
Abstract
Electrochemical CO2 conversion to methane, powered by intermittent renewable electricity, provides an entrancing opportunity to both store renewable electric energy and utilize emitted CO2. Copper-based single atom catalysts are promising candidates to restrain C-C coupling, suggesting feasibility in further protonation of CO* to CHO* for methane production. In theoretical studies herein, we find that introducing boron atoms into the first coordination layer of Cu-N4 motif facilitates the binding of CO* and CHO* intermediates, which favors the generation of methane. Accordingly, we employ a co-doping strategy to fabricate B-doped Cu-Nx atomic configuration (Cu-NxBy), where Cu-N2B2 is resolved to be the dominant site. Compared with Cu-N4 motifs, as-synthesized B-doped Cu-Nx structure exhibits a superior performance towards methane production, showing a peak methane Faradaic efficiency of 73% at −1.46 V vs. RHE and a maximum methane partial current density of −462 mA cm−2 at −1.94 V vs. RHE. Extensional calculations utilizing two-dimensional reaction phase diagram analysis together with barrier calculation help to gain more insights into the reaction mechanism of Cu-N2B2 coordination structure.
Similar content being viewed by others

Introduction
The immoderate burning of fossil fuels along with the wanton emission of carbon dioxide (CO2) into atmosphere has aroused the global warming effect1,2,3,4. Electrocatalytic CO2 conversion (CO2RR), powered by intermittent renewable electricity, provides an unprecedented possibility to address this global challenge5,6,7,8. Previous studies on CO2RR have reported the generation of various C1-C3 products including hydrocarbons and oxygenates9,10,11,12,13,14,15,16,17. Methane (CH4) possesses the largest heating value among hydrocarbons and is an important raw material for the manufacture of many other chemical products including aromatic hydrocarbon. Also, CH4 as the main component of natural gas, possesses a good compatibility with the existing infrastructure for storage, distribution, and consumption18,19,20. Then, electrocatalytic conversion of CO2 into CH4 offers an entrancing opportunity to both storing renewable electric energy and utilizing CO2 emissions. In general, starting with the CO* intermediates of CO2RR, protonation of CO* to CHO* leads to CH4, whereas the competitive CO* dimerization generates C2 products21,22,23,24. On the other hand, the non-optimized binding of CO* intermediates will result in release of gaseous CO, further suppressing the CH4 selectivty25. To perform CO2-to-CH4 economically at scale, a catalyst capable of mediating the efficient formation of CH4 with high selectivity and productivity is a prerequisite. Copper (Cu) has been extensively noted for their high catalytic activity towards hydrocarbons for CO2RR26,27,28,29. However, selective production of CH4 using Cu catalysts can be difficult owing to the sluggish eight electron transfer steps of CO2-to-CH4 and the inevitable C-C coupling process on bulk Cu30,31.
Single-atom catalysts (SACs) with adjustable and isolated active sites have been applied to electrochemical catalysis32,33,34, showing potential in expelling C-C coupling in CO2RR. Particularly, the unique 3d transition-metal-four nitrogen (M-N4) configuration consisting of an isolated single metal atom coordinated by four N atoms in carbon matrix has been demonstrated to be favorable for CO2 electroreduction13,25,35. However, the performance of Cu-N4 materials in methane production is still unsatisfactory, specifically, manifesting selectivity to CO at a less cathodic potential (<−1.0 V versus reversible hydrogen electrode, vs. RHE)36,37,38 and sluggish kinetics to CH4 at a more cathodic potential39. Previous theoretical calculations revealed that relatively weak adsorption of CO* over Cu-N4 results in feasible CO* desorption instead of further protonation to CH440. As such, modulation of the electronic structure of Cu-N4 sites, by substituting relatively weak electronegativity functionalities, e.g., boron (B), is promising to facilitate the adsorption of key intermediates and thus steer the CO2RR selectivity towards CH4.
Here, we firstly conducted theoretical simulations to predict the adsorption of intermediates and thermodynamic trend over a series of sites with different concentration of boron in Cu-NxBy. It showed that, by introducing boron atoms into the coordination structure of Cu-N4, the binding of CO* and CHO* intermediates are promoted significantly, indicating the facile generation of CH4. Inspired by the theoretical predictions, we then managed to manipulate the nearest neighbor structure of isolated Cu sites with boron dopant (BNC-Cu). Comprehensive analysis of XAS data revealed that atomically dispersed Cu atoms in BNC-Cu possessed the B-doped Cu-Nx structure (Cu-NxBy) mainly in the form of Cu-N2B2. Electrochemical measurements displayed a huge boost in CH4 production, with BNC-Cu showing a high CH4 Faradaic efficiency (FE) of 73% and partial CH4 current density (jCH4) of −292 mA cm− 2 at −1.46 V vs. RHE. The divergence in CO2RR performance between boron-doped and undoped Cu sites validated the theoretical predictions, demonstrating that doping B into Cu-N4 structure serves as an effective way to enhance deep reduction activity in CO2RR. We further studied several Cu-N2B2-containing sites with the application of two-dimensional (quasi) activity and selectivity map along with reaction phase diagram, and the microkinetic simulation of selectivity also matched well with experimental data.
Results and discussion
Thermodynamic trend prediction of CO2-to-CH4 on Cu-NxBy
We first built a series of Cu-NxBy structures with different boron concentrations (Supplementary Fig. 1) and performed density functional theory (DFT) calculations to obtain the adsorption energies of all intermediates in CO2-to-CH4 (Supplementary Fig. 2). Based on the scaling relations shown in Supplementary Fig. 3, the adsorption free energy of intermediate CHO*, Gad(CHO*), was chosen as the primary descriptor to evaluate the (thermodynamic) activity trend of CO2 RR to CH4. The trend was established considering optimal paths with globally minimal (limiting) energies.
As schematically shown in Fig. 1a, a reaction path consists of many elementary steps. For given paths (black and red), they are not limited by the steps at the beginning of reaction. Instead, rA and rC are the limiting factors. All competing reaction paths were enumerated by the CatRPD code41, prior to energetic comparison, as explicitly described in our previous work41,42. Briefly, for the internal comparison of a path, the most difficult steps were first determined, such as the rA, rB, and rC from these three different paths in Fig. 1a. Then, the external comparison between all paths can derive the optimal step and energy, defined as ∆GRPD-limiting step and energy. In this case, blue path consisting of rB with the global minimum ΔG is considered as the actual path while rB is ∆GRPD-limiting step. As the ∆GRPD-limiting steps and energies are evolutional with the change of descriptor value, the (quasi) activity and selectivity trend for CO2RR can be established, named as reaction phase diagram (RPD) analysis.

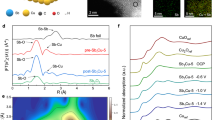
a A scheme of global energy optimization to determine the optimal path. b Thermodynamic trend for CO2RR to CH4 at −1.2 V vs. RHE, as a function with Gad(CHO*). c–f Projected density of states (PDOS) for adsorbed CHO* over Cu-N4 (c), Cu-N3B (d), Cu-N2B2 (e), Cu-NB3 (f), and Cu-N4B4 (g), where the electronic states of 3d-Cu, 2p-C (of CHO*), and 2p-B are shown in orange, gray, and blue, respectively.
Compared to pristine Cu-N4, the boron dopants can effectively enhance the reactivity of Cu sites and the adsorption energies. For instance, the more stable COOH* and CHO* can promote the protonation of CO2 and CO, resulting in promoted production of CH4 over Cu-NxBy sites at various applied potential (Fig. 1b and Supplementary Fig. 4). Projected density of states (PDOS) of CHO* adsorbed on different Cu-NxBy sites explain well more stable adsorption energies (Fig. 1c–g and Supplementary Fig. 5). Different from the localized 2p-C states of adsorbed CHO* on Cu-N4 (Fig. 1c), it is more delocalized as the C atom (of CHO*) simultaneously binds with Cu and B at Cu-NxBy sites, which indicates much stronger electronic interaction between CHO* and Cu-NxBy sites. Besides, the hybridized peaks (i–x, as marked in Fig. 1d–g and Supplementary Fig. 5, respectively) below Fermi level show the strong electronic resonance between 2p-C (of CHO*) and 3d-Cu/2p-B of Cu-N3B, Cu-N2B2, Cu-NB3, and Cu-N4B4, suggesting enhanced reactivity on different Cu-NxBy sites. This is also supported by analyzing the crystal orbital Hamilton population (COHP) (Supplementary Fig. 6). The COHP analysis showed that, the intermediate CHO* adsorbs on CuN4 through C atom bonding with Cu (C-Cu), where the antibonding states of C-Cu bond is partially occupied. With B substituting in Cu-N4 motif, the C-B bonds are much stronger than C-Cu, indicating the enhancement of CHO* adsorption at Cu-NxBy sites. Besides, the marked peaks below Fermi level (b-e) refer to the strong electronic resonance between 2p-C (of CHO*) and 2p-B of Cu-NxBy sites. Both PDOS and COHP analysis indicated that B substituting in Cu-N4 motif can promote the adsorption of key intermediates, providing an opportunity to boost the intrinsic activity of CO2-to-CH4.
Synthesis and characterization of Cu single atom catalysts
Encouraged by these promising theoretical findings, we sought to construct an isolated Cu catalyst in which boron and nitrogen atoms are coordinated with Cu center. The lack of molecules containing Cu-N and Cu-B bond simultaneously limited the ball milling synthesis of a well-defined catalyst with a precise coordination configuration43,44. Hence, we managed to introduce B via a one-pot carbonization process (see details in “Methods”). Nevertheless, the high-temperature pyrolysis process may lead to the co-existence of several structures containing Cu-NxBy motifs. We also prepared the Cu-N4 catalyst (NC-Cu) similarly but without boron precursor (Supplementary Figs. 7 and 8). Transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images indicated that the BNC-Cu catalyst possessed a tubular structure with a mean external diameter of ~100 nm (Fig. 2a, b). X-ray diffraction (XRD) pattern and Raman spectrum only exhibited fingerprint peaks corresponding to graphitic carbon without Cu/CuOx/CuBx signals, suggesting the formation of highly dispersed Cu species (Supplementary Figs. 9 and 10). The uniform distribution of Cu, B, C, and N was verified by energy-dispersive X-ray spectroscopy (EDS) elemental mapping (Supplementary Fig. 11), confirming the formation of B and N co-doped carbon support. The bright spots marked with red circles in high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) image represented isolated Cu atoms dispersed in BNC support (Fig. 2c). Cu particles were not observed in HAADF-STEM images (Fig. 2c and Supplementary Fig. 11). Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis revealed Cu loadings to be 2.5 wt.% for BNC-Cu and 2.7 wt.% for NC-Cu, respectively. X-ray photoelectron spectroscopy (XPS) results indicated the elemental composition of BNC-Cu with N: 17.2%, C: 38.0%, B: 16.9% (Supplementary Fig. 12a), demonstrating heavy doping of N and B into carbon matrix with similar doping-level. The high-resolution XPS spectrum for B 1 s (Supplementary Fig. 12b) suggested the existence of B-C, B-N, and B-O structure. N 1 s spectrum (Supplementary Fig. 12c) also displayed obvious component contribution corresponding to N-B45,46,47. The detailed information for high-resolution XPS spectra of NC-Cu could be found in Supplementary Fig. 13. Comparison between N 1 s spectra of BNC-Cu and NC-Cu (Supplementary Fig. 14) also indicated that different N species were generated from general interaction between B and N atoms.

a–c TEM image (a), SEM image (b), and HAADF-STEM image (c) of BNC-Cu. d, e B K-edge (d), N K-edge (e) XANES spectra of BNC-Cu. The B K-edge XANES for BNC-Cu showed the spectral fingerprints of B (sp2)-N (sp2) bonds with three B 1 s → π* resonances and one 1 s → σ* resonance. The resonance of B-C bonds could also be seen. The generally-existed interaction between B and N in BNC-Cu was further identified by signals of N 1 s → π* resonance and N 1 s → σ* resonance of B-N bonds in N K-edge XANES results, π* and σ* resonances of C = N were also observed. f WT-EXAFS plots of Cu foil, NC-Cu, and BNC-Cu. g FT-EXAFS spectra at Cu K-edge of BNC-Cu, CuO, and Cu foil. h, The corresponding EXAFS fitting curves of BNC-Cu at R space. The inset of h is the schematic model of the fitting result, Cu (orange), N (blue), B (pink).
We further employed X-ray absorption near-edge structure (XANES) spectroscopy to accurately identify the nitrogen and boron species in BNC-Cu. The B, N, and C K-edges XANES (Fig. 2d, e and Supplementary Fig. 15) for BNC-Cu exhibited the fingerprint peaks from B-C, B-N, and C-N, demonstrating the strong orbital interaction, via direct bonding, of arbitrary pairs from B, N, and C atoms45,48,49. Fourier transform infrared (FTIR) spectrum of BNC-Cu (Supplementary Fig. 16) also confirmed the existence of similar structures B-N, C-N, and B-C50. Meanwhile, the broadened N K-edge spectrum was associated with multiple forms of N species, corresponding well with XPS results. These results strongly supported that boron and nitrogen dopants were integrated into carbon matrix simultaneously, and interaction generally existed between such co-doped boron and nitrogen atoms. We then conducted XAS measurements (Fig. 2f–h and Supplementary Figs. 17–20) of Cu K-edge to reveal the geometric/electronic structure of isolated Cu center in BNC-Cu and NC-Cu. The Fourier transform extended X-ray absorption fine structure (FT-EXAFS) (Fig. 2g) of both samples showed only one main peak with similar radial distance at ~1.46 Å which was ascribed to Cu-N/Cu-B scattering path, different from typical Cu-O coordination at 1.57 Å in CuO. No characteristic peak of Cu-Cu contribution at 2.20 Å could be seen, confirming the atomic dispersion of Cu atoms on BNC support. To further investigate the first layer atomic coordination environment of Cu single atoms on BNC and NC support, we performed wavelet transform (WT) of the Cu K-edge EXAFS oscillations (Fig. 2f and Supplementary Fig. 19). The WT spectra for Cu foil and CuO showed intensity peaks at 7.90 and 6.40 Å–1, attributed to typical Cu-Cu and Cu-O coordination, respectively. However, for NC-Cu catalyst, the WT spectrum showed one dominated peak, with smaller k value than those of Cu-Cu and Cu-O coordination, indicating the sole formation of Cu-N bonds (Fig. 2f). We further observed the slight WT peak shift of BNC-Cu sample compared to NC-Cu, as shown in Fig. 2f, suggesting the subtle change of atomic coordination environment between BNC-Cu and NC-Cu. A positive shift of absorption edge in BNC-Cu compared with NC-Cu, as shown in Supplementary Fig. 17, indicated that the electronic structure of Cu atoms changed with the doping of B atoms. EELS point spectrum obtained at the edge of substrate (white box in Supplementary Fig. 21) on a small area, containing three Cu single atoms (bright dots), showed the colocation of B and N atoms around single Cu atoms within the carbon matrix. Considering the angstrom resolution of the electron probe, the signals in EELS point spectrum comes from the closest neighboring atoms of Cu single atoms51,52, suggesting a high possibility of Cu–N/B direct coordination in BNC-Cu53,54,55. Shift of XPS N 1 s spectrum of BNC-Cu compared with NC-Cu (Supplementary Fig. 13), due to the general interaction between B and N, also indicated the ubiquitous nearby boron atoms around nitrogen. Thus, the more positive Cu valence in BNC-Cu was very likely due to boron atoms that substituted nitrogen atoms and directly coordinated with Cu. Hence, taken above together, we could speculate that boron has been incorporated into the first coordination shell of isolated Cu, forming Cu-NxBy motif. Furthermore, we fitted the EXAFS spectra of BNC-Cu and NC-Cu to illustrate the most possible local configuration of isolated Cu center, as shown in Fig. 2h, Supplementary Fig. 20, and Supplementary Tables 1 and 2. The best direct fitting results of FT-EXAFS showed that Cu center in NC-Cu mainly possessed Cu-N4 configuration, while the coordination numbers of Cu-N and Cu-B in BNC-Cu were 2.1 and 2.2, respectively, suggesting that the atomically dispersed Cu sites in BNC-Cu were most likely coordinated with two nitrogen atoms and two boron atoms. Considering that the resolved coordination numbers were not exactly 2, which was very likely due to both fitting error and the existence of other coordination structures in low content, we further conducted an inverse analysis by simulating the XANES spectra of different Cu-NxBy centers with each optimized DFT models. The simulation result for Cu-N2B2 model showed the best alignment with experimental curve of BNC-Cu towards the main observed features, while the subtle mismatch of experimental spectrum and simulation result was probably ascribed to other Cu-NxBy centers with a low proportion. Though we could not completely exclude the formation of other Cu-NxBy centers, determined by the thermodynamic distribution, especially under a rapid and harsh pyrolysis condition, calculations of the formation energy among different coordination structures confirmed Cu-N2B2 as the most stable structure in BNC-Cu (Supplementary Fig. 22), again validating the majority of Cu-N2B2 center, consistent with the XAS analysis.
Catalytic performance for CO2RR
The electrocatalytic properties of BNC-Cu and NC-Cu towards CO2RR were measured in a flow reactor where 0.5 M KHCO3 aqueous solution was used as electrolyte (see details in “Methods”). We performed bulk electrolysis over BNC-Cu under different current density with potential range of -0.93 to −2.06 V vs. RHE (Fig. 3a) and over NC-Cu with potential range of −1.18 to −1.96 V vs. RHE (Supplementary Fig. 25). Gaseous and aqueous products were quantified by gas chromatography (GC) and 1H Nuclear Magnetic Resonance (NMR) (Supplementary Figs. 26 and 27), respectively. We found that, compared with NC-Cu, BNC-Cu exhibited much increased CO2RR current density under similar applied potentials. In addition, B-doped Cu-Nx active site in BNC-Cu demonstrated significant lower overpotential for CO2-to-CH4 conversion compared to Cu-N4 by in situ differential electrochemical mass spectrometry (DEMS) analysis (Fig. 3b and Supplementary Fig. 28). More specifically, defining the potential, where S/N of m/z = 15 signal is 5, as the onset potential for CH4 production, we could extrapolate the CH4 onset potential of −1.11 V vs. RHE for NC-Cu whereas that of BNC-Cu was −0.82 V vs. RHE (Supplementary Fig. 29). Such difference indicated that introduction of boron ligand into Cu-N4 motif did further promote its intrinsic activity for CO2-to-CH4.

a Total current density of BNC-Cu and NC-Cu at different applied potentials. b In situ DEMS data of BNC-Cu and NC-Cu. The total current densities (i), CO2 ionic currents (m/z = 44) (ii), and CH4 ionic currents (m/z = 15) (iii) for BNC-Cu and NC-Cu electrodes in CO2-saturated 0.5 M KHCO3 as measured in the negatively going potential sweep from −0.5 to −2.0 V vs. RHE at 1 mV s−1. c Total current densities and faradaic efficiencies for various products of BNC-Cu at different applied potentials, the error bars of FEs are calculated based on three independent measurements. d Faradaic efficiencies and partial current densities for CH4 of BNC-Cu and NC-Cu as a function of cathodic potentials. Red curve for partial current density and black curve for methane FE. The error bars of FECH4 and jCH4 are calculated based on three independent measurements. e Stability test of CO2 to CH4 during an 8-h electrolysis under the current density of −200 mA cm−2. Red star for methane FE and black line for potential curve. f FT-EXAFS spectra of BNC-Cu at Cu K-edge under different applied potentials.
For BNC-Cu, the hydrogen evolution reaction (HER) firstly dominated the cathodic reaction under low applied potentials, and then retreated dramatically with increased bias while CO2RR became the main cathodic reaction (Fig. 3c). The FECH4 of BNC-Cu kept above 60% at a wide potential range from −1.23 to −1.83 V vs. RHE. The highest FECH4 of 73% reached at −1.46 V vs. RHE with a jCH4 of −292 mA cm–2, as shown in Fig. 3d. The jCH4 continuously grew with increasing cathodic potential, reaching to an impressive value of −462 mA cm–2 at −1.94 V vs. RHE. In stark contrast, NC-Cu showed very mediocre performance for CH4 production. The FE for CH4 rarely surpassed 30% at tested potential range while the FE for H2 basically kept above 60% (Supplementary Fig. 25). Comparison of FEH2 and FECH4 between such two catalysts at various similar potentials also confirmed the boost in CH4 production with BNC-Cu (Supplementary Figs. 30 and 31). H2-free selectivity of CH4 in CO2RR products under various potentials further confirmed the preferential production of CH4 over BNC-Cu (Supplementary Fig. 32). The contribution of substrates to CH4 production was rationally excluded, with both BNC and NC showing dominant HER activity and negligible CH4 production (Supplementary Figs. 33 and 34). Stability test of BNC-Cu manifested that the FE for CH4 maintained around 70% after 8-hour’s continuous electrolysis at −200 mA cm–2, without obvious potential fluctuation (Fig. 3e). Post-analysis of the cycled catalyst further demonstrated the structural stability of the formed Cu-NxBy geometry for CO2-to-CH4 (Supplementary Fig. 20 and Supplementary Table 1). For the concern of intensely dynamic aggregation of atomic-dispersed Cu species to clusters during electrolysis, as proposed and demonstrated by other researchers56,57, in situ XAS experiments for BNC-Cu and NC-Cu at Cu K-edge under different applied potentials were also carried out. No visible signal of Cu-Cu coordination could be seen in both catalysts when applied with different negative potentials ranging from −0.3 to −1.5 V vs. RHE (Fig. 3f and Supplementary Fig. 35), indicating that the atomically dispersed Cu atoms remained as dominant Cu species during electrolysis. The above results revealed that BNC-Cu catalyst performed much better than the boron-free counterpart NC-Cu for converting CO2 to CH4, validating the theoretical prediction results.
Insight into CO2-to-CH4 conversion mechanism
To further elucidate the CO2RR to CH4 mechanism over BNC-Cu, in situ attenuated total reflectance surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) measurements were conducted for BNC-Cu, as well as the NC-Cu control (Fig. 4a and Supplementary Fig. 36). As depicted in Fig. 4a, on applying potential from −0.8 to −1.5 V vs. RHE over BNC-Cu, two peaks could be well noted. Both peaks showed a slight red-shift with increasing applied potential, indicating that such observed signals originated from the in situ formed CO2RR intermediates58. We ascribed the detected two peaks at around 2100 and 1720 cm–1 to CO*59,60 and the key intermediate to produce CH421,22,23,61, CHO*62, respectively. On scanning to a negative potential, with the gradual disappearance of CO* signal, we observed the accumulation of CHO* intermediate, revealing the rapid transformation of CO* into CHO* at high applied overpotential. On the other hand, no visible CHO* peak was observed for NC-Cu (Supplementary Fig. 36), while only the CO* fingerprint peak appeared and the intensity remained almost constant over the tested potential range, revealing higher barrier for CO* hydrogenation to CHO* on NC-Cu. Such a difference on CO*-to-CHO* transition may explain the CO2RR performance gap between BNC-Cu and NC-Cu. Besides, the ATR-SEIRAS displayed another two strong broad peaks at around 1650 and 3400 cm–1, which could be ascribed to the interfacial H2O63,64. We noted the intensity of these peaks increased with increasing applied potential, suggesting that water molecules tended to adsorb on the surface of BNC-Cu during CO2RR (Fig. 3a) 65. Such readily adsorbed water molecules could provide adequate hydrogen source for CH4 production, but this trend was not obvious for NC-Cu (Supplementary Fig. 36).

a In situ ATR-IR spectra for BNC-Cu. b Two dimensional (quasi) activity and selectivity map for CO2RR and HER at −1.46 V vs. RHE, shown with two independent descriptors: Gad(CHO*) and Gad(CO*). c–e Calculated charge transfer (Δq) and Φ on electrochemical interface at the initial states (IS), transition states (TS), and final states (FS) for CO* + H2O → CHO* + (OH−e−) (c), CH3* protonation (d), and Heyrovsky steps (e) over Cu-N2B2-2, while others are shown in Supplementary Figs. 37 and 38. f Reaction free energy diagram at −1.46 V vs. RHE for CO2RR and HER (inset) over Cu-N2B2-2 and Cu-N4B4-3 sites. g Comparison between computational and experimental Faradaic efficiencies at −1.46 V vs. RHE.
The observed signals of CHO* further corroborated the use of CHO* as the target in the aforementioned computational screening over a series of Cu-NxBy sites (Supplementary Figs. 2 and 3). Based on the resolving results of Cu center in BNC-Cu, it is reasonable to speculate the boost of CH4 production was very likely due to the dominant Cu-N2B2 structure, and we thus performed further theoretical investigations to understand such high selectivity towards CH4 with barrier calculations in consideration of several structures containing Cu-N2B2 motif (Cu-N2B2−1, Cu-N2B2−2, Cu-N2B2−3, Cu-N4B4−1, and Cu-N4B4−3, illustrated in Supplementary Figs. 2 and 3). On the basis of one dimensional (quasi) activity map using Gad(CHO*) as the primary descriptor (Fig. 1b), we chose Gad(CO*) as the second descriptor as the adsorption strength of CO* is tightly correlated with CO production, thus forming a two-dimensional (quasi) activity and selectivity map that clearly shows the selectivity trend of Cu-N4, Cu-N2B2, and Cu-N4B4 sites (Fig. 4b). At −1.46 V vs. RHE, the maximum FECH4 of BNC-Cu observed in experiments, HER is thermodynamically favorable at sites with weak surface reactivity, for instance, Cu-N4 site. It again demonstrates how corporation of B atoms into Cu-N4 structure augments CO2RR to CH4. However, CO2RR is the main reaction on sites with strong adsorption of intermediates, where relatively weak CO* adsorption is prone to desorb and produce CO, otherwise to CH4 production. Accordingly, the site Cu-N2B2−2, as well as Cu-N4B4−3, are more likely to contribute in CO2 to CH4 process among other structures.
To further make comparison between Cu-N2B2−2 and Cu-N4B4−3, electrochemical barriers of CO2RR and HER over the two structures were calculated and shown in Fig. 4c–f. The electrochemical barriers of proton-coupled electron transfer reactions were calculated via a capacitor model66,67. According to “charge-extrapolation” method67,68, the amount of electron transfer (Δq) from water to the electrode surface is linearly correlated with the relative work function (Φ) at the initial states (IS), transition states (TS), and final states (FS). Figure 4c–e show such linear correlations of the critical steps for products selectivity. Illustrated by Fig. 4c, the TS of CO* protonation step to CHO* is IS-like, giving rise to a small charge transfer coefficient (β). A median TS of the ∆GRPD-limiting step for CO2RR, CH3* protonation (Fig. 4d), leads to a moderate β. However, the β of Heyrovsky step, the ∆GRPD-limiting step for HER, is larger due to its FS-like TS (Fig. 4e). It accurately predicts the high FE of H2 at very negative potentials. Based on calculated kinetic barriers, a detailed free energy landscape of CO2RR over Cu-N2B2−2 and Cu-N4B4−3 sites is shown in Fig. 4f (HER in subfigure). The CO* protonation has a barrier of 0.43 eV on Cu-N2B2−2, while such process is barrier-less on Cu-N4B4−3. However, for CH3* protonation, the larger barrier on Cu-N4B4−3 of 1.40 eV than that on Cu-N2B2−2 of 0.48 eV supports well the significance of global energy optimization. Specifically, Cu-N2B2−2 shows lower barriers of the most energetically difficult steps than Cu-N4B4−3 for CO2RR, which is consistent with the (quasi) activity map in Fig. 4b. Thus, Cu-N2B2−2 site is expected to show prominent CH4 production during CO2RR. The following microkinetic modeling was conducted for Cu-N2B2−2 at −1.46 V vs. RHE (Fig. 4g). The similarity of selectivity between computational and experimental data inversely suggested that Cu-N2B2−2 sites are probably the dominant sites among various possible Cu-containing sites. The experimental FECH4 value was 75%, which was lower than the simulated result of 80%. This discrepancy was due to the existence of other sites that were present in small proportions and did not have as high a methane productivity. The comparable product distribution also inversely validated our speculation that the majority of Cu-containing sites was Cu-N2B2, resolved by multiple XAS-relative studies. Compared with the pristine Cu-N4, the enhanced surface reactivity and appropriate adsorption energies over Cu-N2B2−2 is thus proposed as the chemical origin of the high selectivity towards CH4 for as-synthesized BNC-Cu catalyst.
In summary, by virtue of thermodynamic trend and global energy optimization analysis, refined manipulation of experimental synthesis and characterizations, theoretical microkinetic modeling, this work showcased a general way to design Cu single atom catalysts towards methane production via rationally regulating the nearest coordination environment. By modifying the Cu-N4 sites with partial B substitution, the enhanced adsorption of CO* and CHO* intermediates are theoretically proven to be beneficial for CH4 generation. Experimentally, as-obtained B-doped Cu-Nx active sites exhibited a high CH4 FE of 73% at −1.46 V vs. RHE and a maximal jCH4 of −462 mA cm–2 at −1.94 V vs. RHE, respectively. This work implicates a valuable avenue for bolstering selectivity of Cu towards a specific product.
Methods
Computational details
All density functional theory (DFT) calculations in this work were performed via the Vienna Ab-initio Simulation Package (VASP)69,70. The generalized gradient approximation (GGA)71 with the revised Perdew–Burke–Ernzerhof (rPBE) functional72 (GGA-rPBE) was employed to describe the electron interactions. The projected augmented wave (PAW)73,74 was employed to describe the valence electrons with a plane wave basis sets and the kinetic energy cutoff was set to 400 eV. Structural optimizations were performed with the residual force and electronic energy differences smaller than −0.05 eV Å–1 and 10–5 eV, respectively. In addition, dispersion effects have been taken into account by DFT-D3 method. Locating transition states were conducted by the climbing image nudged elastic band (CI-NEB)75 and dimer76,77 methods, where the convergence force was set as smaller than 0.1 eV Å−1. Moreover, a Gaussian smearing with a width of 0.2 eV and a Monkhorst–Pack k-point mesh grid of 2 × 2 × 1 were used. For atomic model construction, Cu-N4 was embedded in a graphene structure of 17.16 × 14.87 Å2 as the active center of NC-Cu (Supplementary Fig. 1), where a vacuum of 15 Å was introduced to avoid the interaction of adjacent layers. Several typical structures with different amounts of boron incorporated were also optimized (Supplementary Fig. 1).
The adsorption energy (Ead) of intermediates refers to the energies of gas-phase CO(g), H2(g), and H2O(g), the chemical potential of single H, O, C atom is \({E}_{{{{{{\rm{H}}}}}}}=\frac{1}{2}{E}_{{{{{{{\rm{H}}}}}}}_{2}}\), \({E}_{{{{{{\rm{O}}}}}}}={E}_{{{{{{{\rm{H}}}}}}}_{2}{{{{{\rm{O}}}}}}}-{E}_{{{{{{{\rm{H}}}}}}}_{2}}\), and \({E}_{{{{{{\rm{C}}}}}}}={E}_{{{{{{{\rm{CO}}}}}}}}-{E}_{{{{{{\rm{O}}}}}}}\). The intermediates adsorption energy (Ead) can be computed by the following equation:
where Ebare and Etot are the energies of bare catalysts and the catalysts with adsorbates, respectively. The numbers of carbon, hydrogen and oxygen atoms in intermediates are described by coefficients α, β, and γ. Furthermore, free energy corrections were conducted in this work at the temperature of 298 K, with the scheme described in the previous work78.
The formation energy of CuNxB4-x sites (x = 0–3) are computed via the following equation:
where ni and μi refer to the number and chemical potential of element i, respectively. The energies of Cu and C are referenced to the Cu bulk and graphene, respectively. Besides, the chemical potentials of N and B are calculated via the energies of HNO3, H3BO3, NH2CN, and H2O.
The elementary steps of electrochemical CO2RR and HER
We have considered three whole reactions:
In which several elementary reactions may be involved, as listed in the following:
Theoretical XANES spectrum calculations
The Cu K-edge XANES simulation was conducted with the FDMNES code in multiple scattering mode (Green) using the muffin-tin potential. The energy dependent exchange-correlation potential was calculated in the real Hedin–Lundqvist scheme, and then the spectra convoluted using a Lorentzian function with an energy-dependent width to account for the broadening due both to the core-hole width and to the final state width79,80. Parameter optimization was performed by comparing the theoretical and experimental spectra to acquire the most appropriate convolution parameters. The calculated models were built based on DFT calculations to avoid manual bias.
Chemicals
All chemicals were used as received without further purification. Copper (II) nitrite trihydrate (Cu(NO3)2·3H2O, AR, 99%), cyanamide (NCNH2), boric acid (H3BO3, GR, 99.8%) and polyethylene glycol-2000 (PEG-2000, average Mn = 2000) were purchased from Shanghai Macklin Biochemical Co., Ltd (Shanghai, China). Sustainion® XA-9 Alkaline Ionomer (5 % in Ethanol) was purchased from Dioxide MaterialsTM. All the chemicals were used without further purification.
Materials synthesis
BNC-Cu was synthesized through a one-pot carbonization process, copper nitride, boric acid, cyanamide, PEG-2000 acting as copper, boron, nitrogen and carbon source and soft-template respectively. In a typical synthesis, copper (II) nitrite trihydrate (0.15 g), boric acid (0.6 g), cyanamide (14.0 g), and PEG-2000 (2.0 g) were dissolved in 120 mL of deionized water under ultrasonication for 15 min. The homogeneous solution was then heated to 120 °C for 12 h under reflux and continued stir. A gray crystalline powder was obtained upon rotary evaporation of the solvent. The obtained solid mixture was annealed at 900 °C for 6 h with a heating rate of 5 °C min−1 under the protection of argon, and the obtained final product was BNC-Cu. The NC-Cu catalyst was synthesized similarly with 0.1 g copper (II) nitrite trihydrate input and without boric acid. The pure-BNC substrate was also synthesized without copper precursor, while pure NC substrate was synthesized without both coppern precursor and boric acid.
Characterization techniques
The as-synthesized BNC-Cu and NC-Cu were characterized by various analytical techniques. X-ray diffraction (XRD) was performed on a Philips X’Pert Pro Super diffractometer with Cu-Kα radiation (λ = 1.54178 Å). The morphology of the samples was observed by scanning electron microscopy (SEM, Zersss Supra 40) and transmission electron microscopy (TEM, Hitachi H-7650). HAADF-STEM images, energy-dispersive X-ray Spectroscopy (EDS) elemental mapping, and electron energy loss spectroscopy (EELS) were carried out on JEOL ARM-200F field-emission transmission electron microscope operating at an accelerating voltage of 200 kV using Mo-based TEM grids. Raman spectra were taken on a Raman microscope (Renishaw®) excited with a 785 nm excitation laser. X-ray photoelectron spectroscopy (XPS) measurements were performed on a VG ESCALAB MK II X-ray photoelectron spectrometer with Mg Kα = 1253.6 eV as the exciting source. Soft X-ray absorption spectra (B K-edge, N K-edge, and C K-edge) were carried out at the Catalysis and Surface Science Endstation at the BL11U beamline in the National Synchrotron Radiation Laboratory (NSRL) in Hefei, China.
Electrochemical measurements
The electrochemical measurements were conducted with an electrochemical workstation (CHI 660E, Shanghai CH Instruments). The Ag/AgCl wire in saturated KCl solution was adopted as the reference electrode, and the counter anodic reaction was oxygen evolution reaction over a Ni foam. All potentials were converted to the RHE reference scale using the relation ERHE = EAg/AgCl + 0.197 + pH × 0.059. Solution resistance was determined by potentiostatic electrochemical impedance spectroscopy at frequencies ranging from 0.1 Hz to 200 kHz and compensated by 85%. To prepare a catalyst cathode in a flow reactor, the catalyst ink was prepared at first with a constant composition ratio for each sample, 10 mg of catalyst, mixed with 25 μL of ionomer in 2 mL of isopropanol. Such ink was then airbrushed onto a 2 cm × 1.5 cm carbon gas diffusion layer (39BC) under the heating of a heat stage set in 80 °C. The mass loading for each catalyst was determined by weighing the mass of carbon paper before and after sprayed with catalyst ink and controlled to be ~1.0 mg cm−2. Such two electrodes were then placed on opposite sides of two polytetrafluoroethylene (PTFE) sheets with 0.4 cm × 1.5 cm channels. The geometric surface area of catalysts was controlled as 0.6 cm2. A Nafion 115 membrane (Fuel Cell Store) was sandwiched between the two PTFE sheets to separate the chambers. On the cathode side, a titanium gas flow chamber supplied 30 s.c.c.m. CO2 (monitored by an Alicat Scientific mass flow controller). 0.5 M KHCO3 electrolyte was pumped through cathode chambers with a constant rate of 0.75 mL min−1, while 1 M KOH served as anodic electrolyte was circulated around the anode with a rate of 25 mL min−1.
CO2 reduction product analysis
To quantify the gas products obtained during CO2 electrolysis, pure CO2 gas was delivered into the cathodic compartment at a constant rate and vented into a gas chromatograph (PerkinElmer Clarus® 690) equipped with a thermal conductivity detector and a flame ionization detector. The liquid products were quantified using a 400 MHz NMR spectrometer. Typically, after electrolysis, 600 μL electrolyte was mixed with 100 μL D2O (Sigma Aldrich, 99.9 at.% D) and 0.05 μL dimethylsulfoxide (Sigma Aldrich, 99.9%) as internal standard.
Ex situ and in situ XAS experiments
The ex situ X-ray absorption spectroscopy (XAS) spectra of Cu K-edge were obtained using beamline 44 A of Taiwan Photon Source (TPS) at National Synchrotron Radiation Research Center, Taiwan. All XAS data of Cu K-edge were collected in fluorescence mode using 7-element SDD detector and the incident photon energy were calibrated using standard Cu foil. In situ XAS spectra of Cu K-edge were obtained using beamline BL11B and BL14W at the Shanghai Synchrotron Radiation Facility (SSRF), Shanghai advanced Research Institute, Chinese Academy of Sciences. All XAS data of Cu K-edge were collected in fluorescence mode and the incident photon energy were calibrated using standard Cu foil. A self-design organic glass electrochemical cell was set in a three-electrode configuration and employed for our in situ XAS experiments. A rectangular orgonic glass cap was used to cover the cell and to keep the cell at fixed position of optical path. Several holes on the cap are used for CO2 bubbling and ensuring a fixed distance between working and reference electrodes for all experiment. A graphite rod and an Ag/AgCl electrode were used as the counter electrode and reference electrode, respectively. The working cell has flat walls with a single circular hole of 2 cm in diameter as a window of contact between electrolyte and catalysts, and a beam of synchrotron radiation X-ray light irradiated within the circular area during the in situ XAS experiments. To prepare a catalyst working electrode, 18 mg of catalyst, mixed with 45 μL of ionomer in 4 mL of isopropanol, such ink was then airbrushed onto a 2.5 cm × 2.5 cm carbon gas diffusion layer (39BC) with a mass loading of ~1.0 mg cm−2. Catalyst coated carbon paper was in contact with a slip of copper with the catalyst layer facing inward. Then 20 mL 0.5 M KHCO3 solution pre-saturated with CO2 was poured into the cell. The solution was not stirred and CO2 was bubbled into the solution bottom through the hole on that orgonic glass cap during the experiment. The flow rate of CO2 was 10 s.c.c.m. monitored by an Alicat Scientific mass flow controller. The cell was connected to an electrochemical station by making electrical contact to the copper tape slip that protruded from the side of the working cell. Before the in situ XAS experiments, XAFS spectra were recorded at different positions on the electrode to check the homogeneity of the catalyst. During the in situ XAS experiments, the potential on working electrode started from ocp to a series of cathodic potentials, and back to ocp. At each potential, the system was allowed to equilibrate for 10 min before recording a spectrum, then scans at the Cu K-edge were recorded. Data reduction, data analysis, and EXAFS fitting for XAS analysis in this work were performed with the Athena, Artemis, and IFEFFIT software packages. For quantitative analysis, phase shifts and backscattering amplitudes were generated by the FEFF calculations based on crystal structures of Cu, and were then calibrated through performing the FEFFIT of the EXAFS data of the reference samples, mainly to obtain the amplitude reduction factor (S02) values. With S02 known, the EXAFS data of the catalyst materials were fitted with such generated phase shifts and amplitudes.
In situ DEMS experiments
The “probe-type” DEMS was applied for the detection of volatile CH4 and H2 produced during the CO2RR, as well as the reactant CO2 consumed. The whole tests were conducted in a flow-cell system with a gas diffusion electrode as working electrode. To prepare a catalyst working electrode, 10 mg of catalyst, mixed with 25 μL of ionomer in 2 mL of isopropanol, such ink was then airbrushed onto a 2 cm × 1.5 cm carbon gas diffusion layer (39BC) with a mass loading of ~1.0 mg cm−2. Catalyst coated carbon paper was in contact with a slip of copper with the catalyst layer facing inward to the electrolyte. The effective area of the working electrode is a circle of 1 cm in diameter. A sampling probe approached the working electrode at a distance of ca. 20 μm, and a peristaltic pump replaced the solution near the working electrode at a flow rate of 1.25 mL min–1. The onset potential for each product is defined as the potential where the S/N of corresponding m/z signal is 5.
In situ attenuated total reflection surface-enhanced IR absorption spectroscopy (ATR-SEIRAS) measurements
In situ ATR-SEIRAS spectrum was gathered by a FT-IR spectrometer (Thermo Scientific Nicolet iS50) equipped with MCT-A detector. The catalyst inks were prepared by mixing 10 mg electrocatalysts, 5 mL ethanol, and 25 μL of ionomer. 10 μL of ink solution was dropped onto the central area (confined by an O-ring with Φ = 8 mm) of an Au film deposited on the basal plane of a hemicylindrical Si prism by evaporation. The Si prism was assembled in a spectro-electrochemical cell with Pt wire as a counter electrode, Ag/AgCl wire in saturated KCl solution as reference electrode, and 0.5 M KHCO3 solution pre-saturated and continuously bubbled with CO2 as electrolyte. All spectra are collected at a resolution of 4 cm−1 and each single-beam spectrum is an average of 200 scans. A CHI 660e electrochemistry workstation (Shanghai CH Instruments, Inc.) was used for potential control.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Chu, S. & Majumdar, A. Opportunities and challenges for a sustainable energy future. Nature 488, 294–303 (2012).
Davis Steven, J. et al. Net-zero emissions energy systems. Science 360, eaas9793 (2018).
Nitopi, S. et al. Progress and perspectives of electrochemical CO2 reduction on copper in aqueous electrolyte. Chem. Rev. 119, 7610–7672 (2019).
Millar, R. J. et al. Emission budgets and pathways consistent with limiting warming to 1.5 °C. Nat. Geosci. 10, 741–747 (2017).
Seneviratne, S. I., Donat, M. G., Pitman, A. J., Knutti, R. & Wilby, R. L. Allowable CO2 emissions based on regional and impact-related climate targets. Nature 529, 477–483 (2016).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364, eaav3506 (2019).
Lin, S. et al. Covalent organic frameworks comprising cobalt porphyrins for catalytic CO2 reduction in water. Science 349, 1208–1213 (2015).
Liu, C., Colón Brendan, C., Ziesack, M., Silver Pamela, A. & Nocera Daniel, G. Water splitting–biosynthetic system with CO2 reduction efficiencies exceeding photosynthesis. Science 352, 1210–1213 (2016).
Mariano Ruperto, G., McKelvey, K., White Henry, S., & Kanan Matthew,W. Selective increase in CO2 electroreduction activity at grain-boundary surface terminations. Science 358, 1187–1192 (2017).
Wakerley, D. et al. Gas diffusion electrodes, reactor designs and key metrics of low-temperature CO2 electrolysers. Nat. Energy 7, 130–143 (2022).
Xia, C. et al. Continuous production of pure liquid fuel solutions via electrocatalytic CO2 reduction using solid-electrolyte devices. Nat. Energy 4, 776–785 (2019).
Zheng, T. et al. Copper-catalysed exclusive CO2 to pure formic acid conversion via single-atom alloying. Nat. Nanotechnol. 16, 1386–1393 (2021).
Wu, Y., Jiang, Z., Lu, X., Liang, Y. & Wang, H. Domino electroreduction of CO2 to methanol on a molecular catalyst. Nature 575, 639–642 (2019).
Manthiram, K., Beberwyck, B. J. & Alivisatos, A. P. Enhanced electrochemical methanation of carbon dioxide with a dispersible nanoscale copper catalyst. J. Am. Chem. Soc. 136, 13319–13325 (2014).
Wang, X. et al. Efficient electrically powered CO2-to-ethanol via suppression of deoxygenation. Nat. Energy 5, 478–486 (2020).
Dinh, C.-T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783–787 (2018).
Huang Jianan, E. et al. CO2 electrolysis to multicarbon products in strong acid. Science 372, 1074–1078 (2021).
Howarth, R. W., Ingraffea, A. & Engelder, T. Should fracking stop? Nature 477, 271–275 (2011).
Connolly, B. M. et al. Tuning porosity in macroscopic monolithic metal-organic frameworks for exceptional natural gas storage. Nat. Commun. 10, 2345 (2019).
Gan, Y. et al. Carbon footprint of global natural gas supplies to China. Nat. Commun. 11, 824 (2020).
Wang, X. et al. Efficient methane electrosynthesis enabled by tuning local CO2 availability. J. Am. Chem. Soc. 142, 3525–3531 (2020).
Xu, Y. et al. Low coordination number copper catalysts for electrochemical CO2 methanation in a membrane electrode assembly. Nat. Commun. 12, 2932 (2021).
Zhang, H. et al. Computational and experimental demonstrations of one-pot tandem catalysis for electrochemical carbon dioxide reduction to methane. Nat. Commun. 10, 3340 (2019).
De Luna, P. et al. Catalyst electro-redeposition controls morphology and oxidation state for selective carbon dioxide reduction. Nat. Catal. 1, 103–110 (2018).
Zhang, X. et al. Molecular engineering of dispersed nickel phthalocyanines on carbon nanotubes for selective CO2 reduction. Nat. Energy 5, 684–692 (2020).
Li, F. et al. Molecular tuning of CO2-to-ethylene conversion. Nature 577, 509–513 (2020).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
García de Arquer, F. P. et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2. Science 367, 661–666 (2020).
Kuhl, K. P., Cave, E. R., Abram, D. N. & Jaramillo, T. F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 5, 7050–7059 (2012).
Appel, A. M. et al. Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem. Rev. 113, 6621–6658 (2013).
Vasileff, A., Xu, C., Jiao, Y., Zheng, Y. & Qiao, S.-Z. Surface and interface engineering in copper-based bimetallic materials for selective CO2 electroreduction. Chem 4, 1809–1831 (2018).
Wang, A., Li, J. & Zhang, T. Heterogeneous single-atom catalysis. Nat. Rev. Chem. 2, 65–81 (2018).
Yang, H. B. et al. Atomically dispersed Ni(I) as the active site for electrochemical CO2 reduction. Nat. Energy 3, 140–147 (2018).
Gu, J., Hsu, C.-S., Bai, L., Chen Hao, M. & Hu, X. Atomically dispersed Fe3+ sites catalyze efficient CO2 electroreduction to CO. Science 364, 1091–1094 (2019).
Ren, S. et al. Molecular electrocatalysts can mediate fast, selective CO2 reduction in a flow cell. Science 365, 367–369 (2019).
Jiao, L. et al. Single-atom electrocatalysts from multivariate metal-organic frameworks for highly selective reduction of CO2 at low pressures. Angew. Chem. Int. Ed. 59, 20589–20595 (2020).
Chang, Q. et al. Metal-coordinated phthalocyanines as platform molecules for understanding isolated metal sites in the electrochemical reduction of CO2. J. Am. Chem. Soc. 144, 16131–16138 (2022).
Cheng, H. et al. Construction of atomically dispersed Cu-N4 sites via engineered coordination environment for high-efficient CO2 electroreduction. Chem. Eng. J. 407, 126842 (2021).
Guan, A. et al. Boosting CO2 electroreduction to CH4 via tuning neighboring single-copper sites. ACS Energy Lett. 5, 1044–1053 (2020).
Li, J. et al. Volcano trend in electrocatalytic CO2 reduction activity over atomically dispersed metal sites on nitrogen-doped carbon. ACS Catal. 9, 10426–10439 (2019).
Fu, X., Li, J., Long, J., Guo, C. & Xiao, J. Understanding the product selectivity of syngas conversion on ZnO surfaces with complex reaction network and structural evolution. ACS Catal. 11, 12264–12273 (2021).
Guo, C. et al. Toward computational design of chemical reactions with reaction phase diagram. WIREs Comput. Mol. Sci. 11, e1514 (2021).
Deng, D. et al. A single iron site confined in a graphene matrix for the catalytic oxidation of benzene at room temperature. Sci. Adv. 1, e1500462 (2015).
Cui, X. et al. Room-temperature methane conversion by graphene-confined single iron. Atoms. Chem. 4, 1902–1910 (2018).
Min, Y. et al. Integrating single-cobalt-site and electric field of boron nitride in dechlorination electrocatalysts by bioinspired design. Nat. Commun. 12, 303 (2021).
Gu, Y. et al. Atomic Pt embedded in BNC nanotubes for enhanced electrochemical ozone production via an oxygen intermediate-rich local environment. ACS Catal. 11, 5438–5451 (2021).
Li, X. et al. Carbon doped hexagonal BN as a highly efficient metal-free base catalyst for Knoevenagel condensation reaction. Appl. Catal. B 239, 254–259 (2018).
Zhang, X. et al. Methanol conversion on borocarbonitride catalysts: Identification and quantification of active sites. Sci. Adv. 6, eaba5778 (2020).
Chen, S. et al. Designing boron nitride islands in carbon materials for efficient electrochemical synthesis of hydrogen peroxide. J. Am. Chem. Soc. 140, 7851–7859 (2018).
Dong, J. et al. Reaction-induced strong metal–support interactions between metals and inert boron nitride nanosheets. J. Am. Chem. Soc. 142, 17167–17174 (2020).
Li, J. et al. Atomically dispersed manganese catalysts for oxygen reduction in proton-exchange membrane fuel cells. Nat. Catal. 1, 935–945 (2018).
Chung, H. T. et al. Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst. Science 357, 479–484 (2017).
Xia, C. et al. General synthesis of single-atom catalysts with high metal loading using graphene quantum dots. Nat. Chem. 13, 887–894 (2021).
Wu, Z. Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 2870 (2021).
Zhao, D. et al. Atomic-level engineering Fe1N2O2 interfacial structure derived from oxygen-abundant metal–organic frameworks to promote electrochemical CO2 reduction. Energy Environ. Sci. 15, 3795–3804 (2022).
Weng, Z. et al. Active sites of copper-complex catalytic materials for electrochemical carbon dioxide reduction. Nat. Commun. 9, 415 (2018).
Creissen, C. E. & Fontecave, M. Keeping sight of copper in single-atom catalysts for electrochemical carbon dioxide reduction. Nat. Commun. 13, 2280 (2022).
Wei, X. et al. Highly selective reduction of CO2 to C2+ hydrocarbons at copper/polyaniline interfaces. ACS Catal. 10, 4103–4111 (2020).
Moradzaman, M. & Mul, G. Infrared analysis of interfacial phenomena during electrochemical reduction of CO2 over polycrystalline copper electrodes. ACS Catal. 10, 8049–8057 (2020).
Kim, C. et al. Achieving selective and efficient electrocatalytic activity for CO2 reduction using immobilized silver nanoparticles. J. Am. Chem. Soc. 137, 13844–13850 (2015).
Li, Y. et al. Promoting CO2 methanation via ligand-stabilized metal oxide clusters as hydrogen-donating motifs. Nat. Commun. 11, 6190 (2020).
Ma, W. et al. Electrocatalytic reduction of CO2 to ethylene and ethanol through hydrogen-assisted C–C coupling over fluorine-modified copper. Nat. Catal. 3, 478–487 (2020).
Nihonyanagi, S., Yamaguchi, S. & Tahara, T. Counterion effect on interfacial water at charged interfaces and its relevance to the Hofmeister series. J. Am. Chem. Soc. 136, 6155–6158 (2014).
Katayama, Y. et al. An in situ surface-enhanced infrared absorption spectroscopy study of electrochemical CO2 reduction: selectivity dependence on surface C-bound and O-bound reaction intermediates. J. Phys. Chem. C 123, 5951–5963 (2019).
Ge, W. et al. Dynamically formed surfactant assembly at the electrified electrode–electrolyte interface boosting CO2 electroreduction. J. Am. Chem. Soc. 144, 6121–6622 (2022).
Fang, Y.-H., Wei, G.-F. & Liu, Z.-P. Constant-charge reaction theory for potential-dependent reaction kinetics at the solid–liquid interface. J. Phys. Chem. C 118, 3629–3635 (2014).
Chan, K. & Nørskov, J. K. Electrochemical barriers made simple. J. Phys. Chem. Lett. 6, 2663–2668 (2015).
Chan, K. & Nørskov, J. K. Potential dependence of electrochemical barriers from ab initio calculations. J. Phys. Chem. Lett. 7, 1686–1690 (2016).
Kresse, G. & Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269 (1994).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P. et al. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 46, 6671–6687 (1992).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Blöchl, P. E., Jepsen, O. & Andersen, O. K. Improved tetrahedron method for Brillouin-zone integrations. Phys. Rev. B 49, 16223–16233 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Henkelman, G., Uberuaga, B. P. & Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 113, 9901–9904 (2000).
Henkelman, G. & Jónsson, H. A dimer method for finding saddle points on high dimensional potential surfaces using only first derivatives. J. Chem. Phys. 111, 7010–7022 (1999).
Kästner, J. & Sherwood, P. Superlinearly converging dimer method for transition state search. J. Chem. Phys. 128, 014106 (2008).
Li, H. et al. Toward a comparative description between transition metal and zeolite catalysts for methanol conversion. Phys. Chem. Chem. Phys. 22, 5293–5300 (2020).
Joly, Y. X-ray absorption near-edge structure calculations beyond the muffin-tin approximation. Phys. Rev. B 63, 125120 (2001).
Bunău, O. & Joly, Y. Self-consistent aspects of X-ray absorption calculations. J. Phys. Condens. Matter. 21, 345501 (2009).
Acknowledgements
J.Z. acknowledges National Key Research and Development Program of China (2021YFA1500500, 2019YFA0405600), CAS Project for Young Scientists in Basic Research (YSBR-051), National Science Fund for Distinguished Young Scholars (21925204), NSFC (U19A2015, 22221003, 22250007), Fundamental Research Funds for the Central Universities, Provincial Key Research and Development Program of Anhui (202004a05020074), K.C. Wong Education (GJTD−2020−15), and the DNL Cooperation Fund, CAS (DNL202003). This work was partially carried out at the USTC Center for Micro and Nanoscale Research and Fabrication. C.X. acknowledges the National Key Research and Development Program of China (2022YFB4102000), NSFC (22102018 and 52171201), the Natural Science Foundation of Sichuan Province (2022NSFSC0194), the “Pioneer” and “Leading Goose” R&D Program of Zhejiang (No. 2023C03017), the Huzhou Science and Technology Bureau (2022GZ45), the Hefei National Research Center for Physical Sciences at the Microscale (KF2021005), and the University of Electronic Science and Technology of China for startup funding (A1098531023601264). J. Xiao acknowledges the National Key R&D Program of China (No. 2021YFA1500702), the National Natural Science Foundation of China (No. 22172156), the AI S&T Program of Yulin Branch, Dalian National Laboratory For Clean Energy, CAS, Grant No.DNL-YLA202205.
Author information
Authors and Affiliations
Contributions
The project was conceptualized and supervised by J.Z., C.X., and J. Xiao, Y.D. and C.W. prepared the catalysts and performed the catalytic tests. H.L. conducted theoretical calculations. Y.D., C.W., W.X., M.Z., D.Z., J.L., and X.L. performed the catalyst characterizations. J. Xue, L.L., and C.L. performed the XAFS measurements. P.C. carried out fitting of EXAFS data. Q.J., T.Z., S.G., and Y.Z. helped in the analysis of data. Y.D., H.L., and C.W. wrote the paper with the input from all authors. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dai, Y., Li, H., Wang, C. et al. Manipulating local coordination of copper single atom catalyst enables efficient CO2-to-CH4 conversion. Nat Commun 14, 3382 (2023). https://doi.org/10.1038/s41467-023-39048-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-39048-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
