
Abstract
Here, we report an asymmetric electrochemical organonickel-catalyzed reductive cross-coupling of aryl aziridines with aryl iodides in an undivided cell, affording β-phenethylamines in good to excellent enantioselectivity with broad functional group tolerance. The combination of cyclic voltammetry analysis of the catalyst reduction potential as well as an electrode potential study provides a convenient route for reaction optimization. Overall, the high efficiency of this method is credited to the electroreduction-mediated turnover of the nickel catalyst instead of a metal reductant-mediated turnover. Mechanistic studies suggest a radical pathway is involved in the ring opening of aziridines. The statistical analysis serves to compare the different design requirements for photochemically and electrochemically mediated reactions under this type of mechanistic manifold.
Similar content being viewed by others
Introduction
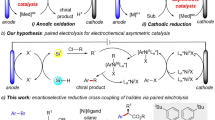
With the renaissance of organic electrolysis1,2,3,4,5,6,7,8, asymmetric electrocatalysis involving anodic oxidation has attracted extensive interest in recent years9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28. However, less attention has been paid to asymmetric electrochemical reduction reactions29,30,31,32. In 1997, Durandetti developed the first example of asymmetric electrochemical cross-coupling using chiral auxiliaries29. In 2019, a Ni/Box catalytic system for the cross-coupling of alkenyl and benzyl halides without super-stoichiometric metal powder reductants was reported by Reisman and coworkers (Fig. 1a)30. Our group described a Ni/Pyrox catalyzed electrochemical reductive homocoupling of aryl bromides, allowing the readily scalable synthesis of enantioenriched axially chiral BINOL derivatives31. Very recently, Baran and coworkers demonstrated an electrochemical Nozaki-Hiyama-Kishi coupling reaction with a chiral sulfonamide ligand, providing access to chiral alcohols (Fig. 1b)32. The scarcity of asymmetric electrochemical reduction reactions can probably be ascribed to the intolerance of chiral catalysts to reductive electrochemical conditions.

a Pioneering work on enantioselective electrochemical reductive cross-couplings by Reisman. b Enantioselective electrochemical NHK coupling by Reisman, Blackmond, and Baran. c This work: Enantioselective electrochemical reductive cross-coupling of aryl aziridines to aryl iodides.
Inspired by the pioneering work of Hillhouse33, Wolfe34, Alper, and others35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53, we wished to develop an electrochemical reductive approach for the cross-coupling of aziridines. Specifically, they showed that transition metal-catalyzed ring expansions35,36,37,38 and ring opening39,40,41,42,43,44,45,46,47,48,49,50,51,52,53 of aziridines could enable the mild synthesis of β-lactams, β-phenethylamine, and β-amino acids. Aryl aziridines are versatile synthetic intermediates for the preparation of optically pure 2,2-diarylethylamine scaffolds, which are prevalent in bioactive molecules and pharmaceuticals, including dopamine receptor agonists54,55. Although highly regioselective transformations of aziridines have been achieved, enantioselective transformations are challenging and less explored, probably due to the propensity for stereospecificity in transition metal-catalyzed nucleophilic ring openings46. Until now, only one example of asymmetric reduction of racemic aryl aziridines and aryl iodides has been realized, namely by Doyle and coworkers, using a nickel/BiOx system in the presence of Mn as the reductant51. However, 3–4 days are required to complete the transformation due to the slow turnover-limiting reduction of the Ni catalyst by Zn56,57. This important precedent indicated that accelerating the reduction of the catalyst is key to improving the efficiency of this reaction58,59,60,61,62,63,64,65,66. The efficacy of electrochemistry in the reduction of transition metal catalysts67,68,69,70,71,72,73,74, as part of our program31,75,76,77,78, encouraged us to develop transition metal-catalyzed electrochemical reactions.
Herein, we report the first example of enantioselective Ni/BiIM-catalyzed electrochemical reductive cross-coupling of aryl aziridines to aryl iodides (Fig. 1c). Specifically, the pre-activation procedure and requirement of an activating agent for the metal reductant in traditional reduction cross-couplings were avoided by using electrons to turn over the catalyst. Moreover, the limited compatibility of chiral catalysts in an electrochemical cell was resolved by identifying a ligand scaffold that is both selective and stable.
Results
Optimization studies
Initially, different chiral ligands were examined for the coupling of 2-phenyl-1-tosylaziridine (1a) to 1-iodo-4-methoxybenzene (2a) (Fig. 2A). The use of L1, which was previously employed by Doyle and coworkers, afforded 3a in less than 5% yield with 81% enantioselectivity (entry 1)51. Other chiral ligands such as pyridine bisoxazoline, pyridine oxazoline, and bisoxazoline all gave low yield and enantioselectivity of 3a (entries 2–4). To our delight, BiIM ligands L5 and L6 improved the yield of 3a to 39 and 70%, respectively (entries 5 and 6)53,79,80,81,82,83. The desired 2,2-diarylethylamine 3a could be obtained in 97% yield and 88% ee using L7 as the ligand (entry 7), while L8 resulted in a diminished yield (entry 8). In addition, when the reaction was conducted at 0 °C with an electric current of 2 mA for 18 h, 3a was isolated in 95% yield with 90% ee (entry 9). At this point, the cathodic potential was measured to be −2.20 V vs. Fc/Fc+ (Fig. 2B, black line). Using Pt as the anode instead of Zn was also tested (entry 10), and the cathodic potential was too positive to reduce the catalyst (Fig. 2C). A low yield of 3a was observed by replacing Zn with reticulated vitreous carbon (RVC, entry 11), which offers a sufficiently high but unstable anode potential (Supplementary Fig. 10). Evaluation of other anode materials in this transformation also revealed that the cathode potential could be tuned by different anode materials (Supplementary Fig. 7–9).

A Ligand evaluation. B, C Voltaic profiles of the Zn/Pt anode (red) and RVC cathode (black). D Cyclic voltammograms were recorded on a glassy carbon WE and Pt CE. DMAc containing 0.1 M n-Bu4NPF6 and 5 mM [Ni] and 5 mM Ln. Potentials were calibrated with Fc as an internal standard. E NPA 1, the partial charge of the carbon atom bridge, represents a threshold parameter able to distinguish between the BiIM and BiOx ligands. The y axis represents the values of NPA 1 and the x axis is the scope of ligands tested.
The improved performance of BiIM ligands compared to BiOx ligands in this system is strikingly different from the trend observed for the system developed by Doyle and coworkers51 using the Mn reductant. We hypothesized that the stronger binding of electron-rich BiIM ligands was required for obtaining stable electrochemically active nickel species. Namely, the color in the solution of NiBr2•glyme and L7 in DMAc changed from light green to reddish-brown within 30 min, whereas the mixture with L1 remained colorless (Supplementary Fig. 4). Furthermore, the electron-rich nature of L7 and the stability of the complexes formed with such ligands was also supported by the facile ligand exchange of L1 ligated nickel complex to L7 ligated nickel complex (Fig. 2D, and Supplementary Figs. 5 and 6). To support this claim, we sought to identify a molecular feature that could reveal a correlation with the yield observed with different ligands. We were specifically intrigued by the fact that all BiOx ligands led to <5% yield, whereas only one BiIM led to such a low value. We found that the electronic character of the carbon bridge, which likely represents the push-pull balance of the ligand, correlated well with the yield (Fig. 2E and Supplementary Fig. 19).
Substrate scope
With the optimized reaction conditions in hand, we investigated the generality and limitations of this electrochemical reductive cross-coupling reaction. As shown in Fig. 3, para-(2a–2k) and meta-substituted (2l–2u) aryl iodides with a variety of functional groups such as ether (2a, 2f, 2s), alkyl (2b–2d, 2l), ester (2e, 2n, 2o, 2t), aryl (2g, 2m), trifluoromethyl (2h, 2p), halogen (2i, 2j, 2q, 2r), and amino (2k, 2u) groups were well tolerated under the electrochemical reductive cross-coupling conditions, affording the corresponding β, β-diarylethylamines in good yields and high enantioselectivities. Multi-substituted iodobenzenes were converted to the corresponding products (3v and 3w) in excellent yields and enantioselectivities, which could be further derivatized to other biologically active intermediates84,85. Hindered 2-naphthoiodobenzene was also effectively coupled in this reaction to give 3x in 68% yield with 90% enantioselectivity. Heterocyclic iodoarenes were also suitable coupling partners, affording 3y–3ac in good yields, albeit with generally slight reductions in enantioselectivities. Alkyl iodides such as (2-Iodoethyl)benzene and other alkyl halides were examined under the electrochemical conditions, and no desired products were detected (Supplementary Table 13). Aryl aziridines bearing various functional groups on the arene were also well-suited for this electrochemical reductive cross-coupling reaction, affording 3ad–3ag in good yields and ee values. Notably, 3a could be synthesized in a gram-scale reaction with 85% yield and 90% enantioselectivity. Heteromatic, 1,2-disubstituted, poly-substituted, and alkyl aziridines were not efficient in this transformation, 2-(naphthalen-2-yl)–1-tosylaziridine and 2-benzyl-1-tosylaziridine were covert to the corresponding ring-opening product in 31 and 28% yield, respectively (Supplementary Table 11 and 12). The absolute configuration of this ring-opening product 3a was determined by single-crystal X-ray diffraction.

aAll yields refer to isolated product. b3.4 mmol scale.
Discussion
On/off experiments illustrated the crucial role of electricity and show the chemo-reduction of nickel catalyst by Zn does not occur in the background (Fig. 4A). The reaction proceeded more efficiently under electrochemical conditions than Mn powder activated by TMSCl, which further demonstrates the relative efficacy of electroreduction for turning over the nickel catalyst (Fig. 4B). To gain insights into the reaction mechanism, we conducted another series of cyclic voltammetric (CV) analyses. The mixture of NiBr2•glyme and L7 in a ratio of 1:1 exhibits two quasi-reversible reductive peaks at −1.60 V and −2.20 V vs. Fc/Fc+ in dimethylacetamide, which may be attributed to the reductive potential of NiII/NiI and NiI/Ni0, respectively (Fig. 4C, red line). No significant increase in the reduction peak was observed by the addition of 1a (Ep = −2.71 V) into the mixture of NiBr2•glyme and L7 (see Supplementary Fig. 2). In contrast, the reduction peaks of NiII/NiI and NiI/Ni0 were dramatically increased by the addition of 2a (Ep = −2.69 V, see Supplementary Fig. 2) to the mixture, along with the loss of the oxidation peak (Fig. 4D). These results demonstrate that the catalytic system of NiBr2•glyme and L7 can oxidatively add to 2a but not 1a51. Tuning the potential (Fig. 5A): at −1.0 V (NiII), 3a was not detected; at −1.8 V (NiI), 3a was detected in 10% yield; at −2.2 V (Ni0), 3a was formed in 68% yield. These constant potential electrolysis experiments illustrate that the oxidative addition of aryl iodides is predominantly caused by Ni(0) species.

A On/off experiment. B Time course experiment. C CV analysis on the interaction of 1a with the catalyst. D CV analysis on the interaction of 2a with the catalyst.

A Constant potential electrolysis. B Control experiment. C Ring-opening experiments of aziridine. D Transformation of the proposed intermediate. E Radical scavenging experiments and stereochemical studies. F Proposed mechanism.
Since the oxidative addition of 1a to nickel species33 was not observed by CV analysis (Fig. 4C)51, we carried out the reaction in the absence of the catalyst: no desired product 3a was detected, whereas 2-vinylnaphthalene and p-toluenesulfonamide (TsNH2) were formed with 70 and 68% yield, respectively (Fig. 5B). We presumed that the nucleophilic attack of iodine anion on aziridine leads to its ring opening and the proton formed via oxidation of Et3N86,87 could act as Brønsted acid to promote the ring-opening process. Treatment of 1a with Et3N•HI or HI, 2-Iodo-2-phenylmethyl-4-methylbenzenesulfonamide (4a) was formed in 81 and 87% yield, respectively (Fig. 5C). Upon subjecting 4a to our reaction conditions in the absence of the catalyst TsNH2 was formed in 58% yield (Fig. 5D). In the presence of the catalyst, 4a was readily converted to product 3a in 67% yield with 90% enantioselectivity (Fig. 5D), which indicates that 4a is a potential intermediate in this process. To clarify the mechanism of this reaction, we performed the coupling in the presence of radical scavengers (Fig. 5E, entries 1 and 2). Consequently, no reaction occurred in the presence of 2 equivalents of 2,2,6,6‐tetramethylpiperidine-1‐oxyl (TEMPO) or bis(pinacolato)diboron (B2pin2). Furthermore, both racemic and enantioenriched aziridines afford the enantioenriched product 3a with equal selectivity (Fig. 5E, entries 3–5). The stereoconvergence of this transformation also supports a radical-based mechanism.
Based on these studies and previous reports51, a plausible mechanism is presented in Fig. 5F. A [Ni0] species (B) may be formed via the cathodic reduction of a [NiII] precatalyst. Oxidative addition of aryl iodide to B generates Ar[NiII] species C. Concomitantly, nucleophilic iodide ring-opening of 1a could afford E, which can undergo single electron transfer (SET) to create radical F. The way of nickel(I) activates E by single electron transfer can not be ruled out at this stage. This radical intermediate could be trapped by C to give [NiIII] intermediate D. Reductive elimination could generate the cross-coupled product 3a and [NiI].
Given this mechanistic proposal, we sought to better understand the role of the ligand structure in the induction of selectivity. We were particularly intrigued by the difference in ligand requirements between this reaction and a mechanistically related photocatalytic reaction developed by the Doyle group (vide infra)53. To this end, enantioselectivity values for all tested ligands (Supplementary Fig. 15, a total of 21 ligands) were collected to identify a multivariable model based on the structural features of the ligand. According to the proposed mechanism, intermediate C (Fig. 5F) was selected for the extraction of structural features across the set of ligands because it directly precedes the enantioselectivity determining step. The computed Ni complex was taken to be square planar, and for simplicity, the aryl group was set as unsubstituted phenyl. The model with the strongest statistical fit that we identified is presented in Fig. 6B. The parameters in this model are both related to the substituents, namely, the distances between the carbons connecting the ligand core to its substituents. We consider this to be a stereoelectronic parameter because it reflects the steric nature of the substituent and the ability of the ligands’ core to push or pull electron density from the substituents.

A Examples of ligands used in both studies. B Model for the electrochemical reaction. C Model for the photochemical reaction. D 3D representation of the ligand.
Recently, the Doyle group reported the use of a photocatalyst coupled with a Ni catalyst for the reductive cross-coupling between styrene oxides and aryl iodides53. Although they propose a similar mechanism to the one proposed herein (Fig. 5F), the features affecting enantioselectivity in both cases are distinct. The Doyle group identified a model that is mainly influenced by electronic features, which stands in contrast to the more steric nature of the model identified for our electrocatalytic reaction (Fig. 6B, C). To verify that the use of a different parameter set in our work is not at the root of this discrepancy, we set out to identify a model for the enantioselectivity outcomes obtained by the Doyle group using our parameter set. The model with the strongest statistical fit identified for this set is presented in Fig. 6C. As anticipated and in accordance with the model identified by the Doyle group, the parameters featured in this model are mainly electronic in nature. Moreover, the parameters are focalized on the metal center. Specifically, the NPA charge of the metal and its distance from the iodine and nitrogen all play a similar role in the prediction of enantioselectivity.
Indeed, the parameters that describe the photocatalytic reaction reveal a much more electronic character. We assume that the enantioselectivity divergence using the same ligands in these two reactions (see ligands highlighted in light blue, Fig. 6B, C) stems from these distinct features. Thus, it seems that the selection of optimal ligands in both cases should be guided by different considerations. Moreover, the range of enantioselectivity values is different, revealing a much broader spread for the electrochemical reaction, which may allude to a strong dependence on catalyst structure. Indeed, once the optimal ligand was identified, a broad scope was attained, and the enantioselectivity remained fairly high throughout the whole set. Ultimately, this comparison highlights differences between these seemingly innocuous methods for SET and the utility of having both in the synthetic toolbox.
In summary, we have presented the first example of an enantioselective nickel-catalyzed electrochemical reductive cross-coupling of aryl aziridines to aryl iodides in an undivided cell, affording a wide range of β-phenethylamines under mild reaction conditions. The utilization of an electron-rich BiIM ligand is crucial to promoting this transformation. More importantly, using electrons to turn over catalysts showed high efficiency, which provided an alternative method to a combination of TMSCl with a metal reductant system. The role of the ligand structure in improving the reactivity and selectivity of this reaction was elucidated by statistical analysis and was compared to the role of the ligand structure in a mechanistically related photochemical reaction.
Methods
General procedure for the electrolysis
In the glovebox, an oven-dried electrochemical cell with a stir bar was charged with aziridine (0.2 mmol, 55 mg, 1 equiv.) and aryl iodide (0.6 mmol, 141 mg, 3 equiv.), NiBr2·glyme (0.02 mmol, 6.2 mg, 10 mol%), ligand L7 (0.03 mmol, 16.1 mg, 15 mol%), NaI (0.4 mmol, 60 mg, 2 equiv.), triethylamine (0.4 mmol, 2 equiv.), H2O (0.6 mmol, 3 equiv.), 4 mL of DMAc. The tube installed an RVC as the cathode and zinc as the sacrificial anode. The mixture was stirred at 0 °C for 30 min. The reaction mixture was electrolyzed under a constant current of 2 mA at 0 °C until the complete consumption of the starting materials was monitored by TLC (about 18 h). The resulting mixture was diluted with EtOAc and quenched with sat. NH4Cl solution and the aqueous layer extracted with EtOAc (2 × 20 mL). The combined organic layers were dried over MgSO4, the filtrate was concentrated, and the crude product was purified by automated silica gel column chromatography (EtOAc/hexanes).
More experimental procedures and a photographic guide for Ni-catalyzed enantioselective electrochemical reductive cross-couplings are provided in the Supplementary Information.
Data availability
The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC) under deposition number CCDC 2161740 (3a). The data can be obtained free of charge from The Cambridge Crystallographic Data Centre [http://www.ccdc.cam.ac.uk/data_request/cif]. The data supporting the findings of this study are available within the article and its Supplementary Information files. The complete datasets used in this paper and the XYZ files are also available at: https://github.com/Milo-group. Data is available from the corresponding authors upon request.
References
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Yuan, Y. & Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution reactions. Acc. Chem. Res. 52, 3309–3324 (2019).
Xiong, P. & Xu, H.-C. Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res. 52, 3339–3350 (2019).
Roeckl, J. L., Pollok, D., Franke, R. & Waldvogel, S. R. A decade of electrochemical dehydrogenative C, C-coupling of aryls. Acc. Chem. Res. 53, 45–61 (2020).
Jiao, K.-J., Xing, Y.-K., Yang, Q.-L., Qiu, H. & Mei, T.-S. Site-selective C–H functionalization via synergistic use of electrochemistry and transition metal catalysis. Acc. Chem. Res. 53, 300–310 (2020).
Siu, J. C., Fu, N. & Lin, S. Catalyzing electrosynthesis: a homogeneous electrocatalytic approach to reaction discovery. Acc. Chem. Res. 53, 547–560 (2020).
Zhu, C., Ang, N. W. J., Meyer, T. H., Qiu, Y. & Ackermann, L. Organic electrochemistry: molecular syntheses with potential. ACS Cent. Sci. 7, 415–431 (2021).
Yuan, Y., Yang, J. & Lei, A. Recent advances in electrochemical oxidative cross-coupling with hydrogen evolution involving radicals. Chem. Soc. Rev. 50, 10058–10086 (2021).
Lin, Q., Li, L. & Luo, S. Asymmetric electrochemical catalysis. Chem. Eur. J. 25, 10033–10044 (2019).
Ghosh, M., Shinde, V. S. & Rueping, M. A review of asymmetric synthetic organic electrochemistry and electrocatalysis: concepts, applications, recent developments and future directions. Beilstein J. Org. Chem. 15, 2710–2746 (2019).
Yamamoto, K., Kuriyama, M. & Onomura, O. Anodic oxidation for the stereoselective synthesis of heterocycles. Acc. Chem. Res. 53, 105–120 (2020).
Chang, X., Zhang, Q. & Guo, C. Asymmetric electrochemical transformations. Angew. Chem. Int. Ed. 132, 12712–12722 (2020).
Wang, X., Xu, X., Wang, Z., Fang, P. & Mei, T. Advances in asymmetric organotransition metal-catalyzed electrochemistry. Chin. J. Org. Chem. 40, 3738–3747 (2020).
Minato, D., Nagasue, Y., Demizu, Y. & Onomura, O. Efficient kinetic resolution of racemic amino aldehydes by oxidation with N-iodosuccinimide. Angew. Chem. Int. Ed. 47, 9458–9461 (2008).
Fu, N. et al. New bisoxazoline ligands enable enantioselective electrocatalytic cyanofunctionalization of vinylarenes. J. Am. Chem. Soc. 141, 14480–14485 (2019).
Zhang, Q., Chang, X. & Guo, C. Asymmetric Lewis acid catalyzed electrochemical alkylation. Angew. Chem. Int. Ed. 58, 6999–7003 (2019).
Huang, X., Zhang, Q., Lin, J., Harms, K. & Meggers, E. Electricity-driven asymmetric Lewis acid catalysis. Nat. Catal. 2, 34–40 (2019).
Dhawa, U. et al. Enantioselective pallada-electrocatalyzed C–H activation by transient directing groups: expedient access to helicenes. Angew. Chem. Int. Ed. 59, 13451–13457 (2020).
Chang, X., Zhang, J., Zhang, Q. & Guo, C. Merging electrosynthesis and bifunctional squaramide catalysis in the asymmetric detrifluoroacetylative alkylation reactions. Angew. Chem. Int. Ed. 59, 18500–18504 (2020).
Jensen, K. L., Franke, P. T., Nielsen, L. T., Daasbjerg, K. & Jørgensen, K. A. Anodic oxidation and organocatalysis: direct regio- and stereoselective access to meta-substituted anilines by α-arylation of aldehydes. Angew. Chem. Int. Ed. 49, 129–133 (2010).
Fu, N., Li, L., Yang, Q. & Luo, S. Catalytic asymmetric electrochemical oxidative coupling of tertiary amines with simple ketones. Org. Lett. 19, 2122–2125 (2017).
Li, L., Li, Y., Fu, N., Zhang, L. & Luo, S. Catalytic asymmetric electrochemical α-arylation of cyclic β-ketocarbonyls with anodic benzyne intermediates. Angew. Chem. Int. Ed. 59, 14347–14351 (2020).
Lu, F.-Y. et al. Highly enantioselective electrosynthesis of C2-quaternary indolin-3-ones. Chem. Commun. 56, 623–626 (2020).
Wang, Z.-H. et al. TEMPO-enabled electrochemical enantioselective oxidative coupling of secondary acyclic amines with ketones. J. Am. Chem. Soc. 143, 15599–15605 (2021).
Zhang, Q., Liang, K. & Guo, C. Asymmetric electrochemical arylation in the formal synthesis of (+)-amurensinine. CCS Chem. 3, 338–347 (2021).
Liang, K., Zhang, Q. & Guo, C. Nickel-catalyzed switchable asymmetric electrochemical functionalization of alkenes. Sci. Adv. 8, eadd7134 (2022).
Zhang, Q., Liang, K. & Guo, C. Enantioselective nickel-catalyzed electrochemical radical allylation. Angew. Chem. Int. Ed. 61, e202210632 (2022).
Xiong, P., Hemming, M., Ivlev, S. I. & Meggers, E. Electrochemical enantioselective nucleophilic α-C(sp3)–H alkenylation of 2-acyl imidazoles. J. Am. Chem. Soc. 144, 6964–6971 (2022).
Durandetti, M., Périchon, J. & Nédélec, J.-Y. Asymmetric induction in the electrochemical cross-coupling of aryl halides with α-chloropropionic acid derivatives catalyzed by nickel complexes. J. Org. Chem. 62, 7914–7915 (1997).
DeLano, T. J. & Reisman, S. E. Enantioselective electroreductive coupling of alkenyl and benzyl halides via nickel catalysis. ACS Catal. 9, 6751–6754 (2019).
Qiu, H. et al. Enantioselective Ni-catalyzed electrochemical synthesis of biaryl atropisomers. J. Am. Chem. Soc. 142, 9872–9878 (2020).
Gao, Y. et al. Electrochemical Nozaki-Hiyama-Kishi coupling: scope, applications, and mechanism. J. Am. Chem. Soc. 143, 9478–9488 (2021).
Lin, B. L., Clough, C. R. & Hillhouse, G. L. Interactions of aziridines with nickel complexes: oxidative-addition and reductive-elimination reactions that break and make C–N bonds. J. Am. Chem. Soc. 124, 2890–2891 (2002).
Ney, J. E. & Wolfe, J. P. Synthesis and reactivity of azapalladacyclobutanes. J. Am. Chem. Soc. 128, 15415–15422 (2006).
Alper, H., Urso, F. & Smith, D. J. H. Regiospecific metal-catalyzed ring expansion of aziridines to β-lactams. J. Am. Chem. Soc. 105, 6737–6738 (1983).
Calet, S., Urso, F. & Alper, H. Enantiospecific and stereospecific rhodium(I)-catalyzed carbonylation and ring expansion of aziridines. Asymmetric synthesis of β-lactams and the kinetic resolution of aziridines. J. Am. Chem. Soc. 111, 931–934 (1989).
Baeg, J.-O., Bensimon, C. & Alper, H. The first enantiospecific palladium-catalyzed cycloaddition of aziridines and heterocumulenes. Novel synthesis of chiral five-membered ring heterocycles. J. Am. Chem. Soc. 117, 4700–4701 (1995).
Piotti, M. E. & Alper, H. Inversion of stereochemistry in the Co2(CO)8-catalyzed carbonylation of aziridines to β-lactams. the first synthesis of highly strained trans-bicyclic β-lactams. J. Am. Chem. Soc. 118, 111–116 (1996).
Huang, C.-Y. & Doyle, A. G. The chemistry of transition metals with three-membered ring heterocycles. Chem. Rev. 114, 8153–8198 (2014).
Takeda, Y., Sameera, W. M. C. & Minakata, S. Palladium-catalyzed regioselective and stereospecific ring opening cross-coupling of aziridines: experimental and computational studies. Acc. Chem. Res. 53, 1686–1702 (2020).
Huang, C.-Y. & Doyle, A. G. Nickel-catalyzed Negishi alkylations of styrenyl aziridines. J. Am. Chem. Soc. 134, 9541–9544 (2012).
Nielsen, D. K., Huang, C.-Y. & Doyle, A. G. Directed nickel-catalyzed Negishi cross-coupling of alkyl aziridines. J. Am. Chem. Soc. 135, 13605–13609 (2013).
Jensen, K. L., Standley, E. A. & Jamison, T. F. Highly regioselective nickel-catalyzed cross-coupling of N-tosylaziridines and alkylzinc reagents. J. Am. Chem. Soc. 136, 11145–11152 (2014).
Huang, C.-Y. & Doyle, A. G. Electron-deficient olefin ligands enable generation of quaternary carbons by Ni-catalyzed cross-coupling. J. Am. Chem. Soc. 137, 5638–5641 (2015).
Duda, M. L. & Michael, F. E. Palladium-catalyzed cross-coupling of N-sulfonylaziridines with boronic acids. J. Am. Chem. Soc. 135, 18347–18349 (2013).
Takeda, Y., Ikeda, Y., Kuroda, A., Tanaka, S. & Minakata, S. Pd/NHC-catalyzed enantiospecific and regioselective Suzuki–Miyaura arylation of 2-arylaziridines: synthesis of enantioenriched 2-arylphenethylamine derivatives. J. Am. Chem. Soc. 136, 8544–8547 (2014).
Takeda, Y., Kuroda, A., Sameera, W. M. C., Morokuma, K. & Minakata, S. Palladium-catalyzed regioselective and stereo-invertive ring-opening borylation of 2-arylaziridines with bis(pinacolato)diboron: experimental and computational studies. Chem. Sci. 7, 6141–6152 (2016).
Yu, X.-Y. et al. Dual photoredox/nickel-catalyzed regioselective cross-coupling of 2-arylaziridines and potassium benzyltrifluoroborates: synthesis of β-substitued amines. Org. Lett. 20, 421–424 (2018).
Takeda, Y., Matsuno, T., Sharma, A. K., Sameera, W. M. C. & Minakata, S. Asymmetric synthesis of β2-aryl amino acids through Pd-catalyzed enantiospecific and regioselective ring-opening Suzuki–Miyaura arylation of aziridine-2-carboxylates. Chem. Eur. J. 25, 10226–10231 (2019).
Davies, J. et al. Ni-catalyzed carboxylation of aziridines en route to β-amino acids. J. Am. Chem. Soc. 143, 4949–4954 (2021).
Woods, B. P., Orlandi, M., Huang, C.-Y., Sigman, M. S. & Doyle, A. G. Nickel-catalyzed enantioselective reductive cross-coupling of styrenyl aziridines. J. Am. Chem. Soc. 139, 5688–5691 (2017).
Steiman, T. J., Liu, J., Mengiste, A. & Doyle, A. G. Synthesis of β-phenethylamines via Ni/photoredox cross-electrophile coupling of aliphatic aziridines and aryl iodides. J. Am. Chem. Soc. 142, 7598–7605 (2020).
Lau, S. H. et al. Ni/photoredox-catalyzed enantioselective cross-electrophile coupling of styrene oxides with aryl iodides. J. Am. Chem. Soc. 143, 15873–15881 (2021).
Zhang, A., Neumeyer, J. L. & Baldessarini, R. J. Recent progress in development of dopamine receptor subtype-selective agents: potential therapeutics for neurological and psychiatric disorders. Chem. Rev. 107, 274–302 (2007).
Zhang, J., Xiong, B., Zhen, X. & Zhang, A. Dopamine D1 receptor ligands: where are we now and where are we going. Med. Res. Rev. 29, 272–294 (2009).
Lin, Q. & Diao, T. Mechanism of Ni-catalyzed reductive 1,2-dicarbofunctionalization of alkenes. J. Am. Chem. Soc. 141, 17937–17948 (2019).
Lin, Q., Fu, Y., Liu, P. & Diao, T. Monovalent nickel-mediated radical formation: a concerted halogen-atom dissociation pathway determined by electroanalytical studies. J. Am. Chem. Soc. 143, 14196–14206 (2021).
Arendt, K. M. & Doyle, A. G. Dialkyl ether formation by nickel-catalyzed cross-coupling of acetals and aryl iodides. Angew. Chem. Int. Ed. 54, 9876–9880 (2015).
Kadunce, N. T. & Reisman, S. E. Nickel-catalyzed asymmetric reductive cross-coupling between heteroaryl iodides and α‑chloronitriles. J. Am. Chem. Soc. 137, 10480–10483 (2015).
Johnson, K. A., Biswas, S. & Weix, D. J. Cross-electrophile coupling of vinyl halides with alkyl halides. Chem. Eur. J. 22, 7399–7402 (2016).
Cheung, C.-W., Ploeger, M. L. & Hu, X.-L. Nickel-catalyzed reductive transamidation of secondary amides with nitroarenes. ACS Catal. 7, 7092–7096 (2017).
Poremba, K. E., Kadunce, N. T., Suzuki, N., Cherney, A. H. & Reisman, S. E. Nickel-catalyzed asymmetric reductive cross-coupling to access 1,1-diarylalkanes. J. Am. Chem. Soc. 139, 5684–5687 (2017).
Cheung, C. W., Ma, J. A. & Hu, X. Manganese-mediated reductive transamidation of tertiary amides with nitroarenes. J. Am. Chem. Soc. 140, 6789–6792 (2018).
Wang, X. et al. Ni-catalyzed reductive coupling of electron-rich aryl iodides with tertiary alkyl halides. J. Am. Chem. Soc. 140, 14490–14497 (2018).
Cheung, C.-W., Ploeger, M. L. & Hu, X.-L. Amide synthesis via nickel-catalysed reductive aminocarbonylation of aryl halides with nitroarenes. Chem. Sci. 9, 655–659 (2018).
Ploeger, M. L., Darù, A., Harvey, J. N. & Hu, X.-L. Reductive cleavage of azoarene as a key step in nickel-catalyzed amidation of esters with nitroarenes. ACS Catal. 10, 2845–2854 (2020).
Claraz, A. & Masson, G. Recent advances in C(sp3)–C(sp3) and C(sp3)–C(sp2) bond formation through cathodic reactions: reductive and convergent paired electrolyses. ACS Org. Inorg. Au 2, 126–147 (2022).
Li, C. et al. Electrochemically enabled, nickel-catalyzed amination. Angew. Chem. Int. Ed. 56, 13088–13093 (2017).
Truesdell, B. L., Hamby, T. B. & Sevov, C. S. General C(sp2)–C(sp3) cross-electrophile coupling reactions enabled by overcharge protection of homogeneous electrocatalysts. J. Am. Chem. Soc. 142, 5884–5893 (2020).
Zhang, L. & Hu, X. Nickel catalysis enables convergent paired electrolysis for direct arylation of benzylic C–H bonds. Chem. Sci. 11, 10786–10791 (2020).
Luo, J., Hu, B., Wu, W., Hu, M. & Liu, T. L. Nickel-catalyzed electrochemical C(sp3)–C(sp2) cross-coupling reactions of benzyl trifluoroborate and organic halides. Angew. Chem. Int. Ed. 60, 6107–6116 (2021).
Zhang, H.-J. et al. Chemoselective, scalable nickel-electrocatalytic O-arylation of alcohols. Angew. Chem. Int. Ed. 60, 20700–20705 (2021).
Li, Z. et al. Electrochemically enabled, nickel-catalyzed dehydroxylative cross-coupling of alcohols with aryl halides. J. Am. Chem. Soc. 143, 3536–3543 (2021).
Hamby, T. B., LaLama, M. J. & Sevov, C. S. Controlling Ni redox states by dynamic ligand exchange for electroreductive Csp3–Csp2 coupling. Science 376, 410–416 (2022).
Liu, D., Ma, H.-X., Fang, P. & Mei, T.-S. Nickel-catalyzed thiolation of aryl halides and heteroaryl halides through electrochemistry. Angew. Chem. Int. Ed. 58, 5033–5037 (2019).
Jiao, K.-J. et al. Nickel-catalyzed electrochemical reductive relay cross-coupling of alkyl halides to aryl halides. Angew. Chem. Int. Ed. 59, 6520–6524 (2020).
Liu, D. et al. Nickel-catalyzed N-arylation of NH-sulfoximines with aryl halides via paired electrolysis. Angew. Chem. Int. Ed. 60, 9444–9449 (2021).
Liu, D. et al. Paired electrolysis-enabled nickel-catalyzed enantioselective reductive cross-coupling between α-chloroesters and aryl bromides. Nat. Commun. 13, 7318 (2022).
Cheng, X., Lu, H. & Lu, Z. Enantioselective benzylic C–H arylation via photoredox and nickel dual catalysis. Nat. Commun. 10, 3549–3556 (2019).
Cheng, X., Li, T., Liu, Y. & Lu, Z. Stereo- and enantioselective benzylic C–H alkenylation via photoredox/nickel dual catalysis. ACS Catal. 11, 11059–11065 (2021).
He, Y., Liu, C., Yu, L. & Zhu, S. Enantio- and regioselective NiH-catalyzed reductive hydroarylation of vinylarenes with aryl iodides. Angew. Chem. Int. Ed. 59, 21530–21534 (2020).
He, Y., Song, H. & Zhu, S. NiH-catalyzed asymmetric hydroarylation of N-acyl enamines to chiral benzylamines. Nat. Commun. 12, 638–644 (2021).
Wang, D. & Xu, T. A pivotal role of chloride ion on nickel-catalyzed enantioselective reductive cross-coupling to perfluoroalkylated boronate esters. ACS Catal. 11, 12469–12475 (2021).
Ishibashi, H. et al. A convenient synthesis of 1, 3, 4, 5,-tetrahydro-2H-3-benzazepin-2-ones by acid-catalyzed cyclization of N-(2-arylethyl)-N-methyl-2-sulfinylacetamides. Chem. Pharm. Bull. 37, 939–943 (1989).
Zára-Kaczián, E., György, L., Deák, G., Seregi, A. & Dóda, M. Synthesis and pharmacological evaluation of some new tetrahydroisoquinoline derivatives inhibiting dopamine uptake and/or possessing a dopaminomimetic property. J. Med. Chem. 29, 1189–1195 (1986).
Constantin, T. et al. Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides. Science 367, 1021–1026 (2020).
Yang, D.-T. et al. Direct electrochemical carboxylation of benzylic C-N bonds with carbon dioxide. ACS Catal. 9, 4699–4705 (2019).
Acknowledgements
This work was financially supported by the National Key R&D Program of China (No. 2021YFA1500100), the NSF of China (Grants 21821002, 21772222, and 91956112), the S&TCSM of Shanghai (Grants 18JC1415600 and 20JC1417100), Syngenta (UK), and Bayer AG (Germany). The authors thank Abigail G. Doyle (University of California, Los Angeles) for her advice.
Author information
Authors and Affiliations
Contributions
Y.-Z.W. and Z.-H.W. designed and performed the experiments. I.L.Z. contributed to the theoretical calculation part. A.M. and T.-S.M. directed the project. B.S., D.L., and Y.-C.G. revised the manuscript. Y.-Z.W., Z.-H.W., A.M., and T.-S.M. wrote the manuscript with input from all authors. All authors analyzed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, YZ., Wang, ZH., Eshel, I.L. et al. Nickel/biimidazole-catalyzed electrochemical enantioselective reductive cross-coupling of aryl aziridines with aryl iodides. Nat Commun 14, 2322 (2023). https://doi.org/10.1038/s41467-023-37965-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37965-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
