predicted by HeliQuest. d Composition of α-helix conformation calculated from the ellipticity value at 222 nm using the relation (% α-helix = ([θ222]−3000)/(−36000−3000)). The ratio [θ]222/[θ]208 obtained by CD spectroscopy is defined as R2 value. R2 >1 is the hallmark of coiled-coil α-helical structure with >80% helical content (32)
By qualitatively evaluating the spectra, we conclude that DRGN-1 and VK25 showed a random coil conformation in aqueous solution (10 mM phosphate buffer) and 60 mM SDS solution. The α-helical content of DRGN-1 and VK25 were 22.3 and 32.8% in 50% TFE, respectively, with the percent contribution to secondary structure was measured using methods determined by Raussens et al.28 Both DRGN-1 and VK25 have a very low hydrophobic moment (0.013 and 0.088, respectively) compared with the highly α-helical LL-37 (0.521), suggesting that DRGN-1 and VK25 are not α-helical (Table 2). Nevertheless, DRGN-1 (0.64) displays a slightly larger R2 value, the hallmark of the coiled-coil α-helical structure in 50% TFE, than VK25 (0.44). It has been shown by several groups that TFE can induce and support α-helical structures in peptides.29, 30 These results suggest that the reversal of amino acids at the N-terminal contributes to an overall conformational change.
Efficacy of DRGN-1 on in vivo wound healing
We have demonstrated that DRGN-1 peptide significantly inhibited biofilms of P. aeruginosa and S. aureus separately, and a mixed biofilm. Since biofilm may protect bacteria during infection, we sought to test whether application of DRGN-1 to an infected wound would promote clearance and healing.
To evaluate the impact of a mixed biofilm infection on an open wound, we developed a wound model using BALB/c mice. We have previously characterized P. aeruginosa and S. aureus-mixed biofilms for their heterogeneity and composition;19 these biofilms were generated and applied to a mouse wound. In our model, we will test whether wound healing in P. aeruginosa and S. aureus biofilm-infected skin wounds is significantly improved following peptide treatment compared to an untreated, infected wound, following the design of other published studies.31
We have previously characterized a mixed biofilm of P. aeruginosa and S. aureus,19 which is applied to the wound as the source of infection. Using this model, we evaluated the potential clinical application of DRGN-1 against P. aeruginosa/S. aureus biofilm-infected wounds (Fig. 5). Full-thickness round wounds of 6 mm in diameter were made between the shoulder blades of mice and overlaid with a mixed biofilm of P. aeruginosa/S. aureus grown on agar for 2 days.19 The kinetics of wound closure was evaluated by measuring the original wound area before and after treatment with peptides. DRGN-1, VK25, and LL-37 (20 μg/wound) were applied topically to their specified groups every other day until day 6. Reconstitution buffer was used as a control in this experiment. Treatment with DRGN-1 on a mixed biofilm-infected wound significantly reduced wound size compared to the controls as soon as day 4 post treatment. When the wound dressing was taken off on day 6, wound healing sped up in both treated and untreated conditions as observed at day 9.

Efficacy in wound healing of peptides in a mixed biofilm-infected mouse skin wound model. a Schematic experimental design that includes the timing of infection, drug treatments, dressing off. b Effect of peptides on closure of P. aeruginosa/ S. aureus-infected wounds in mice. Wounds shown are representative of the group. Six-millimeter-diameter excisions were made on the back of mice. Each wound was infected with a mixed biofilm (P. aeruginosa/ S. aureus) grown on a polycarbonate membrane for 2 days. DRGN-1, VK25 or LL-37 (20 µg) was topically treated every 2 days in 1% hypromellose (n = 6 mice per group). Mice were lightly anesthetized with isofluorane and photographed on the indicated days following treatment. c, d Assessment of bacterial colonization in P. aeruginosa/ S. aureus-infected wounds. Wound tissue samples were harvested on day 6 after treatment, and the number of CFU/wound was counted in a selective medium for each species. The median value in each group is shown as a horizontal bar (n = 6; **p < 0.01 and ***P < 0.001)
Wounds treated with DRGN-1 were consistently smaller than wounds treated with phosphate buffered saline (PBS) buffer, VK25, or LL-37 (Fig. 5b, Fig. S3). The results showed that the difference in size was apparent by day 4 after the first treatment. By day 11, the wounds treated with DRGN-1 were completely healed, while the wounds treated with PBS, VK25, and LL-37 were not. After 6 days, the first treatment (three treatments), the bacterial counts of S. aureus and P. aeruginosa were significantly reduced in the DRGN-1-treated and LL-37-treated groups compared to the control group (Fig. 5c, d). In contrast, there was no significant reduction in S. aureus and only a slight reduction in P. aeruginosa in VK25-treated wound areas. These results indicate that DRGN-1 accelerates skin wound closure and healing, and reduction of bacterial counts in the wounds of P. aeruginosa/ S. aureus biofilm-infected mice. When the wound dressings were taken off on day 6, wound healing was accelerated in both treated and untreated conditions due to the air exposure, as observed at day 9 and 11 post treatment. In addition, hematoxylin–eosin staining of skin at day 11 clearly demonstrated that the skin layers were completely rehabilitated in the DRGN-1-treated wounds (Fig. S4).
To precisely dissect the effect of DRGN-1 on re-epithelialization and new tissue formation in wound, we then investigated the in vivo wound closure activity of DRGN-1 in a mouse full-thickness skin (uninfected) wound model. In this study, we used a silicone splint32 to avoid contraction of an extensive subcutaneous striated muscle layer, which is specific to mice, called the panniculus carnosus that is largely absent in humans.33 Full-thickness round wounds of 6 mm in diameter were made between the shoulder blades on mice, and the wound closure was evaluated by measurement of original wound area (%). When DRGN-1 or VK25 (20 μg/wound) was applied topically every 2 days post injury, DRGN-1-treated wounds were consistently smaller than wounds treated with PBS or VK25 (Fig. 6). At day 6, the wounds treated with DRGN-1 were smaller in area (41% compared to original wound area) than the control (65%) or VK25-treated (85%) wound. These results indicate that DRGN-1 peptide directly accelerates skin wound closure and healing in mice. In summary, wound healing was significantly enhanced by DRGN-1 in both uninfected and mixed biofilm (P. aeruginosa and S. aureus)-infected murine wounds.

Wound healing of DRGN-1 in a mouse full-thickness skin (uninfected) wound model. a Schematic experimental design that includes the timing of drug treatments. b Effect of DRGN-1 on closure of full-thickness excisional wounds (6-mm-diameter) in mice. Each wound was treated with DRGN-1 or VK25 every 48 h (n = 5 mice per group). The same animal was lightly anesthetized and photographed on the indicated days following injury. c Changes in percentage of wound area at each time point in comparison to the original wound area. *P < 0.05 and **P < 0.01
Efficacy of DRGN-1 on keratinocyte migration in a scratch-wound closure assay
Some CAMPs also have host-directed functions and thus CAMPs are also called host-defense peptides. One of the potential functions of CAMPs is the activation of keratinocyte migration resulting in the promotion of skin wound healing,9, 34, 35 which may contribute to the in vivo results we observed. Therefore, we tested the capacity of DRGN-1 to induce wound closure via this mechanism.
We first investigated whether CAMPs induced the migration of HEKa cells into a wound area using a scratch wound closure assay. The migration of cells into the wounded area was significantly increased in the presence of DRGN-1, as well as LL-37, compared to migration in the control within 7 and 24 h after wounding (Fig. 7a, b). There, significantly less wound closure activity was observed in the presence of VK25, which was slightly more than the control treatment (peptide reconstitution buffer). This suggests that part of the wound-healing benefit of DRGN-1 peptide is the promotion of keratinocyte migration. Subsequently, in order to determine whether the wound closure was influenced by an increased proliferation of keratinocytes upon their exposure to DRGN-1, the wound healing assay was carried out in the presence of 20 μM mitomycin C to block cell proliferation. As shown in Fig. 7c, d, mitomycin C strongly inhibited the migratory activity of keratinocytes induced by DRGN-1 (10 μg/ml). This suggests that the proliferation of HEKa cells highly contributes to the wound-healing effect produced by DRGN-1. To further understand the pathway by which DRGN-1 activates keratinocyte migration and proliferation, epidermal growth factor receptor (EGFR) was selected as a potentially activated kinase, because it has been reported to be central to the regulation of keratinocyte migration and proliferation.35,36,37 Previous studies have shown that LL-37 peptide can induce transactivation of the EGFR.35, 38

'Scratch' assay for HEKa cells. a Confluent cells in medium with HGKS were wounded by a scratch with a pipette tip, and ‘gap’ closure in the presence of peptides (10 µg/ml) was monitored by light microscopy at 0, 7 and 24 h post peptide treatment. b Relative wound closure of separate experiments using either 1 or 10 µg/ml of peptide as indicated was calculated from micrographs using ImageJ software. The results shown are combined from three independent experiments; error bars represent the mean ± SD. The dotted lines indicate the control values for easier comparison to the other bars. c HEKa cells were pre-incubated with 20 µM mitomycin C for 90 min or 0.2 µM AG1478 for 10 min and subsequently treated or not with 10 μg/ml DRGN-1, as indicated. In parallel, cells treated with the peptide alone were included for comparison. Cells incubated with medium served as a control (Ctrl). d Relative wound closure. All data are the means of at least three independent experiments ± SE. The levels of statistical significance between groups are indicated as follows: *P < 0.05
AG1478 is an inhibitor of EGFR tyrosine kinase, which blocks the activation of EGFR.38, 39 Interestingly, pretreatment of HEKa cells with 0.2 μM AG1478 prevented cell migration induced by DRGN-1. To examine potential EGFR pathway activation by DRGN-1, we investigated EGFR phosphorylation by Western blotting with an anti-phospho-EGFR antibody. DRGN-1 treatment led to phosphorylation of the EGFR at 5 min and the phosphorylation persisted for 20 min, which was not observed in AG1478-treated cells (Fig. 8a–c). The amount of EGFR protein did not change during this time period and treatment with human keratinocyte growth supplements (HKGS) containing 0.2 ng/ml of human epidermal growth factor (EGF) was used as a positive control. EGFR signaling pathway initiates several signal transduction cascades, such as STAT3 (Signal Transducer and Activator of Transcription) and Akt with the functional consequence of enhanced cell migration.40 STAT3 was also phosphorylated 10–30 min after the addition of DRGN-1 (Fig. 8d–f). From these results, we conclude that DRGN-1 induces keratinocyte migration via EGFR-STAT3 pathway activation.

Activation of EGFR-STAT pathway by DRGN-1. a, b, d, e Subconfluent keratinocytes were starved for 2 h in HGKS-free medium and stimulated with 10 µg/ml DRGN-1. b, e For EGFR inhibition, cells were preincubated with 0.2 µM AG1478 for 10 min and subsequently treated with 10 μg/ml DRGN-1. The cells were harvested into lysis buffer at the indicated times. a, b, d, e The phosphorylation of EGFR (p-EGFR) and STAT3 (p-STAT3) was analyzed by Western blotting. EGFR indicates total EGFR protein. STAT3 indicates total STAT3 protein. c, f Densitometric analyses of the expression of phospho-protein relative to un-phosphorylated proteins from panels a & b, d & e above
Discussion
In this study, we evaluated the biological activities of the histone H1-derived peptide, VK25 identified from Komodo dragon blood and the N-terminally modified synthetic peptide DRGN-1. The peptide DRGN-1 has broad-spectrum activity, and is highly potent in antimicrobial and anti-biofilm assays. The bactericidal properties of a few histone-derived fragments (mainly H1 or H2B) have been recently reported in the literature.41,42,43,44,45 Some histone H1-derived CAMPs are found in various animals including fish44, 46 as well as humans.47, 48 We previously identified a potential histone H1-derived CAMP from Komodo dragons by a novel peptide-identification bioprospector platform.13 VK25, a peptide containing a native sequence from Komodo histone H1, did not display significant antimicrobial activity despite the predictive algorithm indicating a high probability of antimicrobial activity (Table 2). When the two N-terminal amino acids were switched from “SP” to “PS” in a new synthetic peptide that we named DRGN-1, it exhibited potent antimicrobial activity. Thus, we sought to investigate why this reversal induced the activity.
One possibility is that this switch may induce overall structural changes, thus enabling the peptides to exert differential activity.5 We determined the secondary structures of the peptides DRGN-1 and VK25 using CD spectroscopy. This data suggested that both peptides have a similar secondary structure, displaying typical random coil structure in aqueous solution. In the presence of 50% TFE, the peptides underwent the conformational changes to α-helical structure. However, the R2 ([θ]222:[θ]208 ratio) of DRGN-1 in 50% TFE was somewhat higher than VK25, suggesting a slightly more organized structure of DRGN-1 in this solution.49 When we examined the structure of CAMPs in a membrane-mimicking environment (SDS), there was no conformational difference between the peptides, supporting the idea that these CAMPs have a random coil structure in aqueous solutions. Although CD spectra suggest a slight conformational difference under TFE conditions, this may not be sufficient to explain the significantly different antimicrobial activity of these peptides.
Another potential explanation of VK25’s relative inactivity is peptide degradation by the “N-end rule” pathway, which is mediated by the molecular chaperones ClpXP or ClpS-ClpAP systems in bacteria. Our data suggested the hypothesis that the switching of the proline–serine sequence of the original VK25 peptide may contribute to DRGN-1 peptide’s antimicrobial activity, perhaps by the “N-end rule”. Notably, the DRGN-1 peptide displayed activities comparable to that of the “classic” AMP human cathelicidin LL-37 (EC50 = 0.24–2.7 µg/ml; 0.05–0.60 µM).3, 4 The “N-end rule” states that the half-life of a protein is determined by the nature of its N-terminal residue.50 Mogk et al. suggest that in prokaryotic cells, there is a “proteasome-like” machinery that includes an ATPase molecule (either ClpA, or ClpX) working with a proteolytic molecule such as Lon that is either covalently attached or such as ClpP, which is diffusible. This protein complex mediates the proteolysis of misfolded or specially tagged proteins in the bacterial cytosol.50 In particular, ClpXP system is able to recognize 5 motifs at the terminal regions: two located at the C terminus of proteins, and three N-terminal motifs.51 The N-terminal regions of Dps, σS and GlcB that start with serine (Ser) contain sequences that are necessary (and sufficient) to target those proteins for degradation by the ClpXP system.51 In this aspect, we speculate that since VK25 starts with Ser at the N-terminus, it may be a substrate for ClpXP. DRGN-1 starts with proline (pro) at the N-terminus and exhibits significant antimicrobial activity, suggesting it is resistant to ClpXP-mediated degradation. In addition, the addition of Pro at the N-terminus of the cell-permeable antimicrobial peptide Buforin II led to greater efficacy of this CAMP against P. aeruginosa and S. aureus (Chung and van Hoek, unpublished data). This implies alteration of CAMP degradation (thus improving the CAMP stability) by the “N-end rule” pathway. Further studies will be done to determine the contribution of the “N-end rule” to improve the activity of the DRGN-1 AMPs, to address remaining questions such as, the cytosolic location of ClpXP vs. the membrane-targeting activities of the AMPs.
To study the modes of antimicrobial action of DRGN-1 peptide, we first measured the intrinsic permeabilization of the bacterial membrane by the peptides with EtBr uptake and dysregulation of the membrane potential with DiSC3(5). EtBr uptake data indicated that DRGN-1, much more than VK25, moderately disrupts the cytoplasmic membrane causing leakage of intracellular components (although less than LL-37). In contrast, DRGN-1 induced a slow dissipation of the membrane potential when an environment-sensitive fluorescent probe DiSC3(5) was used. The results showed that DRGN-1 could induce bacterial membrane disruption, but does not drastically alter the proton motive force, potentially causing structural damage to the cytoplasmic membrane. Due to its random coil structure, DRGN-1 is likely to be a pore-forming and membrane-permeable peptide. Thus, it seems that the target sites of DRGN-1 are likely to be both the cell membrane itself and potentially the nucleic acids inside of the cell. A DNA binding assay (results not shown) supported this hypothesis, showing that DRGN-1 binds tightly to DNA, presumably resulting in inhibition of the macromolecular synthesis of the cell once inside the bacteria, similar to the mechanism of Buforin II52 and PR-39.53 Peptides such as DRGN-1 that have multiple modes of action against bacteria are of significant interest for potential future development, as it is less likely that bacteria will be able to generate resistance mechanisms against multiple modes of action.
DRGN-1 exhibited wound closure activity both in vitro and in vivo through the proposed mechanism of promotion of keratinocyte cell migration and proliferation. Thus, we have demonstrated that in addition to its antimicrobial activities, DRGN-1 has host-directed activities. DRGN-1 possesses potent, direct wound closure activity in an in vitro wound scratch assay using human keratinocytes. DRGN-1 elicited significantly stronger wound closure activity than VK25 by inducing more HEKa cell migration (similar to LL-37), accompanied by activation of EGFR. DRGN-1 treatment also induced phosphorylation of EGFR and STAT3 in HEKa cells. A recent study suggested that LL-37 can activate EGFR and lead to phosphorylation of STAT3, leading to keratinocyte migration38 through a mechanism of “transactivation”. That study demonstrated that LL-37 activates a metalloproteinase, which then cleaves the extracellular domain of heparin-binding EGF (HB-EGF). The soluble-form of HB-EGF then binds to and phosphorylates EGFR.38 Therefore, we postulate that DRGN-1 may act through EGFR, similar to the transactivation mechanism of LL-37, which is mediated by HB-EGF. We are continuing to determine the mechanism of DRGN-1 action on host cells.
In addition, topical application of DRGN-1 accelerated closure and healing of full-thickness excisional wounds in mice. Consistent with the in vitro scratch wound assay, there was no significant wound reduction in VK25-applied wound areas compared to the control wound. These results suggest that DRGN-1, significantly more than VK25, is active for wound closure in the in vivo wound environment, although the precise mechanistic differences between our two peptides towards the EGFR pathway has not yet been elucidated.
Anti-biofilm activity is another biological function exhibited by DRGN-1. Biofilms are structured multicellular communities of microorganisms that are associated with surfaces and resistant to antibiotic treatment.54 Our results showed that our short synthetic peptide DRGN-1 significantly inhibited biofilms of P. aeruginosa and S. aureus separately, as well as in mixed biofilms, similar to the human cathelicidin LL-37.3, 4, 19 Since the formation of biofilm protects bacteria during infection and allows for survival in a hostile environment,55 the inhibition of biofilm formation by DRGN-1 in a wound may serve as an additional mechanism to prevent bacterial survival in the host and promote healing.6, 31
To evaluate in the infected wounds the impact of DRGN-1 on a mixed biofilm infection of a wound, we developed a mouse model using BALB/c mice. We have previously characterized P. aeruginosa and S. aureus-mixed biofilms,19 which were applied to the wound as a source of infection. In our model, wound healing in P. aeruginosa and S. aureus biofilm-infected skin wounds was significantly delayed compared to an uninfected wound. Treatment with DRGN-1 on a mixed biofilm-infected wound significantly reduced wound size compared to the controls as soon as day 4 post treatment, and appeared to promote clearance of the biofilm from the wound. When the wound dressing was taken off on day 6, wound healing was faster in both treated and untreated conditions as observed at day 9. This observation supports a previously described hypothesis that oxygen is another essential component of wound healing.56 Thus, treatment with DRGN-1 significantly promoted the healing of mixed-biofilm-infected wounds in vivo.
One potential limitation of these experiments is that these studies are performed on the mouse wound model, as a first demonstration of in vivo activity prior to advancing towards clinical studies. Another potential limitation of this study is that our model of polymicrobial biofilm infection has two organisms, P. aeruginosa and S. aureus, not the full repertoire of all six ESKAPE pathogens that are known to potentially infect wounds. However, these studies suggest that DRGN-1 is a good candidate for additional studies and possible development as a topical therapeutic agent for infected wounds.
Taken together, our data on the antibacterial, anti-biofilm, and wound closure activities suggest that DRGN-1, a synthetic derivative of the histone H1-derived peptide VK25 from the Komodo dragon, might be a potent wound healing agent in polymicrobial infected wounds, with host-directed effects accomplished through the EGFR-STAT pathway as well as through anti-biofilm activity against the bacteria. Furthermore, DRGN-1 did not affect the viability of human keratinocytes and erythrocytes. These features suggest that DRGN-1 might be developed as a topical agent for infected wound treatment.
Methods
Bacterial strains and peptides
Pseudomonas aeruginosa ATCC 9027, P. aeruginosa PAO1 ATCC 15692, Escherichia coli O157:H7 ATCC 51659, and Staphylococcus aureus ATCC 25923 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). F. novicida U112 NR-13 was obtained from BEI Resources (Manassas, VA, USA). P. aeruginosa PAO1 cells that constitutively expressed GFP (PAO1 pTDK-GFP) and S. aureus SH1000 that expressed RFP (SH1000 pAH9-RFP) were kindly provided by Dr. Weibel57 and Dr. Boles,58 respectively. DRGN-1 and VK25 were synthesized by the conventional solid-phase method by ChinaPeptides (Suzhou, China). The crude compounds were purified to chromatographic homogeneity in the range of >95% by using reversed-phase high-performance liquid chromatography with a mass spectrometer by the company. LL-37 was purchased from AnaSpec (Fremont, CA, USA). All peptides were reconstituted in a buffer consisting of 20 mg bovine serum albumin and 10 µl acetic acid in 50 ml PBS.59 Table 2 summarizes characteristics of peptides used in this study.
Assays for antimicrobial and anti-biofilm activity
P. aeruginosa and S. aureus were cultured in nutrient broth and mannitol salt broth, E. coli was cultured in Luria broth (LB), and F. novicida was cultured in tryptic soy broth containing 0.1% cysteine overnight at 37 °C. Overnight-cultured bacteria were numerated by a standard curve using optical density vs. colony forming units (CFU) per ml. The culture was resuspended in 10 mM sodium phosphate buffer (PB) (pH 7.4) and adjusted to a final amount of 2 × 106 CFU/ml. For EC50 determination, peptides were used at variable concentrations (0.1–100 μg/ml) from a stock solution. Fifty microliters of each concentration of peptide solution was added to each corresponding well of a 96-well plate (BD Falcon, USA) and 50 μl of bacteria was added and incubated for 3 h at 37 °C. After dilution in the buffer, the bacteria were spotted on an agar plate of the corresponding media and colonies were numerated. Anti-biofilm activity was measured as previously described19 with the following modifications. For the mixed biofilm inhibition assays in the 96 well plate format, a 99:1 ratio of S. aureus and P. aeruginosa was used as the starting inoculation. Two-fold diluted peptides in TSB medium were incubated with bacteria (1:50 dilution in the same medium) overnight. A crystal violet assay was used for biofilm formation as described.60 A tube-based biofilm assay was performed as previously described.61
For confocal microscopy, bacteria (P. aeruginosa PAO1 pTDK-GFP and S. aureus SH1000 pAH9-RFP) were grown on the Lab-Tek II chamber slide (Thermo Scientific) for 24 h in TSB and incubated with the indicated amount of peptide. Biofilms attached to the glass were observed with a 40× objective using a Nikon TE2000-U confocal laser scanning microscope equipped with an argon ion laser.
Cell infection
Adult human epidermal keratinocytes (HEKa) (Invitrogen) cells were seeded in 24-well plates (Corning) and grown until they reached confluence (48 h) in EpiLife medium supplemented with HKGS containing 0.2% v/v bovine pituitary extract, 5 µg/ml bovine insulin, 0.18 μg/ml hydrocortisone, 5 µg/ml bovine transferrin, and 0.2 ng/ml human EGF (Invitrogen). The bacterial strain P. aeruginosa ATCC 9027 was grown in brain heart infusion (BHI) media at 37 °C with mild shaking and then harvested by centrifugation. Bacterial suspension (MOI 10) in EpiLife medium was co-incubated with keratinocytes for 2 h at 37 °C and 5% CO2. The medium was aspirated and the cells were washed three times with PBS to remove non-adherent bacteria. In order to kill extracellular bacteria, cells were incubated for 1 h in EpiLife without HKGS supplemented with 100 μg/ml of gentamicin. Afterwards, the medium was aspirated and the infected cells were washed three times, as described above. Three hundred microliters of peptide solution (30 µg/ml) in EpiLife without HKGS was added to each well and the plate was incubated for 2 h at 37 °C and 5% CO2. Then, the peptide solution was removed and the cells were washed again with PBS three times and lysed with 100 μl of 0.1% Triton X-100 in PBS for 5 min at room temperature. The resulting bacterial suspension was sonicated in a water bath for 10 min, to break up possible clumps, and appropriate dilutions were plated on agar plates for colony counts; the CFU were counted after 24 h at 37 °C.
Membrane permeability
For intrinsic permeabilization of bacterial membrane, EtBr uptake was in performed 10 mM potassium phosphate buffer (pH 7.2) as described62 with some modifications. Overnight cultures of P. aeruginosa ATCC 9027 in BHI were transferred to fresh medium and grown to OD600 values of 0.5–0.6 (log phase). Cells were harvested and resuspended in 10 mM potassium phosphate buffer (pH 7.2) at a final OD600 of 0.12–0.15. The cells (40 µl) were added to 50 μl of 12 µM EtBr, followed by the peptide (20 µg/ml final) samples after 30 s. Excitation and emission wavelengths were set at 545 and 600 nm, respectively. The increase in fluorescence as a result of partitioning of EtBr into the cytosol was measured 20 min after the addition of peptides using a fluorescence spectrophotometer (Tecan Safire2 Multi-detection Microplate Reader). Membrane potential was measured by DiSC3(5) as described.63 Briefly, mid-log phase bacteria were suspended in 5 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer containing 20 mM glucose (OD = 0.05) and DiSC3(5) (1 µM final) was added for 1 h at room temperature. Then KCl was added at 0.1 M of final concentration. After addition with peptide (20 µg/ml) or TX-100 (final 0.1%), fluorescence was excited at 622 nm (bandwidth 5 nm) and monitored at 670 nm for emission (bandwidth 20 nm).
Circular dichroism (CD) analysis
CD spectra of the peptides were collected using a Jasco J-1500 spectropolarimeter as previously described.3 Data was collected in a 0.1 cm path length cuvette, with a chamber temperature of 25 °C. The spectra were collected from 190 to 260 nm with 0.1-nm intervals. Three scans per sample were averaged. All peptides were analyzed at 100 μg/ml concentration in three different mediums: 10 mM PB (pH 7), 50% (v/v) TFE in 10 mM PB (pH 7), and 60 mM SDS in 10 mM PB (pH 7). The ellipticity at 222 nm is used as a means of providing a rough estimate of the degree of peptide helicity under the three conditions investigated (10 mM PB, 60 mM SDS, and 50% TFE) consistent with the work reported by Raussens, et al.28 and as previously reported by our group.22 We performed the CD scan across the wavelengths shown in Fig. 4. In 10 mM PB and 60 mM SDS conditions, the peptides appeared to show very low helical content, however the results in TFE suggest higher helical character. Percent contribution to secondary structure was measured using methods determined by Raussens et al.28 We have applied a second approach to estimating helicity for the peptides in TFE. We have used the ratios of the signals at 222 and 208 nm (R2 = [Θ]222 nm:[Θ]208 nm), as has been reported by Graddis, et al.49 and Juba, et al., to provide a second means of estimating potential for helical structure.
In vitro scratch wound closure assays
In vitro wound closure was assayed in confluent cell monolayers of HEKa (Invitrogen) as described.34 Cells were seeded in 6-well plates (Corning, NY, USA) and grown until they reached confluence (48 h) in EpiLife medium supplemented with HKGS. Cells were starved for 18 h in EpiLife without HKGS and then scratched once vertically with a 200-μl pipette tip to create an artificial wound. For inhibitor studies, mitomycin C (20 µM final) and AG1478 (0.2 µM final) were added 90 and 10 min prior to scratch generation. After being washed twice with PBS to remove cellular debris, cells were treated with 10 µg (Fig. 7a) or 1 and 10 µg (Fig. 7b) DRGN-1, VK25 or LL-37. PBS was used as a negative control. Cells were photographed before peptide treatment and 7 and 24 h after peptide treatment using an EVOS® Cell Imaging Systems (Invitrogen). The same image fields were captured and the wound areas were estimated by ImageJ software.64
MTT assay
The toxic effect of the peptides on adult HEKa cells was evaluated using the MTT dye colorimetric method.65 MTT’s intensity of the color is directly proportional to the number of metabolically active cells. Keratinocytes were plated in triplicate wells of a 96-well plate, at 4 × 104 cells/well in EpiLife medium supplemented with HKGS. After overnight incubation at 37 °C in a 5% CO2 atmosphere, the plate was incubated with the peptides at different concentrations for 2 h, and 0.5 mg/ml of MTT was added. After 4 h of incubation, the formazan crystals were dissolved by adding 100 μl of acidified isopropanol, and the absorbance of each well was measured at 570 nm using a microplate reader (PowerWave X, BioTek).
Hemolytic assay
The hemolytic activities of peptides was determined using sheep erythrocytes66 in an assay adapted to a microtiter plate format. Briefly, erythrocytes were prepared by centrifuging fresh defibrinated blood, re-suspending the pelleted cells in sterile PBS. The cells were washed with PBS three times. In the final wash, the cells were resuspended in 0.75 ml PBS. A 2% erythrocyte suspension was prepared for the assay and incubated with the indicated amount of peptide.
Western blotting
Semi-confluent HEKa cells were serum-starved for 3 h and then stimulated with 10 µg/ml DRGN-1 or HKGS for 5–30 min at 37 °C. AG1478 (0.2 µM final) was added 10 min prior to DRGN-1 or HKGS treatment. Cells were lysed with 2× Laemli sample buffer (Bio-Rad) containing 5 mM sodium fluoride and 2 mM sodium vanadate. Cell lysates were separated on a 4–12 % SDS-PAGE gel and transferred to polyvinylidene difluoride membrane using an iBlot system (Invitrogen). The membranes were probed with antibodies specific to phospho-EGFR and EGFR (Cell Signaling). The membranes were then treated with horseradish peroxidase-conjugated donkey anti-rabbit IgG (Abcam) and developed by SuperSignal West Femto Chemiluminescent Substrate (Fisher Scientific) under GelDoc Imaging System (Bio-Rad).
Animal model of wound healing
Female BALB/c mice (12 weeks old) were purchased from Jackson Laboratory (Bar Harbor, ME, USA). The Institutional Animal Care and Use Committee at George Mason University and ACURO approved all protocols (Protocol No. 0271). The study adhered to the NIH Guide for the Care and Use of Laboratory Animals. The mice were anesthetized with isoflurane and the dorsal skin was shaved to remove hair. The next day, a superficial wound was created between the shoulder blades of the mice using a 6-mm biopsy punch. The wounds were topically treated with 20 μl of a gel containing 20 μg DRGN-1 or VK25 in 1% hypromellose (hydroxy propyl methyl cellulose, HPMC, Sigma) every 48 h (n = 5 animals per group) and covered with a Tegaderm-type semiocclusive dressing (3M, St. Paul, MN) attached to a silicone disc (0.5 mm thick, Life Technologies) by Mastisol liquid (Eloquest Healthcare) for study involving only peptides. Each wound site was digitally photographed at the indicated time intervals and wound areas were determined on photographs using ImageJ. Changes in the wound areas over time were expressed as the percentage of the original wound areas.
For infected wounds, S. aureus ATCC 25923 and P. aeruginosa ATCC 9027 were mixed at an initial ratio of 1000:1 and grown on a 6-mm polycarbonate filter on Nutrient agar plates overnight as previously described.19 The resulting biofilm was applied to the biopsy wound and covered with a Tegaderm dressing adhered to the skin with Mastisol liquid. Mice were housed individually with ad libitum rodent chow and water for 2 days. The wounds (5 mice per group) were topically treated with 20 μl containing 20 μg peptide with 1% hypromellose in 10 mM phosphate buffer every 2 days until 6 days from the first treatment.67 Each wound site was then digitally photographed at the indicated time intervals, and wound areas were determined on photographs using ImageJ software. Changes in wound areas over time were expressed as the percentage of the original wound areas. To assess bacterial colonization, wounds were harvested at day 6 post treatment and homogenized in PBS. Mannitol salt agar was used to selectively culture S. aureus, and tryptic soy agar with triclosan or centrimide agar was used to selectively culture P. aeruginosa.19 At day 11 post-treatment, all mice were sacrificed and tissue samples were harvested in a 4% paraformaldehyde solution. The sections of fixed skin were stained with hematoxylin and eosin for further histological analysis by AML Laboratories (Baltimore, MD, USA).
Statistical analysis
P-values were calculated by one-way ANOVA or Student’s t-test. Statistical significance was set at P < 0.05. Data are presented as arithmetic mean ± SD. Each experiment was repeated at least three times independently.
References
de la Fuente-Nunez, C., Reffuveille, F., Haney, E. F., Straus, S. K. & Hancock, R. E. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog. 10, e1004152 (2014).
Amer, L. S., Bishop, B. M. & van Hoek, M. L. Antimicrobial and antibiofilm activity of cathelicidins and short, synthetic peptides against Francisella. Biochem. Biophys. Res. Commun. 396, 246–251 (2010).
Dean, S. N., Bishop, B. M. & van Hoek, M. L. Natural and synthetic cathelicidin peptides with anti-microbial and anti-biofilm activity against Staphylococcus aureus. BMC Microbiol. 11, 114 (2011a).
Dean, S. N., Bishop, B. M. & van Hoek, M. L. Susceptibility of Pseudomonas aeruginosa biofilm to alpha-helical peptides: D-enantiomer of LL-37. Front. Microbiol. 2, 128 (2011b).
Fjell, C. D., Hiss, J. A., Hancock, R. E. & Schneider, G. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 11, 37–51 (2011).
Duplantier, A. J. & van Hoek, M. L. The human cathelicidin antimicrobial peptide LL-37 as a potential treatment for polymicrobial infected wounds. Front. Immunol. 4, 143 (2013).
Findlay, F., Proudfoot, L., Stevens, C. & Barlow, P. G. Cationic host defense peptides; novel antimicrobial therapeutics against category A pathogens and emerging infections. Pathog. Glob. Health 110, 137–147 (2016).
Mansour, S. C., Pena, O. M. & Hancock, R. E. Host defense peptides: front-line immunomodulators. Trends. Immunol. 35, 443–450 (2014).
Steinstraesser, L. et al. Innate defense regulator peptide 1018 in wound healing and wound infection. PLoS One 7, e39373 (2012).
Cassir, N., Rolain, J. M. & Brouqui, P. A new strategy to fight antimicrobial resistance: the revival of old antibiotics. Front. Microbiol. 5, 551 (2014).
Darville, L. N., Merchant, M. E., Hasan, A. & Murray, K. K. Proteome analysis of the leukocytes from the American alligator (Alligator mississippiensis) using mass spectrometry. Comp. Biochem. Physiol. Part D Genomics Proteomics 5, 308–316 (2010).
Fernandez, L., Breidenstein, E. B., Song, D. & Hancock, R. E. Role of intracellular proteases in the antibiotic resistance, motility, and biofilm formation of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 56, 1128–1132 (2012).
Bishop, B. M. et al. Bioprospecting the American alligator (Alligator mississippiensis) host defense peptidome. PLoS One 10, e0117394 (2015).
Goldstein, E. J. et al. Anaerobic and aerobic bacteriology of the saliva and gingiva from 16 captive Komodo dragons (Varanus komodoensis): new implications for the "bacteria as venom" model. J. Zoo Wildl. Med. 44, 262–272 (2013).
Montgomery, J. M., Gillespie, D., Sastrawan, P., Fredeking, T. M. & Stewart, G. L. Aerobic salivary bacteria in wild and captive Komodo dragons. J. Wildl. Dis. 38, 545–551 (2002).
Park, C. B., Yi, K. S., Matsuzaki, K., Kim, M. S. & Kim, S. C. Structure-activity analysis of buforin II, a histone H2A-derived antimicrobial peptide: the proline hinge is responsible for the cell-penetrating ability of buforin II. Proc. Natl. Acad. Sci. U S A 97, 8245–8250 (2000).
Wiegand, I., Hilpert, K. & Hancock, R. E. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 3, 163–175 (2008).
Dorschner, R. A. et al. The mammalian ionic environment dictates microbial susceptibility to antimicrobial defense peptides. FASEB J. 20, 35–42 (2006).
Dean, S. N., Walsh, C., Goodman, H. & van Hoek, M. L. Analysis of mixed biofilm (Staphylococcus aureus and Pseudomonas aeruginosa) by laser ablation electrospray ionization mass spectrometry. Biofouling 31, 151–161 (2015).
Zhang, L., Dhillon, P., Yan, H., Farmer, S. & Hancock, R. E. Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 44, 3317–3321 (2000).
Nagant, C. et al. Identification of peptides derived from the human antimicrobial peptide LL-37 active against biofilms formed by Pseudomonas aeruginosa using a library of truncated fragments. Antimicrob. Agents Chemother. 56, 5698–5708 (2012).
Barksdale, S. M., Hrifko, E. J., Chung, E. M. & van Hoek, M. L. Peptides from American alligator plasma are antimicrobial against multi-drug resistant bacterial pathogens including Acinetobacter baumannii. BMC Microbiol. 16, 189 (2016).
Nelson, C. A. The binding of detergents to proteins I. The maximum amount of dodecyl sulfate bound to proteins and the resistance to binding of several proteins. J. Biol. Chem. 246, 3895–3901 (1971).
Turner, J., Cho, Y., Dinh, N. N., Waring, A. J. & Lehrer, R. I. Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob. Agents Chemother. 42, 2206–2214 (1998).
Wang, W., Smith, D. K., Moulding, K. & Chen, H. M. The dependence of membrane permeability by the antibacterial peptide cecropin B and its analogs, CB-1 and CB-3, on liposomes of different composition. J. Biol. Chem. 273, 27438–27448 (1998).
Formaggio, F. & Toniolo, C. Electronic and vibrational signatures of peptide helical structures: a tribute to Anton Mario Tamburro. Chirality 22, E30–E39 (2010).
Roccatano, D., Colombo, G., Fioroni, M. & Mark, A. E. Mechanism by which 2,2,2-trifluoroethanol/water mixtures stabilize secondary-structure formation in peptides: a molecular dynamics study. Proc. Natl. Acad. Sci. U S A. 99, 12179–12184 (2002).
Raussens, V., Ruysschaert, J. M. & Goormaghtigh, E. Protein concentration is not an absolute prerequisite for the determination of secondary structure from circular dichroism spectra: a new scaling method. Anal. Biochem. 319, 114–121 (2003).
Hirota, N., Mizuno, K. & Goto, Y. Group additive contributions to the alcohol-induced α-helix formation of melittin: implication for the mechanism of the alcohol effects on proteins. J. Mol. Biol. 275, 365–378 (1998).
Kinoshita, M., Okamoto, Y. & Hirata, F. Peptide conformations in alcohol and water: analyses by the reference interaction site model theory. J. Am. Chem. Soc. 122, 2773–2779 (2000).
Roy, S. et al. Mixed-species biofilm compromises wound healing by disrupting epidermal barrier function. J. Pathol. 233, 331–343 (2014).
Dunn, L., Prosser, H. C., Tan, J. T., Vanags, L. Z., Ng, M. K. & Bursill, C. A. Murine model of wound healing. J. Vis. Exp. 75, e50265 (2013).
Wong, V. W., Sorkin, M., Glotzbach, J. P., Longaker, M. T. & Gurtner, G. C. Surgical approaches to create murine models of human wound healing. J. Biomed. Biotechnol. 2011, 969618 (2011).
Jung Kim, D. et al. Efficacy of the designer antimicrobial peptide SHAP1 in wound healing and wound infection. Amino Acids 46, 2333–2343 (2014).
Yin, J. & Yu, F. S. LL-37 via EGFR transactivation to promote high glucose-attenuated epithelial wound healing in organ-cultured corneas. Invest. Ophthalmol. Vis. Sci. 51, 1891–1897 (2010).
Minder, P., Bayha, E., Becker-Pauly, C. & Sterchi, E. E. Meprinalpha transactivates the epidermal growth factor receptor (EGFR) via ligand shedding, thereby enhancing colorectal cancer cell proliferation and migration. J. Biol. Chem. 287, 35201–35211 (2012).
Niyonsaba, F., Ushio, H., Nakano, N., Ng, W., Sayama, K., Hashimoto, K., Nagaoka, I., Okumura, K. & Ogawa, H. Antimicrobial peptides human beta-defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokines. J. Invest. Dermatol. 127, 594–604 (2007).
Tokumaru, S. et al. Induction of keratinocyte migration via transactivation of the epidermal growth factor receptor by the antimicrobial peptide LL-37. J. Immunol. 175, 4662–4668 (2005).
Hoq, M. I. et al. Human catestatin enhances migration and proliferation of normal human epidermal keratinocytes. J. Dermatol. Sci. 64, 108–118 (2011).
Andl, C. D., Mizushima, T., Oyama, K., Bowser, M., Nakagawa, H. & Rustgi, A. K. EGFR-induced cell migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G1227–G1237 (2004).
Frohm, M. et al. Biochemical and antibacterial analysis of human wound and blister fluid. Eur. J. Biochem. 237, 86–92 (1996).
Hirsch, J. G. Bactericidal action of histone. J. Exp. Med. 108, 925–944 (1958).
Patrzykat, A., Zhang, L., Mendoza, V., Iwama, G. K. & Hancock, R. E. Synergy of histone-derived peptides of coho salmon with lysozyme and flounder pleurocidin. Antimicrob. Agents Chemother. 45, 1337–1342 (2001).
Richards, R. C., O'Neil, D. B., Thibault, P. & Ewart, K. V. Histone H1: an antimicrobial protein of Atlantic salmon (Salmo salar). Biochem. Biophys. Res. Commun. 284, 549–555 (2001).
Robinette, D. et al. Antimicrobial activity in the skin of the channel catfish Ictalurus punctatus: characterization of broad-spectrum histone-like antimicrobial proteins. Cell. Mol. Life Sci. 54, 467–475 (1998).
Fernandes, J. M., Molle, G., Kemp, G. D. & Smith, V. J. Isolation and characterisation of oncorhyncin II, a histone H1-derived antimicrobial peptide from skin secretions of rainbow trout, Oncorhynchus mykiss. Dev. Comp. Immunol. 28, 127–138 (2004).
Jacobsen, F. et al. Transient cutaneous adenoviral gene therapy with human host defense peptide hCAP-18/LL-37 is effective for the treatment of burn wound infections. Gene Ther. 12, 1494–1502 (2005).
Wang, Y., Griffiths, W. J., Jornvall, H., Agerberth, B. & Johansson, J. Antibacterial peptides in stimulated human granulocytes: characterization of ubiquitinated histone H1A. Eur. J. Biochem. 269, 512–518 (2002).
Graddis, T. J., Myszka, D. G. & Chaiken, I. M. Controlled formation of model homo- and heterodimer coiled coil polypeptides. Biochemistry 32, 12664–12671 (1993).
Mogk, A., Schmidt, R. & Bukau, B. The N-end rule pathway for regulated proteolysis: prokaryotic and eukaryotic strategies. Trends. Cell Biol. 17, 165–172 (2007).
Flynn, J. M., Neher, S. B., Kim, Y. I., Sauer, R. T. & Baker, T. A. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 11, 671–683 (2003).
Park, C. B., Kim, H. S. & Kim, S. C. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 244, 253–257 (1998).
Boman, H. G., Agerberth, B. & Boman, A. Mechanisms of action on Escherichia coli of cecropin P1 and PR-39, two antibacterial peptides from pig intestine. Infect. Immun. 61, 2978–2984 (1993).
Costerton, J. W., Stewart, P. S. & Greenberg, E. P. Bacterial biofilms: a common cause of persistent infections. Science 284, 1318–1322 (1999).
Donlan, R. M. & Costerton, J. W. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 15, 167–193 (2002).
Mathieu, D. & Mani, R. A review of the clinical significance of tissue hypoxia measurements in lower extremity wound management. Int. J. Low. Extrem. Wounds 6, 273–283 (2007).
Flickinger, S. T. et al. Quorum sensing between Pseudomonas aeruginosa biofilms accelerates cell growth. J. Am. Chem. Soc. 133, 5966–5975 (2011).
Schwartz, K., Syed, A. K., Stephenson, R. E., Rickard, A. H. & Boles, B. R. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 8, e1002744 (2012).
Fox, M. A., Thwaite, J. E., Ulaeto, D. O., Atkins, T. P. & Atkins, H. S. Design and characterization of novel hybrid antimicrobial peptides based on cecropin A, LL-37 and magainin II. Peptides 33, 197–205 (2012).
Stepanovic, S., Vukovic, D., Dakic, I., Savic, B. & Svabic-Vlahovic, M. A modified microtiter-plate test for quantification of staphylococcal biofilm formation. J. Microbiol. Methods 40, 175–179 (2000).
Hassan, A., Usman, J., Kaleem, F., Omair, M., Khalid, A. & Iqbal, M. Evaluation of different detection methods of biofilm formation in the clinical isolates. Braz. J. Infect. Dis. 15, 305–311 (2011).
Zou, S. B. et al. Loss of elongation factor P disrupts bacterial outer membrane integrity. J. Bacteriol. 194, 413–425 (2012).
Juba, M. L. et al. Large scale discovery and de novo-assisted sequencing of cationic antimicrobial peptides (CAMPs) by microparticle capture and electron-transfer dissociation (ETD) mass spectrometry. J. Proteome Res. 10, 4282–4295 (2015).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Di Grazia, A., Luca, V., Segev-Zarko, L. A., Shai, Y. & Mangoni, M. L. Temporins A and B stimulate migration of HaCaT keratinocytes and kill intracellular Staphylococcus aureus. Antimicrob. Agents Chemother. 58, 2520–2527 (2014).
de Latour, F. A., Amer, L. S., Papanstasiou, E. A., Bishop, B. M. & van Hoek, M. L. Antimicrobial activity of the Naja atra cathelicidin and related small peptides. Biochem. Biophys. Res. Commun. 396, 825–830 (2010).
Desbois, A. P., Sattar, A., Graham, S., Warn, P. A. & Coote, P. J. MRSA decolonization of cotton rat nares by a combination treatment comprising lysostaphin and the antimicrobial peptide ranalexin. J. Antimicrob. Chemother. 68, 2569–2575 (2013).
Gautier, R., Douguet, D., Antonny, B. & Drin, G. HELIQUEST: a web server to screen sequences with specific alpha-helical properties. Bioinformatics 24, 2101–2102 (2008).
Blower, R. J., Barksdale, S. M. & van Hoek, M. L. Snake Cathelicidin NA-CATH and Smaller Helical Antimicrobial Peptides Are Effective against Burkholderia thailandensis. PLoS Negl. Trop. Dis. 9, e0003862 (2015).
Acknowledgements
The authors acknowledge the St. Augustine Alligator Farm and Zoological Park for their contriubtion of samples. We are grateful for technical contributions of Megan Devine and Drs. Paul Russo and Melanie Juba. We acknowledge Ryan Blower for providing the B. thailandensis results, Stephanie Barksdale for Pseudomonas 9027 EC50 data and overall technical assistance, and Andrea George for administrative support. We are also grateful to Dr. Joel Schnur for helpful discussions. This work was funded by the Defense Threat Reduction Agency (contract number HDTRA1-12-C-0039), which also partially supported E.M.C., S.N.D., C.N.P., B.B., and M.V.H. C.N.P was partially supported by a graduate fellowship from Institute for Infectious Animal Diseases via U.S. Department of Homeland Security under Cooperative Agreement Number DHS 2010-ST-061-AG0002.
Author information
Authors and Affiliations
Contributions
M.V.H. and B.B. conceived the study. M.C.C. and M.V.H. designed the experimental studies. M.C.C. and M.V.H. wrote the manuscript. M.C.C., S.N.D. and C.N.P. performed the experiments. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interest
The authors declare that they have no competing interests. We are currently filing a patent on DRGN-1 peptide (“Antimicrobial peptides with wound healing activity”).
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Chung, E.M.C., Dean, S.N., Propst, C.N. et al. Komodo dragon-inspired synthetic peptide DRGN-1 promotes wound-healing of a mixed-biofilm infected wound. npj Biofilms Microbiomes 3, 9 (2017). https://doi.org/10.1038/s41522-017-0017-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-017-0017-2
This article is cited by
-
LysSYL: a broad-spectrum phage endolysin targeting Staphylococcus species and eradicating S. aureus biofilms
Microbial Cell Factories (2024)
-
Antibiotic Resistant Biofilms and the Quest for Novel Therapeutic Strategies
Indian Journal of Microbiology (2024)
-
Cathelicidin AS-12W Derived from the Alligator sinensis and Its Antimicrobial Activity Against Drug-Resistant Gram-Negative Bacteria In Vitro and In Vivo
Probiotics and Antimicrobial Proteins (2024)
-
Antimicrobial strategies for topical biofilm-based wound infections: past, present, and future
Journal of Pharmaceutical Investigation (2023)
-
Effects of Antimicrobial Peptides Gal-13 on the Growth Performance, Intestinal Microbiota, Digestive Enzyme Activities, Intestinal Morphology, Antioxidative Activities, and Immunity of Broilers
Probiotics and Antimicrobial Proteins (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
