
Abstract
Snapdragon (Antirrhinum majus L.), a member of the Plantaginaceae family, is an important model for plant genetics and molecular studies on plant growth and development, transposon biology and self-incompatibility. Here we report a near-complete genome assembly of A. majus cultivar JI7 (A. majus cv.JI7) comprising 510 Megabases (Mb) of genomic sequence and containing 37,714 annotated protein-coding genes. Scaffolds covering 97.12% of the assembled genome were anchored on eight chromosomes. Comparative and evolutionary analyses revealed that a whole-genome duplication event occurred in the Plantaginaceae around 46–49 million years ago (Ma). We also uncovered the genetic architectures associated with complex traits such as flower asymmetry and self-incompatibility, identifying a unique duplication of TCP family genes dated to around 46–49 Ma and reconstructing a near-complete ψS-locus of roughly 2 Mb. The genome sequence obtained in this study not only provides a representative genome sequenced from the Plantaginaceae but also brings the popular plant model system of Antirrhinum into the genomic age.
Similar content being viewed by others


Main
The genus Antirrhinum belongs to the family Plantaginaceae and includes about 20 species with the chromosome number of 2n = 2x = 16. Antirrhinum originated in Europe and is mainly distributed in Europe, Asia and Africa around the Mediterranean coast. Different species in the genus Antirrhinum exhibit differences in flower colour, flower pattern, fragrance and flowering time; interspecific hybridization has also been described. The genus exhibits two major mechanisms that promote outcrossing: insect pollination (entomophily) and self-incompatibility1,2,3. The self-compatible A. majus was domesticated as a garden ornamental over 2,000 years ago1.
Antirrhinum has served as a model system for molecular and developmental genetics for the past three decades1,4. Several key floral genes were first identified in Antirrhinum including founding members of the MADS (DEFICIENS) and TCP (CYCLOIDEA) gene families, MYB genes controlling petal epidermal cell shape (MIXTA) or flower colour (ROSEA and VENOSA) and SLFs (S-Locus F-box) controlling self-incompatibility5,6,7,8,9,10,11,12. Isolation and analysis of genes in Antirrhinum have been facilitated by the availability of endogenous active transposons1,13. For example, five transposable elements (Tam1, Tam2, Tam3, Tam4 and Tam11) 14,15,16,17,18 have contributed to the identification of floral homeotic genes. However, so far these studies have been carried out without the benefit of a genome sequence to provide an overall evolutionary and architectural context for these genes, transposons and traits.
Here we report a near-complete genome assembly of A. majus. We annotated 37,714 protein-coding genes on the basis of expression and homology evidence. The assembly was generated by combining whole-genome shotgun (WGS) sequencing of short reads on the Illumina platform and single-molecule real time (SMRT) long reads on the Pacific Biosciences (PacBio) platform. Most of the assembled sequences were anchored onto chromosomes to form eight pseudomolecules using a genetic map. Comparative analysis based on this sequence reveals that the Plantaginaceae and Solanaceae diverged from their most recent ancestor about 62 Ma, and that a whole-genome duplication (WGD) event occurred around 46–49 Ma. We found that the WGD contributed to the evolution of the TCP gene family related to flower asymmetry in A. majus. We also analysed the near-complete genomic structure of the pseudo (ψ) S-locus of A. majus of roughly 2 Mb, which contained 102 genes including 37 SLF genes. The genome sequence provided here will accelerate genomic and evolutionary studies in this model species.
Results
Genome sequencing, assembly and annotation of A. majus
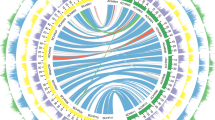
We sequenced a highly inbred Antirrhinum line (A. majus cv. JI7) using a combination of Illumina short-read and PacBio long-read sequencing technologies. The genome size was estimated from k-mer distributions to be about 520 Mb. We obtained 90.85 gigabases (Gb) high-quality Illumina paired-end reads, equivalent to 174-fold sequence coverage of the genome. We used CANU19 to correct and assemble 25.89 Gb PacBio reads into contigs and SSPACE20 for scaffolding with Mate-paired short reads. The assembled genome size was 510 Mb with contig and scaffold N50 (the size above which 50% of the total length of the sequence assembly can be found) sizes of 0.73 and 2.6 Mb, respectively (Fig. 1, Table 1, Supplementary Fig. 1 and Supplementary Table 1–4). The mapping ratio of ×42.22 Illumina data was 99.55% (Supplementary Table 5) and the coverage of contigs using PacBio data was 99.97%. The heterozygosity of the assembled genome was estimated to be 51 single nucleotide polymorphs (SNPs) per 1 Mb (0.0051%) by using Illumina sequencing data.

Roman numerals refer to: I, duplications of genomic paralogous sequences; II, guanine-cytosine (GC) content; III, simple sequence repeats; IV, gene density; V, retroelement density; VI, recombination rate (deep colour shows high recombination rates) and VII, eight Antirrhinum chromosomes with physical distances including low copy number repetitive elements: telomere repeat TTTAGGG (green), 5S recombinant DNA (orange) and pericentromeric repeats CentA1 and CentA2 (purple). A ruler with marks every 1 Mb is drawn on each chromosome.
To anchor the A. majus genome sequence to chromosomes, we created linkage maps by re-sequencing 48 recombinant inbred lines (RILs) derived from A. majus crossed to the self-incompatible species A. charidemi. We identified a total of 4,523,444 homozygous SNPs between the parents on 1,386 contigs and obtained 4,198,995 SNPs on 1,381 contigs for linkage map construction. After validations by known genetic markers21,22, 496.9 Mb (97.12%) of the assembled scaffold sequences were anchored onto eight linkage groups to form pseudomolecules. The pseudomolecules ranged in size from 50.9 to 75.4 Mb. The average recombination rate was 1.798 centimorgans per Mb (Supplementary Table 6 and Supplementary Data Set 1). The relationship between genetic and physical distances revealed significantly lower recombination rates at the centromere regions of all chromosomes and the extended pericentromeric regions of chromosomes 4, 6, 7 and 8. The linkage groups were linked to the physical chromosomes through fluorescence in situ hybridization (FISH) (Supplementary Fig. 2–4 and Supplementary Table 6).
To evaluate the assembled genome quality, we aligned 25,651 expressed sequence tags (ESTs) of Antirrhinum downloaded from National Center for Biotechnology Information (NCBI) nucleotide database to the assembled genome, and found that 96.59% of the ESTs could be mapped. Alignments between the assembled genome and three sequenced Bacterial Artificial Chromosomes (BACs) indicated an average nucleotide accuracy of 99.65% in the assembly. BUSCO23 analysis showed 93.88% complete genes at the genome mode and 93.40% at the protein mode, which suggested that the quality of the assembled Antirrhinum genome sequence was comparable to that of other published plant genomes (Petunia and Arabidopsis) (Supplementary Fig. 5–7 and Supplementary Data Set 2). Taken together, these results suggested that the A. majus genome assembly was both highly accurate and near completion.
We predicted a total of 37,714 protein-coding genes with an average transcript length of 3,166 base pairs (bp) by using a combination of ab initio and evidence-based methods24. We used Antirrhinum EST sequences and RNA-seq data from six major tissues: leaf, root, stem, stamen, pistil and pollen (Supplementary Data Set 3) to confirm the expression of the genes. Approximately 89% of the genes were functionally annotated. The average gene density in Antirrhinum was one gene per 15.5 kilobase (kb), which is about three times lower than Arabidopsis (one gene per 4.5 kb) and slightly higher than tomato (one gene per 25.7 kb). Genes were distributed unevenly, being more abundant towards the ends of the chromosomal arms (Fig. 1). We identified genes encoding 981 transfer RNAs, 800 microRNAs, 10 ribosomal RNAs (18S, 28S, 5.8S and 5S) and 622 small nuclear RNAs. A total of 268.3 Mb (52.6%) of sequences was annotated as repeats, including a wealth of class I (retrotransposon: 182.8 Mb) and class II (DNA transposon: 41.1 Mb) elements (Supplementary Tables 7–10).
We found 95 transposable elements belonging to the En/Spm/CACTA family. Three subfamilies (Tam2, Tam4 and Tam11) had copies with 100% identity, suggesting recent duplication/transposition events. We also identified 166.21 Mb comprising long-terminal repeat (LTR) retrotransposons, with sequence similarity between copies indicating a mean divergence time of ~0.86 Ma. Bursts of Gypsy and Copia retrotransposon insertions were detected at 0.1–0.2 Ma and 120–130 Ma, respectively. These results suggest that the Antirrhinum genome has a long history of active transposition (Supplementary Table 11, Supplementary Fig. 8 and Supplementary Data Set 4).
Comparative genomic analysis of A. majus
Self-alignment analysis revealed duplicated and triplicated regions between and within chromosomes. Paralogous relationships among the eight Antirrhinum chromosomes revealed 45 major duplications and two triplications, collectively containing 1,841 pairs of paralogous genes (Fig. 1 and Supplementary Data Set 5). We performed all-against-all comparisons to identify 2,115 single-copy genes of Antirrhinum with orthologues in nine angiosperm species (A. majus, Arabidopsis thaliana, Amborella trichopoda, Carica papaya, Oryza sativa, Petunia hybrida, Prunus mume, Solanium lycopersicum, Symphytum tuberosum and Vitis vinifera). The resulting phylogenetic tree (Fig. 2a) show that the Antirrhinum lineage split from potato and tomato lineages around 62 Ma, consistent with the results of Bell et al.25.

a, Phylogenetic tree of angiosperm species including their divergence time on the basis of orthologues of single-gene families. The red star highlights the genome duplication in the A. majus lineage. The number in each node indicates Ma between two divergent branches. Green/red numbers indicate expansion and contraction gene families. A. trichopoda was used as an outgroup. Bootstrap values for each node are above 100%. b, Synteny blocks among chromosomes of A. majus, V. vinifera and S. lycopersicum. The numbers represent individual chromosomes. The selected syntenic gene numbers are more than 50 in each block. c, Density distributions of Ks for paralogous genes among A. majus, V. vinifera and S. lycopersicum. d, A Venn diagram of shared orthologues among four species of A. majus, S. lycopersicum, A. thaliana and O. sativa. Each number represents a gene family number shared among the genomes.
For inter-species comparative genomic studies, we examined the synteny of Antirrhinum chromosomes and those of V. vinifera and Solanum lycopersicum. We found only small syntenic blocks between the A. majus and the V. vinifera or S. lycopersicum chromosomes (Fig. 2b). We also compared A. majus with Sesamum indicum, Olea europea, Helianthus annuus and Coffea arabica, which all belong to Lamiales. Large syntenic blocks were found between the Antirrhinum genome and these species, especially between Antirrhinum chromosomes 1, 2, 6 and 8 and C. arabica chromosomes 3, 1/1, 4/7, and 6, respectively (Supplementary Figs. 9 and 10).
We identified the syntenic blocks within the A. majus genome through intragenome comparisons. We calculated the density distribution of synonymous substitution rate per gene (Ks) between the collinear paralogous genes and inferred paleotetraploidy event in Antirrhinum. A peak at around 0.57–0.60 indicated that a WGD, which was Plantaginaceae-specific, occurred around 46–49 Ma (Fig. 2c), clearly after the divergence of Antirrhinum lineage from the lineage of potato and tomato.
We then compared the complexity of gene families between Antirrhinum and other species: 9,503 gene families were shared by Antirrhinum, Arabidopsis, rice and tomato; 6,677 gene families were possibly contracted in Antirrhinum, while the other 3,778 gene families were expanded (Fig. 2d). Gene-set enrichment analysis (GSEA) analysis results showed that gene families encoding protein kinase activity, catalytic activity, transporter activity and ATP-binding activity were most obviously expanded (Supplementary Table 12). Transcription factor gene families, such as AP2, C2H2, GRAS, TCP and Trihelix, were expanded in species belonging to the order Lamiales (A. majus, S. indicum and O. europea) compared with A. thaliana, S. lycopersicum and V. vitis (Supplementary Table 13 and Supplementary Data Set 6).
We analysed the contributions of tandem duplication and WGD to expansions/contractions. We found most F-box domain (PF00646), cytochrome P450 (PF00067) and NB-ARC domain (PF00931) gene families were derived from tandem duplication events (percentage of tandem duplication genes >40%, compared with the genome average 12.1%). The percentage of expanded transcription factor genes caused by WGD (27.5%) was larger than the genome average (16.4%), especially for WRKY (49.2%), MADS-MIKC (43.3%), bZIP (39.1%), TCP (37.5%) and G2-like GARP (36.8%) (Supplementary Data Sets 6 and 7). Thus, both WGD and tandem duplication have played an important role in the expansion of gene families.
Evolution of floral asymmetry and TCP family
A. majus has served as the genetic model of floral symmetry. Previous studies have revealed that floral asymmetry in A. majus is largely controlled by two transcripion factors (TFs) (CYC and DICH) that belong to the TCP gene family7,8,26. To explore their evolution, we analysed the composition of the TCP families in A. majus and several sequenced angiosperms with floral symmetry. The TCP family is divided into two classes, class I (PCF) and II, and class II is further divided into two clades, CIN and CYC/TB1. Both eudicot and monocot share a subfamily containing CYC and DICH genes that belong to the CYC/TB1 clade. However, the basal angiosperm A. trichopoda, which has radially symmetrical flowers, lacks any members of the class II CYC/TB1 clade. Two monocots and several eudicots also have radially symmetrical flowers (Fig. 3). These findings suggest that the TCP class II CYC/TB1 clade26,27 appeared after the emergence of radially symmetrical flowers, and the initial role of CYC/TB1 clade was thus not likely to be involved in the control of floral symmetry (Fig. 3 and Supplementary Data Set 8).

Left, a phylogenetic tree of the flowering plants derived from their divergence time based on orthologues of single-gene families. Three red stars show the whole-genome triplication and two green stars the duplication events (http://chibba.agtec.uga.edu/duplication/). Right, B represents bilateral flower symmetry and R radial flower symmetry; Asterid, Rosid, Basal rosid, Basal eudicot, Monocot and Basal angiosperm represent the clade names, respectively. Total numbers of TCP family genes, Class I PCF, Class II CIN and Class II CYC/TB1 are shown from left to right. The asterisk indicates the sequenced genome of species of Medicago truncatula with flower radial symmetry, but flowers of most Medicago species have bilateral symmetry. The double asterisk indicates Lotus corniculatus in which three TCP genes were identified but a functional TCP gene was not detected in its genome. Four-pointed stars denote flowers with bilateral symmetry with their photos from PPBC (http://www.plantphoto.cn).
We identified a total of 30 putative functional TCP family genes in Antirrhinum: 13 class I genes and 17 class II genes (10 in the CIN clade and 7 in the CYC/TB1 clade) (Supplementary Data Set 8). Syntenic block and Ks analyses of the orthologous gene pairs revealed that both WGD and tandem duplication contributed to the expansion of TCP family members. A previous study showed that CYC and DICH have partial redundancy in the control of flower asymmetry and exhibit only partially similar expression patterns in floral meristems in A. majus, and the two genes act together to establish the flower asymmetry in A. majus8. We found that the CYC and DICH genes reside on a pair of syntenic regions including 79 homologous gene pairs (Supplementary Table 14). The Ks analysis results show that this syntenic block was retained from the Plantaginaceae-specific WGD event. Previous phylogenetic analysis suggested that zygomorphic flowers independently evolved from actinomorphic ancestors more than 25 times28. On the basis of fossil records, it was proposed that clearly zygomorphic flowers emerged in various lineages roughly 50 Ma29, concurrent with the occurring time of the WGD event. These results suggest that the WGD to generate both CYC and DICH genes played a critical role in the evolution of zygomorphic flowers in the Antirrhinum lineage.
Furthermore, two MYB-class genes RAD and DIV, acting downstream of CYC/DICH in the control of floral symmetry, interact with the DRIF gene. The DRIF has homologous copies with similar Ks to CYC/DICH, and they are also located at a WGD-derived syntenic block 30,31,32. These results further support the idea that the key regulators of floral asymmetry were retained from the genes generated by the WGD in Antirrhinum.
Structure of the ψS-locus in A. majus and its gene collinearity in self-incompatible species
In previous cytological investigations, we found that the Antirrhinum S-locus is located in a heterochromatin region on the short arm of chromosome 8 (ref. 33). The cultivated species A. majus is self-compatible, carrying a pseudo (ψ)S-locus. Scanning the A. majus genome for conserved (FBA/FBK domain) of the SLF gene family revealed the presence of 37 SLF genes (SLF1–SLF37) located in the short arm of chromosome 8, probably corresponding to the ψS-locus. The locus covered 874 kb across three scaffolds Sc29, Sc276 and Sc184, possessing a considerably higher number of SLF genes than S. lycopersicum34,35 (Fig. 4a). Six pseudogenes with FBA domains were inferred to be loss-of-function. No S-RNase was found in or near the locus, suggesting it might have been lost during horticultural selection for self-compatible A. majus. The RAD gene was located about 1 Mb upstream of SLF1, consistent with previous studies showing its linkage with the S-locus36. Expression analysis showed that 30 SLF genes were expressed in either pollen or anthers indicating they could play a role in pollen function. The number of SLF genes in the A. majus ψS-locus is more than twice that found in S. lycopersicum (15 SLF genes including 11 pseudogenes)33,35 and that the ψS-locus of A. majus contains the largest number of active SLF genes annotated so far in a plant genome (Fig. 4b and Supplementary Data Sets 9–11).

a, Chromosomal locations of three scaffolds covering the ψS-locus region of A. majus. A genetic recombination map of chromosome 8 is shown on the top panel. The x axis shows its physical distance (Mb) and the y axis its genetic distance (cM). A schematic representation of chromosome 8 is shown in the middle panel with a red dot indicating its centromere. The ψS-locus is depicted as a blue box on its short arm. A vertical red line in the chromosome indicates the RAD gene. The lower panel shows three scaffolds of Sc29, Sc276 and Sc184 covering the ψS-locus region. b, Transcriptional profiles of the ψS-locus and its flanking regions of A. majus. The light orange shadow denotes the predicted ψS-locus region (SLF1–SLF37). This region between RAD and SLF37 contains a total number of 102 annotated genes. The bottom panel is a schematic representation of the SLF genes. Orange squares indicate the ψSLF genes and green arrows the other annotated genes (I: a putative MYB family transcription factor; II and III, putative RNA-binding proteins and IV, a putative phosphate-dependent transferase). c, The synteny of the S-locus regions between A. majus and S1, S2, S4 and S5 haplotypes of A. hispanicum. Different colours indicate syntenic and inversion regions between the ψS-locus and S1 (S1a, S1b and S1c), S2, S4 (S4a, S4b and S4c) or S5 (S5a and S5b) haplotypes of A. hispanicum.
We compared the ψS-locus sequence with nine assembled TAC (transformation-competent artificial chromosome) sequences from four S haplotypes of self-incompatible A. hispanicum. Gene collinearity between S-alleles was revealed in the genomic region extending from AhSLF12 to AhSLF13 (Fig. 4c). An intrachromosome inversion around the S-locus was found to occur in the S2 haplotype of A. hispanicum as described previously33. In contrast to the ψS-locus of A.majus, an S-RNase gene was found in every sequenced A. hispanicum S allele, suggesting that the S-RNase had been deleted in the ψS-locus. Notably, a pseudo-gene AmSLF18 in the A. majus ψS-locus had an orthologue in the S4 haplotype in A. hispanicum, which had a complete coding sequence and was expressed, suggesting the latter is an active gene in the S-locus and the former lost function, possibly following the loss of S-RNase (Fig. 4c and Supplementary Data Set 11). The orthologous SLFs among different haplotypes show a low allelic diversity, consistent with our previous finding34.
The nonsynonymous (Ka) and synonymous (Ks) substitution rates of the 12 collinear SLF gene pairs showed that the values of SLFs are lower than that of S-RNase in Antirrhinum, and the allelic SLF genes showed a ratio of Ka/Ks = 0.41, consistent with a negative frequency-dependent selection detected previously35,37. Only SLF14 appears to be a positively selected gene (Ka/Ks > 1) (Supplementary Data Set 12). The average divergence time of these orthologous SLF genes was estimated to be 4 Ma, similar to an estimated early Antirrhinum species divergence time of less than 5.3 Ma38. However, the average divergence time of the S-RNases of A. hispanicum is estimated to be around 62–120 Ma, similar to the species divergence between Antirrhinum and Solanaceae species estimated in our study (Supplementary Data Set 13). These results suggest that the divergence of S-RNase occurred before the WGD and they were well maintained in the lineage of Antirrhinum. Our results showed that a near-complete ψS-locus A. majus was identified.
Discussion
The genome sequence of A. majus reported here represents a sequenced genome of a species belonging to the family Plantaginaceae, and reveals a WGD specific to this family. Zygomorphy in the Antirrhinum lineage is thought to have arisen in the late Cretaceous period in the fossil record38, as a mechanism facilitating insect-mediated pollination. We show that two key TCP genes controlling zygomorphy are collinear on a syntenic block generated by the WGD of the Plantaginaceae, suggesting that the duplication may have provided a genetic basis for the evolution of this trait. An independent WGD may have been involved in the evolution of zygomorphy in Glycine39, and the missing type I Mβ MADS-box genes family resulted in bilaterally symmetrical flowers in the Orchidaceae40. The timing of the WGD event in the Plantaginaceae corresponds to the age of this family on the basis of the molecular dating approaches25. WGD events have been considered to be catalysts for species diversification and evolutionary novelty in plants41,42,43. The availability of more species with completely sequenced genomes in the Plantaginaceae and their relatives may help clarify the role of the WGD in the expansion and evolution of the family.
The Antirrhinum genome also sheds light on the evolution of self-incompatiblity. The fine genomic structure of the ψS-locus from A. majus reveals a large number of pollen SLFs, probably due to gene duplication, recombination suppression, purifying selection and frequency-dependent selection associated with the S-locus35,37. Relatively low allelic diversity was observed between orthologous SLFs among different haplotypes34, compared with the paralogues within a haplotype, perhaps because extensive divergence would lead to recognition and self-inactivation of S-RNase resulting in loss of self-incompatibility. The deletion of S-RNase in cultivated A. majus could be responsible for the loss of self-incompatibility, giving an essentially irreversible transition. Such deletions may account for why self-compatible species are difficult or almost impossible to revert back to self-incompatible species (Doll’s Law)44. The high microcolinearity of the S-locus between self-incompatible and self-compatible Antirrhinum indicates that the deletion of S-RNase in self-compatible species was a recent event. In fact, some mutated SLF genes in different haplotypes also arose recently34.
The physical size of the S-locus in S. lycopersicum is much larger than that in A. majus (17 Mb compared to 2 Mb)36,45, yet it contains fewer SLF genes (17 compared to 37). Less repetitive sequences are found in the ψS-locus and S loci of Antirrhinum compared with that of Solanum, suggesting that an increase of the gene numbers through unequal crossovers possibly results in the Antirrhinum S-locus, and that repetitive element enrichments could underlie the large physical size and low gene density of the S-locus of Solanum, enhanced perhaps by its centromeric location.
In conclusion, the assembled A. majus sequence provides a reference genome for the Plantaginaceae and will be helpful for genetic, genomic and evolutionary studies in both Antirrhinum and other flowering plants. For example, studies on a natural hybrid zone between Antirrhinum species using this genome sequence as a reference have revealed patterns of selection and gene flow underlying the evolution of flower colour pattern46. We hope the resource will be a useful stimulus to further studies.
Methods
Plant materials
The seeds of cultivated Antirrhinum (A. majus JI7) were surface-sterilized and plated on Murashige–Skoog (1/2 MS) plates (×1/2 MS salts, 0.23% phytagel and ×1 Gamborg’s B5 vitamin mixture, all from Sigma) and grown in growth chambers (160 h/8 h, light/dark) with white fluorescent light (100 μmol m−2 s-1) at 22 °C. After avoiding light for 72 h, we harvested leaf tissues and extracted DNA using the cetyltrimethylammonium bromide (CTAB) method47.
To generate the RILs, A. majus JI7 stock (TA7–7) was crossed to A. charidemi (TA1282). The A. charidemi individual was derived from accession Ac1024 -Y-TES -1, with seed collected from Cabo de Gata Spain. A single F1 hybrid (P107-2) was self-pollinated to produce a total of 195 F2 plants. Each plant was self-pollinated to produce a unique RIL. A total of 48 RILs were eventually developed from single F2 individuals taken through additional rounds of self-pollination through to the F7 to F9 generation. A. hispanicum lines (AhS2S4 and AhS1S5) were maintained by vegetative cuttings as described by Xue et al.48. and were originally sourced from the Gatersleben collection (http://www.ipk-gatersleben.de/en/gbisipk-gaterslebendegbis-i/).
WGS
High-quality genomic DNA was extracted from young leaves of cultivated A. majus JI7 using the CTAB method. According to the manufacturer’s instructions (Illumina HiSeq 2000), we constructed a total of 2 × 100 paired-end sequencing libraries with insert sizes from 170 bp to 20 kb for standard WGS sequencing. For small-insert (<2 kb) libraries, DNA was fragmented, end repaired, ligated to Illumina paired-end adaptors, size selected and purified by PCR amplification. For large-insert (≥2 kb) mate-paired libraries, about 20–50 μg genomic DNA was fragmented, and biotin-labelled adaptors were annealed to the fragment ends before self-ligation to form circularized DNA. This library was re-fragmented and target sequences (that is, the long molecule ends) were enriched using biotin/streptavidin, and then prepared for sequencing. All of the above libraries were sequenced on Illumina Genome Analyzer sequencing platforms. In total, we generated about 90.85 Gb (roughly ×144.24) reads. Using the Pacific Biosciences (PacBio) platform for single-molecule, real-time (SMRT) sequencing we generated a total of 25.89 Gb from 30 SMRT cells, with an average subread length of 5.2 kb and a N50 size of 13.4 kb. The 48 individual RILs were genotyped using the WGS sequencing. We obtained a total of 201.49 Gb sequencing data and the average sequencing depth of each sample was 4.5 Gb (×8.2). 92.40% of the reads could be mapped into the genome.
The genome size was evaluated using the total length of sequence reads divided by sequencing depth as described49. To estimate the sequencing depth, we counted the frequency of each 17-mer from the Illumina WGS sequencing reads and plotted the distribution of copy numbers. The peak value of the frequency curve represents the overall sequencing depth. We used the algorithm (N × (L − K + 1) − B)/D = G, where N is the total sequence read number, L the average length of sequence reads and K the length defined as 17–31 bp here. To minimize the influence of sequencing error, K-mers with low frequency (<4) are discarded. B is the total number of low-frequency K-mers. G denotes the genome size and D is the overall depth estimated from K-mer distribution.
Genome assembly
The assembly was performed on HPC (High Performance Computing) system with 40 nodes, each one having 16 CPU cores and 128 GB of RAM. The operating system was Centos 6.3 64-bit (Linux). We corrected the PacBio long reads using the Canu pipeline19, and then assembled them into contigs (N50 = 733 kb; total length = 510 Mb). The Canu pipeline parameters were: genome size = 600 Mb, error rate = 0.013. We then further polished the PacBio assembled contigs using Quiver49. We used the mate-pair sequences to connect the contig sequences with SSPACE20. Initially, we required 30 connections to support connection of contig sequences into a scaffold. We then repeated this process iteratively using the result of the scaffolding as input but reducing the connection support by five. The final assembly spans were produced with the connection support parameter set to 10.
To construct the linkage map and organize scaffolds into pseudochromosomes, we resequenced individual RILs and their parents. The raw reads generated from the Illumina-Pipeline included low-quality, adaptor contaminated and duplicated reads. Reads were filtered using Trimmomatic50 with default parameters, retaining only reads longer than 50 bp after quality trimming. We used BWA-mem51 (http://bio-bwa.sourceforge.net/) with default settings to align filtered reads to the assembled genome. After alignment, we used SAMtools52 to filter out low-quality (mapping quality <30) alignments and the Genome Analysis Toolkit (GATK)53 (http://www.broadinstitue.org/gatk/) UnifiedGenotypers to call SNPs. The SNPs were filtered using the GATK VariantFiltration program with the following criteria: clusterWindowSize:10, MQ0>=4& ((MQ0/ (1.0* DP)) >0.1), QUAL<50.0, DP< 5. A total of 4,523,444 homozygous SNPs were identified between the parents on 1,386 contigs and used to filter out unmatched SNPs or extremely unevenly distributed SNPs in the RIL population. A total of 4,198,995 filtered SNPs and 2,300 bin markers were obtained on 1,381 contigs for the linkage map construction. Published markers19,20 were used to validate the linkage map (Supplementary Table 5 and Supplementary Data Set 1). Fifty SNP sliding windows were used to create SNP bins to find recombination sites. JoinMap4.1 (https://www.kyazma.nl/index.php/JoinMap/) ML methods were used to cluster the bins into LGs, and then the MstMap54 (http://www.mstmap.org/download.html) Kosambi model was used to compute the order of the bins. The final map anchored 1,280 contigs to eight linkage groups.
To evaluate the assembled genome quality, first we mapped the illumina NGS data to the genome using BWA-mem51 (http://bio-bwa.sourceforge.net/). Then we aligned the EST sequence download from NCBI (http://www.ncbi.nlm.nih.gov/nucest/?term=EST%20Antirrhinum) using BLAT55. Finally, we used BUSCO23 (http://busco.ezlab.org, v3) to examine the gene content with Embryophyta odb9 database and parameters. We also used BWA-mem51 with default settings to align three BAC sequences to the assembled genome. GenBank numbers of the three BACs are AY935269.1, FJ404769.1 and FJ404770.1 with lengths of 85, 51 and 111.3 kb, separately.
Gene structure annotation and functional annotation
The gene annotation in the A. majus genome was performed by a combination of ab initio and evidence-based methods24. The protein sequences from three sequenced plants, namely, A. thaliana, C. papaya and S. tuberosum, were aligned to the genome using TBLASTN55 with an E value cut-off of 1 × 10–5. The homologous genomic sequences were aligned against the matching proteins using GeneWise (https://www.ebi.ac.uk/Tools/psa/genewise/)56 for accurate spliced alignments. For ab initio prediction, Augustus (http://augustus.gobics.de)24 and GlimmerHMM57 were run on the repeat masked genome with parameters trained from the closely related species and partial or small genes that had less than 150 bp coding length were discarded. EST was aligned to the genome using BLAT58 to generate spliced alignments, which were linked according to the overlap using PASA59. Finally, we aligned all the RNA reads to the reference genome by TopHat60 (https://ccb.jhu.edu/software/tophat/index.shtml), assembled the transcripts using Cufflinks61 under default parameters and predicted the open reading frames to get reliable transcripts with HMM-based training parameters. To finalize the gene set, all the predictions were combined using GLEAN62 to produce the consensus gene sets. On the other hand, another gene annotation in the snapdragon genome was performed using the Gramene pipeline63. The evidence included 167 messenger RNAs and 25,310 ESTs of Antirrhinum from the NCBI nucleotide database (https://www.ncbi.nlm.nih.gov), and SwissProt proteins for plants, which were cleaned up by removing redundant sequences with a minimum threshold of 80% for both identity and coverage, which left us with 340,312 sequences. Meanwhile, the mRNAs and ESTs of eudicot species were downloaded from NCBI and filtered to remove redundant sequences with a cut-off of 90% for both identity and coverage, resulting in 2,332,979 complementary DNAs and 152,396 ESTs, and RNA-seq data from six samples of A. majus were downloaded in this study and assembled into contigs using SOAPdenovo-trans v.1.03 (http://soap.genomics.org.cn/SOAPdenovo-Trans.html). The assembled contigs were used as same-species EST evidence. The genes with protein length <100 amino acids and expression level in RNA-seq data <1 RKPM were discarded. In the end, the predicted genes were obtained after selecting the longer ones between overlapping genes.
Gene functions were assigned according to the best match derived from the alignments to the integral database consisting of annotated proteins in Arabidopsis (https://www.arabidopsis.org/download_files/Proteins/TAIR10_protein_lists/TAIR10_pep_20101214) databases and SwissProt proteins using BLASTP55, with 30% minimum identity and coverage as threshold. We annotated motifs and domains using InterProScan64 by searching against publicly available databases, including ProDom65 (http://prodom.prabi.fr/), PRINTS66 (www.bioinf.manchester.ac.uk/dbbrowser/PRINTS/), Pfam67 (http://pfam.xfam.org/), SMART68 (http://smart.embl-heidelberg.de/), PANTHER69 (http://www.pantherdb.org/), SUPERFAMILY70 (http://supfam.org/SUPERFAMILY/), PIR (http://pir.georgetown.edu/) and PROSITE (http://prosite.expasy.org/). Both CPC program71 and gene prediction evidence such as poor coding ability and protein length were used to filter the non-coding genes. All data for the evidence-based prediction were downloaded from corresponding databases on 5 January 2017, with the minimal length of 150 bp per 50 amino acids.
The tRNA genes were identified by tRNA scan-SE72 (https://wiki.gacrc.uga.edu/wiki/TRNAscan-SE) with eukaryote parameters. For rRNA identification, we aligned the A. thaliana rRNA sequences against the A. majus genome by using BLASTN55 with an E value of 1 × 10-5. The snRNA and miRNA predictions were made using INFERNAL73 software (http://eddylab.org/infernal/) and by searching against the Rfam74 database (http://rfam.xfam.org/).
The classification of genes into families was carried out by BLASTP55 all-against-all comparisons of predicted proteins using the duplicate_gene_classifier module integrated within MCScan75 (http://chibba.pgml.uga.edu/mcscan2/) with default parameters. The MCScan software classified the duplicate genes of one genome into whole genome /segmental (≥5 homologous gene pairs in collinear blocks), tandem (consecutive repeat), proximal (in nearby chromosomal region but adjacent within 10 genes) or dispersed (modes other than segmental, tandem and proximal) duplications. Remaining genes were defined as singletons. The iTAK76 database (http://itak.feilab.net/cgi-bin/itak/index.cgi) was used to analyse transcription factor family expansion and to assign genes to specific families.
Genome repeat element identification
Repetitive sequences and transposable elements in the genome were identified using a combination of de novo and homology-based approaches at both the DNA and protein levels. Briefly, we first constructed a de novo repeat library for snapdragon by using LTR_FINDER77 (http://tlife.fudan.edu.cn/ltr_finder/) and passed this to Repeat Modeler78 v.1.08 with default parameters. This library was aligned to the PGSB Repeat Element Database (http://pgsb.helmholtz-muenchen.de/plant/recat/) to generate the classification information for each repeat family. For identification of transposable elements at the DNA level, RepeatMasker was applied using both the repeat database we had built and Repbase79 (http://www.girinst.org/repbase). Next, we executed RepeatProteinMask78 (http://www.repeatmasker.org/) in a WU-BLASTX search against the transposable element protein database to further identify repeat-related proteins. The overlapping transposable elements belonging to the same repeat class were collated and combined according to the coordination in the genome. In addition, we annotated the tandem repeats by using the software Tandem Repeats Finder80 (http://tandem.bu.edu/trf/trf.html).
The full-length Tam sequences were retrieved from the NCBI nucleotide database and used to query the genome with BLASTN (v.4x10, -minIdentity=70). Hits with more than 20% query alignment coverage were retrieved with genomic coordinates. These sequences were then subjected to group analysis with blastclust (-S100-L0.99) integrated within NCBI BLAST (blast-2.2.25). Candidate recent active clusters were further examined by self-BLASTN of member sequences within these clusters and were considered supported where 100% query coverage and 100% identity (without mismatch or indels) were reported. For Tam1, Tam2, Tam4 and Tam11, there was only one full-length Tam sequence detected. For Tam3, BLAST searches using each of five different full-length Tam3 sequence accessions (AB012941, 3,698 bp; AB005454, 3577 bp; AB038403, 3488 bp; AB038404, 3601 bp; AB038404, 3,611 bp) produced similar results to the initial cloned Tam3 (X55078, 3,629 bp) and confirmed that the A. majus genome sequenced in this study did not have a recent active cluster.
Gene family and synteny
To identify gene family groups, we analysed protein-coding genes from nine plant species, A. majus (this study), S. tuberosum (ftp://ftp.ncbi.nlm.nih.gov/genomes/Solanum_tuberosum/)81, P. axillaris (ftp://ftp.solgenomics.net/genomes/Petunia_axillaris/)82, P. inflata (ftp://ftp.solgenomics.net/genomes/Petunia_inflata/)82, S. lycopersicum (http://www.ncbi.nlm.nih.gov/genome/annotation_euk/Solanum_lycopersicum/101/)83, A. thaliana (https://www.arabidopsis.org/, TAIR10)84, C. papaya (http://www.plantgdb.org/CpGDB/, v1. 81)85, P. mume (https://www.rosaceae.org/, v2.0.a1)86, V. vinifera (http://www.genoscope.cns.fr/externe/Download/Projets/Projet_ML/data/12X/)87, O. sativa (http://rice.plantbiology.msu.edu/)88 and A. trichopod (http://www.amborella.org/)89. We performed an all-against-all comparison using BLASTP57 with an E value cut-off of 1 × 10-5, and the OrthoMCL method90 was used to cluster the BLASTP results into paralogous and orthologous clusters.
In total, 2,115 single-copy gene families were used to reconstruct the phylogenetic tree. First, the proteins of single-copy gene families were aligned by MUSCLE91. Following alignment, the protein was reverse-transcribed into the coding sequence and four-fold degenerate sites were extracted from each alignment and concatenated to create one super gene for each species. We used jModelTest to select the best model (http://darwin.uvigo.es)92. PhyML93 was used to construct the phylogenetic tree using the JTT+I+GAMMA model and 1,000 bootstrap replicates. We used a similar method to PGDD (http://chibba.agtec.uga.edu/duplication/) to identify WGD events within the A. majus genome. Proteins were aligned to each other with BLASTP and a filter threshold of 1 × 10-5 was used to identify homologous proteins. MCScanX75 with default parameters (http://chibba.pgml.uga.edu/mcscan2/) was used to find collinear blocks, each containing at least five collinear gene pairs. The Ks value was calculated with the PAML94 yn00 NG model (http://abacus.gene.ucl.ac.uk/software/paml.html). Intragenome dot plot comparison of A. majus was carried out using the SynMap tool from the online CoGe portal95 (http://genomevolution.org/CoGe/). GEvo microsyntenic analysis of each collinear block was performed using SynMap and SynFind also in the CoGe portal. The divergence times of C. papaya–A. thaliana (~55.1–90.6 Ma) and dicot–monocot (~123.9–228.5 Ma) were used for calibration.
Transcriptome analysis
Total RNA was isolated from the leaf, pistil, pollen, root, stamen and stem. For tissues with large biomass (leaf, root and stem), each tissue mixture was obtained from three plants. For the rest of the tissues (pistil, pollen and stamen), each tissue mixture was sampled from at least 10 plants. All plants used in RNA-seq were growth at the environment as that used for genome sequencing and were confirmed with consistent growth. RNA sequencing libraries (300–500 bp fragments) were constructed using the mRNA-Seq Prep Kit (Illumina). Then, we sequenced all libraries using Illumina HiSeq 2000 (2 × 100 bp). FastQC96 qualified reads ware mapped to the genome guided by the final gene model using hisat2 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and the expression level for each gene was calculated by Stringtie97.
Construction of BAC library
High-molecular-weight DNA of over 2 Mb from Antirrhinum (A. majus JI7 line) was extracted from leaf nuclei according to Liu and Whitter98 and partially digested with HindIII. BAC vector preparation, ligation and transformation of TranformMaxTM EP1300TM Escherichia coli (EPICENTRE Biotechnologies) by electroporation followed the described method99. BAC DNA was digested with HindIII and sized-fractioned with a field inversion agarose gel electrophoresis to estimate the insert length. A total of 114,816 clones were selected and stored in 384-well plates.
FISH
Immature 1.5–3.0 mm Antirrhinum flower buds were harvested and fixed in Carnoy’s solution (ethanol:glacial acetic acid, 3:1) and stored at –20 °C. BAC clones were isolated and labelled with digoxigenin-16-dUTP or biotin-11-dUTP by nick translation. FISH was performed on the pachytene chromosomes as described98. Chromosomes were counterstained with 4’6-diamindino-phenylindole (DAPI) in an antifade solution. Chromosomes and FISH signal images were captured with an Olympus BX53 fluorescence microscope conjunct with a micro charge-coupled device camera. Three experiments were performed100.
Evolutionary analysis of TCP family genes
Syntenic block identification and Ks analyses were carried out using MCscanX75 and the PAML94 yn00 NG model, respectively. MEGA7101 was used for the multiple alignment and phylogentic tree construction. Expression pattern was carried out with MeV. TF family annotation was carried out using the website of plantTFDB (http://planttfdb.cbi.pku.edu.cn/prediction.php).
The annotations and sequences of Aquilegia coerulea102 were downloaded from the website https://img.jgi.dofor exampleov/. Phalaenopsis equestris103 was downloaded from http://orchidbase.itps.ncku.edu.tw/.
Petunia axillaris and Petunia inflata were downloaded from https://solgenomics.net/organism/Petunia_axillaris/genome and https://solgenomics.net/organism/Petunia_inflata/genome, respectively.
The functional known protein members in TCP families were downloaded from the original experimental papers and used as marker proteins for TCP subfamily identification. Two putative TCP family genes (Am03g34120 with partial TCP domains and Am01g42140 with two tandem TCP domains) were excluded from phylogenetic analyses. Syntenic block and Ks analysis detected three gene pairs derived from WGD. Am08g22680/Am06g32830 (Ks = 0.99) and Am08g20570/Am06g35450 (Ks = 0.76) were located in a large syntenic block with 79 homologous gene pairs (median Ks = 0.85), while Am08g18340/Am06g39840 (Ks = 0.58) were located in a block with 11 homologous gene pairs (median Ks = 1.01).
Known MYB family TFs involved in zygomorphic flower control, DIV (Q8S9H7), DIVL(AAL78742), RAD(Q58FS3), DRIF-1(AGL11918) and DRIF-2 (AGL11919), were BlastP searched against the proteome. The protein sequences for the top two best hits were retrieved and confirmed by InterPro analysis. Retained duplicated copies derived from the WGD event were confirmed by both syntenic block and Ks analysis. Expression and function analysis identified Lotus japonicus CYC genes LjCYC1 (DQ202475), LjCYC2 (DQ202476), LjCYC3 (DQ202477) and LjCYC5 (DQ202478) that were used to BlastP57 search the L japonicas proteome. All three CYC genes could be detected with ≥98% amino acid identity except for LjCYC2, which was not detected with relaxed alignment criteria nor by Tblastn search of the L. japonicas genome (downloaded from http://chibba.agtec.uga.edu/duplication v.2.5), strongly indicating that the LjCYC2 gene was absent from the current L. japonicas assembly.
Genome-wide search for S-gene candidates in Antirrhinum and related plant species
Except for the genome data set of Antirrhinum, the recently published or revised versions of the other 14 genome data sets were downloaded from their public websites (Supplementary Data Set 14). Published SLF protein sequences of Antirrhinum were separately used to establish the group-specific SLF profiles of Plantaginaceae.
Alignments were verified manually, and a consensus sequence was created for each of the motifs of interest with the help of the Weblogo software package104. This alignment was used to generate an HMM model using the program hmmbuild from the HMMER program suite40. Using hmmsearch, the HMM model was applied in a search against the most recent protein annotations from each plant species. To confirm the presence of both F-box and Kelch/FBA1/FBA3 domains in the obtained sequences (E < 10–30), we further compared the results from hmmsearch and the Pfam databases with the hmmpfam package. Our domains of interest were annotated in Pfam as PF00646 (F-box), PF04300 (FBA1), PF08268 (FBA3) and PF01344 (Kelch domain 1).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Genome assembly data have been deposited at NCBI BioProject ID under accession codes PRJNA227267. The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive105 in the BIG Data Center106, Beijing Institute of Genomics (BIG), Chinese Academy of Sciences, under accession numbers PRJCA000223 and PRJCA001050 that are publicly accessible at http://bigd.big.ac.cn/gsa. We built the Antirrhinum genome website at http://bioinfo.sibs.ac.cn/Am, providing a portal to genome browser, Blast, data download and gene expression functions. All data that support the findings of this study are also available from the corresponding authors upon request.
References
Schwarz-Sommer, Z., Davies, B. & Hudson, A. An everlasting pioneer: the story of Antirrhinum research. Nat. Rev. Genet. 4, 655–664 (2003).
Nettancourt, D. Incompatibility in Angiosperms (Springer, Berlin, 1977).
Franklin-Tong, V. E. Self-Incompatibility in Flowering Plants (Springer, Berlin, 2008).
Coen, E. S., Carpenter, R. & Martin, C. Transposable elements generate novel spatial patterns of gene expression in Antirrhinum majus. Cell 47, 285–296 (1986).
Sommer, H., Beltrán, J. P. & Huijser, P. Deficiens, a homeotic gene involved in the control of flower morphogenesis in Antirrhinum majus: the protein shows homology to transcription factors. EMBO J. 9, 605–613 (1990).
Tröbner, W., Ramirez, L. & Motte, P. GLOBOSA: a homeotic gene which interacts with DEFICIENS in the control of Antirrhinum floral organogenesis. EMBO J. 11, 4693–4704 (1992).
Luo, D., Carpenter, R., Vincent, C., Copsey, L. & Coen, E. Origin of floral asymmetry in Antirrhinum. Nature 383, 794–799 (1996).
Luo, D. et al. Control of organ asymmetry in flowers of Antirrhinum. Cell 99, 367–376 (1999).
Glover, B. J., Perez-Rodrigues, M. & Martin, C. Development of several epidermal cell types can be specified by the same MYB-related plant transcription factor. Development 125, 3497–3508 (1998).
Schwinn, K. et al. A small family of MYB-regulatory genes controls floral pigmentation intensity and patterning in the genus Antirrhinum. Plant Cell 18, 831–851 (2006).
Lai, Z. et al. An F-box gene linked to the self-incompatibility (S) locus of Antirrhinum is expressed specifically in pollen and tapetum. Plant Mol. Biol. 50, 29–42 (2002).
Qiao, H. et al. The F-box protein AhSLF-S2 controls the pollen function of S-RNase-based self-incompatibility. Plant Cell 16, 2307–2322 (2004).
Hudson, A., Critchley, J. & Erasmus, Y. The genus Antirrhinum (snapdragon): a flowering plant model for evolution and development. Cold Spring Harb. Protoc. 3, 1–7 (2008).
Nacken, W. K., Piotrowiak, R., Saedler, H. & Sommer, H. The transposable element Tam1 from Antirrhinum majus shows structural homology to the maize transposon En/Spm and has no sequence specificity of insertion. Mol. Gen. Genet. 228, 201–208 (1991).
Krebbers, E. et al. Molecular analysis of paramutant plants of Antirrhinum majus and the involvement of transposable elements. Mol. Gen. Genet. 209, 499–507 (1987).
Hehl, R., Nacken, W. K., Krause, A., Saedler, H. & Sommer, H. Structural analysis of Tam3, a transposable element from Antirrhinum majus, reveals homologies to the Ac element from maize. Plant Mol. Biol. 16, 369–371 (1991).
Luo, D., Coen, E. S., Doyle, S. & Carpenter, R. Pigmentation mutants produced by transposon mutagenesis in Antirrhinum majus. Plant J. 1, 59–69 (1991).
Galego, L. & Almeida, J. Role of DIVARICATA in the control of dorsoventral asymmetry in Antirrhinum flowers. Genes Dev. 16, 880–891 (2002).
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R. & Phillippy, A. M. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017).
Marten, B. & Walter, P. SSPACE-LongRead: scaffolding bacterial draft genomes using long read sequence information. BMC Bioinformatics 15, 211 (2014).
Schwarz-Sommer, Z. et al. A molecular recombination map of Antirrhinum majus. BMC Plant Biol. 10, 275 (2010).
Feng, X. et al. Evolution of allometry in Antirrhinum. Plant Cell 21, 2999–3007 (2009).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439 (2006).
Bell, C. D., Soltis, D. E. & Soltis, P. S. The age and diversification of the angiosperms re-revisited. Am. J. Bot. 97, 1296–1303 (2010).
Cubas, P., Lauter, N., Doebley, J. & Coen, E. The TCP domain: a motif found in proteins regulating plant growth and development. Plant J. 18, 215–222 (1999).
Cubas, P. in Developmental Genetics and Plant Evolution (eds Cronk, Q. C. B. et al.) Ch. 13 (Taylor & Francis, London, 2002).
Raimundo, J. et al. A subcellular tug of war involving three MYB-like proteins underlies a molecular antagonism in Antirrhinum flower asymmetry. Plant J. 75, 527–538 (2013).
Reardon, W. et al. Different outcomes for the MYB floral symmetry genes DIVARICATA and RADIALIS during the evolution of derived actinomorphy in. Plantago. New Phytol. 202, 716–725 (2014).
Preston, J. C., Martinez, C. C. & Hileman, L. C. Gradual disintegration of the floral symmetry gene network is implicated in the evolution of a wind-pollination syndrome. Proc. Natl Acad. Sci. USA 108, 2343–2348 (2011).
Donoghue, M., Ree, R. & Baum, D. Phylogeny and the evolution of flower symmetry in the Asteridae. Trends. Plant. Sci. 3, 311–317 (1998).
Zhong, J. & Kellogg, E. A. Duplication and expression of CYC2-like genes in the origin and maintenance of corolla zygomorphy in Lamiales. New Phytol. 205, 852–868 (2015).
Yang, Q., Zhang, D., Li, Q., Cheng, Z. & Xue, Y. Heterochromatic and genetic features are consistent with recombination suppression of the self-incompatibility locus in Antirrhinum. Plant J. 51, 140–151 (2007).
Zhou, J. et al. Structural and transcriptional analysis of S-locus F-box genes in Antirrhinum. Sex Plant Reprod. 16, 165–177 (2003).
Kubo, K. et al. Gene duplication and genetic exchange drive the evolution of S-RNase-based self-incompatibility in Petunia. Nat. Plants 1, 14005 (2015).
Schwarz-Sommer, Z. et al. A linkage map of an F2 hybrid population of Antirrhinum majus and A. molle. Genetics 163, 699–710 (2003).
Wright, S. The distribution of self-sterility alleles in populations. Genetics 24, 538–552 (1939).
Vieira, C. P., Vieira, J. & Charlesworth, D. Evolution of the cycloidea gene family in Antirrhinum and Misopates. Mol. Biol. Evol. 16, 1474–1783 (1999).
Jabbour, F., Nadot, S. & Damerval, C. Evolution of floral symmetry: a state of the art. C. R. Biol. 332, 219–231 (2009).
Zhang, G. et al. The Apostasia genome and the evolution of orchids. Nature 549, 379–383 (2017).
Schranz, M. E., Mohammadin, S. & Edger, P. P. Ancient whole genome duplications, novelty and diversification: the WGD Radiation Lag-Time Model. Curr. Opin. Plant. Biol. 15, 147–153 (2012).
Tank, D. C. et al. Nested radiations and the pulse of angiosperm diversification – increased diversification rates often follow whole genome duplications. New Phytol. 207, 454–467 (2015).
Soltis, P. S. & Soltis, D. E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant. Biol. 30, 159–165 (2016).
Igic, B. & Kohn, J. R. The distribution of plant mating systems: study bias against obligately outcrossing species. Evolution 60, 1098–1103 (2006).
Li, W. & Chetelat, R. T. Unilateral incompatibility geneui1.1 encodes an S-locus F-box protein expressed in pollen of Solanum species. Proc. Natl Acad. Sci. USA 112, 4417–4422 (2015).
Bradley, D. et al. Evolution of flower color pattern through selection on regulatory small RNAs. Science 358, 925–928 (2017).
Allen, G. C., FloreVergara, M. A., Krasynanski, S., Kumar, S. & Thompson, W. F. A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 1, 2320–2325 (2006).
Xue, Y., Carpenter, R., Dickinson, H. & Coen, E. Origin of allelic diversity in Antirrhinmm S locus RNases. Plant Cell 8, 805–814 (1996).
Li, R. et al. The sequence and de novo assembly of the giant panda genome. Nature 463, 311–317 (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 30, 2114–2120 (2014).
Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at arXiv: https://arxiv.org/abs/1303.3997 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Wu, Y., Bhat, P. R., Close, T. J. & Lonardi, S. Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet. 4, e1000212 (2008).
Altschul, S. F. & Gish, W. Local alignment statistics. Methods Enzymo. 266, 460–480 (1996).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome Res. 14, 988–995 (2004).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Kent, W. J. BLAT‐‐the BLAST‐like alignment tool. Genome Res. 12, 656–664 (2002).
Campbell, M. A., Haas, B. J., Hamilton, J. P., Mount, S. M. & Buell, C. R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genomics 7, 327 (2006).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010).
Christine, G. E. et al. Creating a honey bee consensus gene set. Genome. Biol. 8, R13 (2007).
Liang, C., Mao, L., Ware, D. & Stein, L. Evidence-based gene predictions in plant genomes. Genome Res. 19, 1912–1923 (2009).
Quevillon, E. et al. InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120 (2005).
Servant, F. et al. ProDom: Automated clustering of homologous domains. Brief. Bioinformatics 3, 246–251 (2002).
Attwood, T. K. et al. The PRINTS Database: a fine-grained protein sequence annotation and analysis resource--its status in 2012. (Oxford, 2012).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016).
Letunic, I., Doerks, T. & Bork, P. SMART: recent updates, new developments and status in 2015. Nucleic Acids Res. 43, D257–D260 (2015).
Mi, H. et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45, D183–D189 (2016).
Gough, J., Karplus, K., Hughey, R. & Chothia, C. Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol. 313, 903–919 (2001).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349 (2007).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Kalvari, I. et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 46, D335–D342 (2017).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40, e49 (2012).
Zheng, Y. et al. iTAK: A program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol. Plant 9, 1667–1670 (2016).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Smit, A. F. A. & Hubley, R. RepeatModeler Open-1.0, V.1.0.11 (Institute for Systems Biology, 2008–2015); http://www.repeatmasker.org
Jurka, J. et al. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome. Res. 110, 462–467 (2005).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Xu, X. et al. Genome sequence and analysis of tuber crop potato. Nature 475, 189–195 (2011).
Bombarely, A. et al. Insight into the evolution of the Solanaceae from the parental genomes of Petunia hybrida. Nat. Plants 2, 16074 (2016).
The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408, 796–815 (2000).
Ming, R. et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452, 991–996 (2008).
Zhang, Q. et al. The genome of Prunus mume. Nat. Commun. 3, 1318 (2012).
Jaillon, O. et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449, 463–467 (2007).
Kawahara, Y. et al. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6, 4 (2013).
Rice, D. W. et al. Horizontal transfer of entire genomes via mitochondrial fusion in the angiosperm. Amborella. Science. 342, 1468–1473 (2013).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Edgar, R. C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113 (2004).
Posada, D. jModelTest: phylogenetic model averaging. Mol. Biol. Evol. 25, 1253–1256 (2008).
Guindon, S., Lethiec, F., Duroux, P. & Gascuel, O. PHYML Online—a web server for fast maximum likelihood‐based phylogenetic inference. Nucleic Acids Res. 33, W557–W559 (2005).
Yang, Z. et al. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556 (1997).
Nelson, A. D. L., Haug-Baltzell, A. K., Davey, S., Gregory, B. D. & Lyons, E. EPIC-CoGe: managing and analyzing genomic data. Bioinformatics 34, 2651–2653 (2018).
Andrews, S. FastQC: a quality control tool for high throughput sequence data. v.0.11.8 (Brabham Bioinformatics, Brabham Institute, 2010); http://www.bioinformatics.babraham.ac.uk/projects/fastqc
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T. & Salzberg, S. L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667 (2016).
Liu, Y. & Whitter, R. Rapid preparation of megabase plant DNA from nuclei in agarose plugs and microbeads. Nucleic Acids Res. 22, 2168–2169 (1994).
Shizuya, H. et al. Cloning and stable maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coli using an F-factor-based vector. Proc. Natl Acad. Sci. USA 89, 8794–8797 (1992).
Jiang, J., Gill, B. S., Wang, G. L., Ronald, P. C. & Ward, D. C. Metaphase and interphase fluorescence in situ hybridization mapping of the rice genome with bacterial artificial chromosomes. Proc. Natl Acad. Sci. USA 92, 4487–4491 (1995).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Filiault, D. et al. The Aquilegia genome: adaptive radiation and an extraordinarily polymorphic chromosome with a unique history. Preprint at bioRxiv https://doi.org/10.1101/264101 (2018).
Cai, J. et al. The genome sequence of the orchid Phalaenopsis equestris. Nat. Genet. 47, 65–72 (2015).
Crooks, G. E., Hon, G., Chandonia, J. M. & Brenner, S. E. WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 (2004).
Wang, Y. et al. GSA: Genome Sequence Archive. Genomics Proteomics Bioinformatics 15, 14–18 (2017).
BIG Data Center Members. The BIG Data Center: from deposition to integration to translation. Nucleic Acids Res. 45, D18–D24 (2017).
Acknowledgements
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences(XDB27010302), the Ministry of Science and Technology of China (2013CB945102), the National Natural Science Foundation of China (31401045 and 31221063) and Y. Luo acknowledges the Youth Innovation Promotion Association of Chinese Academy of Sciences (no. 2017140). We thank F. Zhao and X. Zhou (Institute of Plant Physiology and Ecology, SIBS, CAS) for maintaining the genome website and S. Ou (Michigan State University) for technical help in LTR analysis.
Author information
Authors and Affiliations
Contributions
Y.X., H.Z., D.Z. and M.L. designed the experiments. M.L., Y.L., D.Z., A.W., E.C. and Y.X. wrote the manuscript. Q.G, B.M., C.C., Y.L., Q.L., Y.Z., H.G., J.L., Y.Z., Y.S., L.C., A.W., Y.C., Y.L., M.Q., J.W., Y.C., D.W., J.Z., G.L., B.W., L.Y., C.X., J.L., S.Z., Y.Z, S.H., C.L., Y.Y., E.C. and Y.X. analysed the data and performed the experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Figures 1–10 and Supplementary Tables 1–14.
Supplementary Data
Supplementary Data Set 1–14.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, M., Zhang, D., Gao, Q. et al. Genome structure and evolution of Antirrhinum majus L. Nature Plants 5, 174–183 (2019). https://doi.org/10.1038/s41477-018-0349-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41477-018-0349-9
This article is cited by
-
A chromosome-level genome assembly of Plantago ovata
Scientific Reports (2023)
-
Expansion and adaptive evolution of the WRKY transcription factor family in Avicennia mangrove trees
Marine Life Science & Technology (2023)
-
Molecular mechanisms regulating ornamental traits and scent production in snapdragon (Antirrhinum majus L.)
Horticulture Advances (2023)
-
A CYC–RAD–DIV–DRIF interaction likely pre-dates the origin of floral monosymmetry in Lamiales
EvoDevo (2022)
-
Research advances in and prospects of ornamental plant genomics
Horticulture Research (2021)
