
Abstract
The existence of bound charge transfer (CT) excitons at the interface of monolayer lateral heterojunctions has been debated in literature, but contrary to the case of interlayer excitons in vertical heterostructure their observation still has to be confirmed. Here, we present a microscopic study investigating signatures of bound CT excitons in photoluminescence spectra at the interface of hBN-encapsulated lateral MoSe2-WSe2 heterostructures. Based on a fully microscopic and material-specific theory, we reveal the many-particle processes behind the formation of CT excitons and how they can be tuned via interface- and dielectric engineering. For junction widths smaller than the Coulomb-induced Bohr radius we predict the appearance of a low-energy CT exciton. The theoretical prediction is compared with experimental low-temperature photoluminescence measurements showing emission in the bound CT excitons energy range. We show that for hBN-encapsulated heterostructures, CT excitons exhibit small binding energies of just a few tens meV and at the same time large dipole moments, making them promising materials for optoelectronic applications (benefiting from an efficient exciton dissociation and fast dipole-driven exciton propagation). Our joint theory-experiment study presents a significant step towards a microscopic understanding of optical properties of technologically promising 2D lateral heterostructures.
Similar content being viewed by others

Introduction
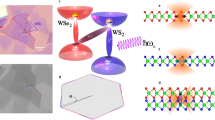
Monolayers of transition metal dichalcogenides (TMD) have attracted much attention due to their remarkable excitonic and optical properties1,2. So far the research has focused on vertical TMD heterostructures obtained by stacking TMD monolayers on top of each other3. These are characterized by spatially separated interlayer excitons forming an out-of-plane dipole and thus allowing, e.g., a gate-controllable exciton transport4,5. In comparison, much less is known about lateral TMD heterostructures6,7,8,9,10,11,12,13,14, where two different TMD monolayer materials are grown sequentially and covalently bond in the plane6,7,8,9,10,11,12 (Fig. 1a). These structures show regular monolayer optics and transport features when optically excited far from the interface12,13. At the interface, however, bound charge transfer (CT) excitons have been theoretically predicted15. Here, the Coulomb interaction binds together electrons and holes that are spatially separated at opposite sides of the junction (cf. Fig. 1a, b). This spatial separation results in an in-plane dipole that is typically larger than in vertical heterostructures15, where the dipole is limited by layer separation. Therefore, the CT exciton binding energy is expected to be smaller compared to interlayer excitons15,16,17. Furthermore, smaller band offsets have been predicted for lateral heterostructures18, suggesting that CT excitons are expected to be energetically close to the intralayer excitons. This reduced energy separation from the bright-exciton energy makes their detection challenging and could explain that so far there have been no clear experimental signatures for the existence of bound CT excitons in lateral TMD heterostructures - in contrast to interlayer excitons in vertical heterostructures3.

a Two TMD monolayers (e.g. MoSe2 and WSe2) are stitched laterally. b They have intrinsic bandgaps \({E}_{{{{{{{{\rm{Mo}}}}}}}}}^{0}\) and \({E}_{{{{{{{{\rm{W}}}}}}}}}^{0}\) while forming conduction and valence band offsets ΔEc, ΔEv around the junction. Spatially separated electrons and holes across the interface form charge-transfer (CT) excitons with the corresponding continuum energy E\({}_{{{{{{{{\rm{CT}}}}}}}}}^{0}={E}_{{{{{{{{\rm{Mo}}}}}}}}}^{0}-{{\Delta }}{E}_{{\rm {v}}}\). c, d Bound CT excitons XCT (red flat line) appear below the energy of intralayer MoSe2 exciton XMo (orange line) for either large band offsets ΔEv (interface engineering) or large dielectric constants ε (dielectric engineering).
In this work, we develop a fully microscopic and material-specific many-particle theory to shed light on the existence of CT excitons in lateral TMD heterostructures. We also perform cryogenic photoluminescence (PL) measurements to directly check the theoretical predictions. Motivated by the recent progress in the growth of lateral heterostructures with atomically sharp interfaces9,14,19,20,21, we theoretically investigate optimal conditions to find CT excitons (i) via interface engineering (interface widths, band offsets) and (ii) dielectric engineering (surrounding substrates). In particular, we address the competition between Coulomb-induced spatial confinement of excitons (Bohr radius) and interface widths. Considering the exemplary case of hBN-encapsulated MoSe2–WSe2 lateral heterostructures14,20, we predict for small junction widths and low temperatures the appearance of an additional low-energy resonance in PL spectra that we assign to a bound CT exciton. To test this, we perform cryogenic PL measurements in hBN-encapsulated MoSe2–WSe2 lateral heterostructures with a high-quality, very narrow junction width of ~2–3 nm14. We find PL emission peaks at the heterojunction in the high-quality samples that are below the MoSe2 and WSe2 intralayer excitons and that present a strong indication for the bound CT excitons predicted by our microscopic theory. Our joint theory–experiment study presents an important advance for a microscopic understanding of lateral TMD heterostructures, as we identify key conditions for the observation of CT excitons in terms of interface and dielectric engineering. Furthermore, we predict CT exciton binding energies of just a few tens of meV as well as extraordinarily large dipole moments for hBN-encapsulated materials. This indicates that lateral heterostructures with ultrathin junctions and weakly bound CT excitons to have also technological relevance for optoelectronic devices due to the expected high exciton mobility13, efficient exciton dissociation, and diode-like exciton transport across the interface14.
Results
We investigate the exemplary case of an hBN-encapsulated MoSe2–WSe2 lateral heterostructure. We start with our microscopic theory and compare then with our cryogenic PL measurements. Figure 1b schematically shows the spatial variation of single-particle energies \({E}_{{{{{{{{\rm{Mo/W}}}}}}}}}^{0}(x)\) in the considered lateral heterostructure. The conduction and valence bands form offsets ΔEc, ΔEv at the interface, typically inducing a type II alignment12,20,22,23,24 with the conduction band minimum located in the MoSe2 layer18. Note that for gate-induced homojunctions25,26,27, the band offsets are the same, i.e. ΔEc = ΔEv, potentially leading to bound excitons for p–i–n junctions confined to a few tens of nanometers28. At the interface, CT excitons can be built (purple oval) with the minimum continuum energy \({E}_{{{{{{{{\rm{CT}}}}}}}}}^{0}={E}_{{{{{{{{\rm{Mo}}}}}}}}}^{0}-{{\Delta }}{E}_{{\rm {v}}}={E}_{{{{{{{{\rm{W}}}}}}}}}^{0}-{{\Delta }}{E}_{{\rm {c}}}\). Here, we focus on bright CT excitons with the hole located at the K valley in the WSe2 layer and the electron located at the K valley in the MoSe2 layer, as this is the energetically lowest CT configuration, cf. the Supplementary material. Dark CT excitons could be important e.g. in lateral WSe2–WS2 heterostructures, where the minimum of the conduction band is located in the WS2 layer18. Importantly, this CT continuum is lower in energy than monolayer bandgaps suggesting a high occupation of these states. To obtain the energy of bound CT excitons, Coulomb interaction needs to be included resulting in excitonic energies (cf. Fig. 1c, d). The two-dimensional nature of TMD monolayers induces a reduced screening of the Coulomb interaction. The weakly screened Coulomb attraction leads in monolayers to quantization in the relative coordinate resulting in the formation of Coulomb-bound electron–hole pairs (excitons) \({X}_{{{{{{{{\rm{Mo/W}}}}}}}}}={E}_{{{{{{{{\rm{Mo/W}}}}}}}}}^{0}-{X}_{{{{{{{{\rm{Mo/W}}}}}}}}}^{{\rm {b}}}\) with large exciton binding energies \({X}_{{{{{{{{\rm{Mo/W}}}}}}}}}^{{\rm {b}}}\). The lowest 1s excitons are characterized by a Bohr radius in the range of one nanometer for hBN-encapsulated TMD monolayers29. At the interface of a lateral heterostructure an additional quantization of the center-of-mass motion can occur. The Coulomb-induced binding of spatially separated electrons and holes can form bound CT excitons that are localized at the interface with the energy \({X}_{{{{{{{{\rm{CT}}}}}}}}}={E}_{{{{{{{{\rm{CT}}}}}}}}}^{0}-{X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\). However, their binding energies \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) are expected to be smaller than in the intralayer case due to the spatial separation between electrons and holes. This reduced binding energy for spatially-separated excitons is qualitatively similar to interlayer excitons in vertical TMD heterostructures16,17, however, the separation of the latter is limited to the interlayer distance of the two TMD layers (although extendable via spacers5). In contrast, the separation of electrons and holes in a CT exciton is not limited by any geometrical constraint and can be principally much larger13,15. As a direct consequence, CT excitons are expected to have smaller binding energies compared to interlayer excitons, but exhibiting a large static electric dipole (cf. the Supplementary Materials). One important goal of this work is to study under what conditions these bound CT excitons XCT can be observed, i.e. when are they clearly below the XMo exciton and have a sufficiently large oscillator strength. To reach this goal we perform interface and dielectric engineering in our calculations, allowing us to shift the relative position of intralayer and CT excitons (cf. Fig. 1c, d).
Methodology and key quantities
To describe the spatially dependent energy landscape in a lateral heterostructure, it is crucial to include both the material-specific single-particle energies (Fig. 1b) as well as the Coulomb interaction that forms excitons (Fig. 1c, d). To this purpose, we investigate the excitonic eigenstates \(\left|{{{\Psi }}}_{n}\right\rangle\) with eigenenergies En of the Schrödinger equation \(H\left|{{{\Psi }}}_{n}\right\rangle={E}_{n}\left|{{{\Psi }}}_{n}\right\rangle\) with the Hamilton operator H including both the spatially dependent single-particle energies Eλ(x) (with the band index λ = c, v) and the Coulomb interaction between electrons and holes by using a generalized Keldysh potential VC(r)30,31. Here r is the in-plane position vector, with x and y denoting the component perpendicular and parallel to the interface, respectively. Exploiting the symmetry along the y direction parallel to the interface and the fact that the total exciton mass M = me + mh is much larger than the reduced mass μ = memh/(me + mh), we can separate the eigenstates in a center-of-mass and a relative part with \({{{\Psi }}}_{n}({{{{{{{\bf{R}}}}}}}},{{{{{{{\bf{r}}}}}}}})={\psi }_{n}({R}_{x}){{\rm {e}}}^{\imath {Q}_{y}{R}_{y}}{\phi }^{{R}_{x}}({{{{{{{\bf{r}}}}}}}})\) with r as the relative coordinate and R and Q as the center-of-mass coordinate and momentum, respectively15. Here, \({\phi }^{{R}_{x}}({{{{{{{\bf{r}}}}}}}})\) and ψn(Rx) are the solutions of the corresponding Schrödinger equations for the relative and the center-of-mass motion:
where \({V}^{{R}_{x}}({{{{{{{\bf{r}}}}}}}})={E}_{{\rm {c}}}^{0}({{{{{{{\bf{r}}}}}}}},\,{R}_{x})-{E}_{{\rm {v}}}^{0}({{{{{{{\bf{r}}}}}}}},\,{R}_{x})\) acts as an interface potential given by the space-dependent band edges \({E}_{{\rm {c,v}}}^{0}\). Note that the quantum numbers n and i describe the quantization in the center-of-mass and relative motion, respectively. In this work, we focus on the energetically lowest excitons corresponding to the i = 1s states. In the case without a junction, there are no band offsets, i.e. ΔEc/v = 0 in Fig. 1b, and Eq. (1) becomes the well-known Wannier equation with a space-independent potential and \({\tilde{E}}_{i}({R}_{x})\equiv {X}_{i}\). In this limit, the center-of-mass equation (Eq. (2)) becomes trivial corresponding to fully delocalized plane waves \({\psi }_{n}({R}_{x})\equiv {{\rm {e}}}^{\imath {Q}_{x}{R}_{x}}\) and resulting in \({E}_{n,i}\equiv {E}_{{Q}_{x},i}={X}_{i}+{\hslash }^{2}{Q}_{x}^{2}/2M\). This implies that the center-of-mass motion of excitons is free and there is no quantization.
Solving Eqs. (1) and (2), two distinct situations can occur for the ground-state energy E0, i.e. either (i) E0 = XMo or (ii) E0 < XMo. In the first case, the regular MoSe2 1s exciton is the lowest state and is expected to dominate the optical response. In the latter case, the CT exciton E0 ≡ XCT is the lowest state and could be principally observed in optical spectra. These CT states can be both bound or unbound and they are separated by the corresponding exciton binding energy \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\), cf. the red and purple lines in Fig. 1c. The conditions for the visibility of the bound CT excitons are a relatively large binding energy (higher than thermal energy to prevent thermal dissociation into unbound states) and that the state is located clearly below the lowest intralayer exciton (XMo for the investigated structure) and thus carrying a sufficiently large occupation.
To optimize the visibility of CT excitons in experiments we need to meet two conditions: (i) sufficiently low temperatures to avoid thermal dissociation of CT excitons and (ii) high sample quality so that the XMo−XCT energy separation is larger than the optical transition linewidth. Note that we recently reported high structural (electron microscopy) and optical quality (exciton transport) at the junction in CVD-grown MoSe2–WSe214. The MoSe2–WSe2 lateral heterostructure offers a small lattice mismatch between MoSe2 and WSe2, while encapsulation of the samples with hBN minimizes the dielectric disorder32 and promotes the intrinsic optical properties of the material in experiments performed at a temperature of T = 4 K. In addition, we will show below that hBN-encapsulation plays an important role for CT exciton optics.
Charge-transfer excitons
To determine the exciton energy landscape, we solve the Schrödinger equation (Eqs. (1) and (2)). We consider hBN-encapsulated samples and start with studying the limit of a relatively small band offset of ΔEv = 100 meV. Here, the energetically deepest excitons are found to be XMo states (cf. Fig. 2a). Momentum-dark exciton states have been neglected, as they are energetically higher than the monolayer exciton XMo (cf. the Supplementary material). The corresponding XW states are located 70 meV above, reflecting the band gap difference of MoSe2 and WSe2 (Fig. 2a). The center-of-mass dispersion is characterized by a parabola, and their wavefunctions ψ(Rx) are confined either on the right- or on the left-hand side of the interface. For small band offsets, the binding energy of monolayer excitons is stronger than the band offset. As a result, we find no bound CT excitons as the energy of the CT continuum is much higher than the intralayer exciton energy XMo (cf. Fig. 1).

a and b Dispersion relation of an hBN-encapsulated MoSe2-WSe2 lateral heterostructure for ΔEv = 100 and 215 meV, respectively. c Wave function of the two lowest bound CT excitons for ΔEv = 215 meV. d The energy of the lowest CT exciton (relative to XMo) and e its in-plane dipole de−h revealing the appearance of bound CT excitons for band offsets larger than ~100 meV. f CT exciton binding energy as a function of junction width w for three different band offset values, revealing that sharp interfaces allow deeply bound CT excitons with \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}} \, \approx\) 30 meV.
The energy landscape changes significantly, when we increase the band offset to ΔEv = 215 meV, which is a realistic value for lateral TMD heterostructures13,18. Interestingly, we find bound CT excitons to be the lowest states (cf. Fig. 2b). They have a flat dispersion indicating localization of excitons, or to put it in other words, there is a quantization of the center-of-mass motion across the junction. These CT exciton states are unquantized along the interface, i.e. forming a one-dimensional CT-exciton channel. We predict two bound CT states and plot their center-of-mass wave functions in Fig. 2c. These are broad in momentum space reflecting a localization in real space around the interface and induced by the Coulomb attraction between the spatially separated electrons and holes. This is in strong contrast to the case of a regular monolayer without a junction, where the center of mass motion is free and the wave functions are very narrow in momentum space and fully delocalized in real space.
We find that the bound states have typically an alternating symmetry, resulting in states with a finite and a negligible component in Qx = 0, respectively (Fig. 2c). The vanishing Qx = 0 component has a direct consequence for their oscillator strength so that only even states can emit light. The oscillator strength is also affected by the relative wavefunction, as the radiative recombination rate is proportional to the probability ∣ϕ(r = 0)∣2 of finding electrons and holes in the same position15 (cf. “Methods” section). Due to the large spatial separation, this is smaller by a factor of almost 35 for CT excitons compared to intralayer states in the situation studied in Fig. 2b. However, being the energetically lowest states, their higher occupation, in particular at low temperatures, could still compensate their smaller oscillator strength and make them visible in optical spectra. In addition, relatively large binding energies are important because they give rise to a larger oscillator strength by increasing ∣ϕ(r = 0)∣2 as the electron–hole separation is reduced.
To sum up, the crucial conditions for the visibility of bound CT states are that they have a relatively large oscillator strength and that they are considerably deeper in energy than the lowest intralayer exciton state. In the following, we investigate interface engineering (variation of band offset and junction width) and dielectric engineering (variation of substrates) to predict optimal conditions for experimental observation of bound CT excitons that have not been demonstrated so far.
Interface engineering
Here, we investigate how the CT exciton energy, its in-plane dipole, and the binding energy depend on the band offset ΔEv and the interface width w (Fig. 1). Note that varying ΔEc gives qualitatively the same results. The band offset can be engineered by growing lateral heterojunctions of different TMD monolayers. The junction width w depends on the exact growth technique and conditions. Recently, there has been an impressive technological development in lateral heterostructures allowing the realization of atomically narrow junctions of just a few nanometers13,14,20,23,24,33,34. In Fig. 2d, e we show the energetically lowest exciton state E0 and its in-plane dipole de−h, respectively. To this end, we solve the Schrödinger equation (Eqs. (1) and (2)) as a function of the band offset ΔEv for three different junction widths w = 2.4, 5, and 12 nm. The lower values correspond to recent experimentally realized sharp interfaces13,14,20,24. Importantly, our calculations show that for band offsets smaller than a critical value of about 100 meV, there are no bound CT excitons, but rather the regular MoSe2 exciton XMo is the lowest state (orange line). Increasing ΔEv, we observe that after a width-dependent critical value (defined as \({{\Delta }}{E}_{v}^{c}\)) bound CT excitons become the lowest states with linearly increasing separation from XMo as ΔEv becomes larger. A similar behaviour is predicted for the free-standing case, but with a much larger \({{\Delta }}{E}_{{\rm {v}}}^{{\rm {c}}} \, \approx \, 200\) meV (cf. the Supplementary material). The binding energy \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) is enhanced for smaller junction widths w (i.e. the red curve is further away from the purple curve in Fig. 2d).
To understand this, we plot the CT binding energy as a function of the junction width (Fig. 2f) for three different values of \({{\Delta }}{E}_{{\rm {v}}} > {{\Delta }}{E}_{{\rm {v}}}^{{\rm {c}}}\). We find that for ΔEv = 165 meV the binding energy decreases by a factor of 3 when going from w = 2.4 to w = 12 nm (X\({}_{\rm {CT}}^{{\rm {b}}} \, \approx\) 23 and 7 meV, respectively). Importantly, only for narrow junction widths, we predict binding energies of the order of the thermal energy also at room temperature. CT excitons with lower binding energy are thermally unstable and are expected to quickly dissociate into continuum states35. In addition, lower binding energies result in a smaller oscillator strength via a reduction of ∣ϕ(r = 0)∣2. We also observe that the binding energy is nearly independent of the band offset (almost overlapping lines in Fig. 2f), in particular for offsets ΔEv much larger than the critical one. For offsets just larger than \({{\Delta }}{E}_{{\rm {v}}}^{{\rm {c}}}\), we predict a monotonic decrease of \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{{{{{{{\rm{b}}}}}}}}}\) with increasing ΔEv15 (cf. the Supplementary material). As a consequence, the energy of the bound CT excitons XCT directly follows the linear decrease of the CT continuum energy (purple line in Fig. 2d) as a function of the band offset.
The abrupt reduction of the CT exciton binding energy for increasing the junction width w (Fig. 2f) induces an increase of the critical band offset \({{\Delta }}{E}_{{\rm {v}}}^{{{{{{{{\rm{c}}}}}}}}}\) from approximately 110–140 meV for junction widths w going from 2.4 to 12 nm (cf. the critical values in Fig. 2d). For the case of \({{\Delta }}{E}_{{\rm {v}}}={X}_{{{{{{{{\rm{Mo}}}}}}}}}^{{\rm {b}}}\) the energy of the MoSe2 exciton XMo exactly coincides with the energy of continuum states \({E}_{{{{{{{{\rm{CT}}}}}}}}}^{0}\) (cf. Fig. 1). For the general case, the critical band offset has to be defined as \({{\Delta }}{E}_{{\rm {v}}}^{{\rm {c}}}={X}_{{{{{{{{\rm{Mo}}}}}}}}}^{{\rm {b}}}-{X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\), such that the bound CT exciton becomes the energetically lowest state. As the binding energy of the monolayer exciton \({X}_{{{{{{{{\rm{Mo}}}}}}}}}^{{\rm {b}}}\) does not depend on the junction width, \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) is the crucial quantity. The latter has been shown to be very sensitive to the junction width (Fig. 2f). This explains why the critical band offset is increased for higher junction widths (i.e. smaller \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\)). This crucial dependence of \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) as a function of the junction width stems from the competition between the junction width w and the Bohr radius rB. The latter provides the spatial scale at which Coulomb-bound electrons and holes can redistribute around a center-of-mass position29. When w ≫ rB, excitons need huge dipoles de−h for their electron/hole constituents to reach the energetically favourable spatial positions. As a result, bound CT excitons show very small binding energy. In the opposite case of w ≲ rB, the CT exciton experiences the maximum band offset already for small spatial separations resulting in large binding energies.
We now investigate the CT-exciton in-plane dipole de−h as a function of the band offset (Fig. 2e). Similarly to the case of CT exciton energy in Fig. 2d, the dipole abruptly increases when the critical band offset \({{\Delta }}{E}_{{\rm {v}}}^{{\rm {c}}}\) is reached, i.e. when bound CT excitons are formed. For larger band offsets, the dipole only weakly increases. The dipole crucially depends on the binding energy of CT excitons: For larger \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) electrons and holes are bound close to the interface, i.e. they have a smaller in-plane distance and thus a smaller dipole. Since \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) depends strongly on w and weakly on ΔEv (Fig. 2f), there is only a small variation of de−h with the band offset (above the critical value \({{\Delta }}{E}_{{\rm {v}}}^{{\rm {c}}}\)), while de−h increases by a factor of three for w going from 2.4 to 12 nm (de−h ≈ 8 and 27 nm, respectively, cf. red and green lines in Fig. 2e). The predicted dipoles are in the range of several nanometers, which is in good agreement with previous studies13,15. The values are much larger than for interlayer excitons in vertical heterostructures, where the electron–hole separation is limited by the layer distance4. The combination of small binding energies and large dipoles is attractive for optoelectronic applications due to efficient exciton dissociation and quick exciton propagation13. From the perspective of exciton optics, this can bring two limitations: First the larger de−h, the smaller is the binding energy \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) (Fig. 2e, f) and the less stable CT excitons are. Second, the increase of the dipole leads to a decrease of ∣ϕ(r = 0)∣2 resulting in a lowering of the oscillator strength with crucial implications for the visibility of CT excitons in experiments.
In a nutshell, by performing interface engineering one can achieve thermally stable bound CT excitons for atomically sharp interfaces. In particular, for the case of hBN-encapsulated MoSe2–WSe2, we predict binding energies of \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}} \, \approx \, 20\!\!-\!\!30\) meV.
Dielectric engineering
Besides interface engineering, Coulomb interaction can be changed by varying the dielectric environment determined by the substrate. We focus again on the lateral MoSe2-WSe2 heterostructure with a narrow junction width of w = 2.4 nm and a band offset of Δv = 215 meV (i.e. above the critical value discussed in Fig. 2). These values are realistic according to the previous studies on lateral heterostructures13,18. Note that in our study we consider the band offset and the interface width to be robust with respect to the change in the dielectric environment.
In Fig. 3a we show the bound and unbound CT energies XCT and \({E}_{{{{{{{{\rm{CT}}}}}}}}}^{0}\) as a function of the dielectric constant ε of the substrate. We focus on CT energies relative to the intralayer MoSe2 exciton XMo, as the occupation of CT excitons is determined by their relative spectral distance to the monolayer exciton. We find a considerable shift to lower energies for increasing ε. The energy separation from XMo of the bound CT exciton XCT is reduced from approximately 6 meV in the free-standing case (ε = 1) to 88 meV for hBN-encapsulated samples. A similar decrease is also found for the unbound CT state \({E}_{{{{{{{{\rm{CT}}}}}}}}}^{0}\). As we are considering only relative energies (with respect to XMo), the dependence of the band gap energy \({E}_{{{{{{{{\rm{Mo}}}}}}}}}^{0}\) on the dielectric screening is cancelled out. Thus the crucial quantities here are the binding energies \({X}_{{{{{{{{\rm{Mo}}}}}}}}}^{{{{{{{{\rm{b}}}}}}}}}\) and \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{{{{{{{\rm{b}}}}}}}}}\) of the monolayer and the bound CT excitons. The latter is very sensitive to the dielectric environment, as shown in Fig. 3b. In particular, the decrease of \({X}_{{{{{{{{\rm{Mo}}}}}}}}}^{{\rm {b}}}\) (orange in Fig. 3b) is responsible for the behaviour found for the energy of unbound CT excitons \({E}_{{{{{{{{\rm{CT}}}}}}}}}^{0}\) (purple in Fig. 3a). Note that for ε ≈ 3, the MoSe2 exciton XMo is shifted above the unbound CT energy resulting in a sign change in the purple line in Fig. 3a.

a Bound and unbound CT exciton energies XCT and \({E}_{{{{{{{{\rm{CT}}}}}}}}}^{0}\) (relative to the intralayer MoSe2 exciton XMo), b CT binding energy \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\), and c the corresponding CT exciton dipole de−h as a function of the dielectric constant of the substrate (for the band offset ΔEv = 0.215 eV and the interface width w = 2.4 nm).
Interestingly, we predict a drastic decrease of \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) resulting in \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) being much smaller than \({X}_{{{{{{{{\rm{Mo}}}}}}}}}^{{\rm {b}}}\) (by approximately a factor of 4 and 7 in the case of SiO2-air and hBN-encapsulation, respectively). This drastic decrease is in contrast to the situation in vertical heterostructures, where the binding energies are comparable for intra- and interlayer excitons16,17. This difference between vertical and lateral heterostructures can be ascribed to the much larger spatial electron-hole separations in CT excitons compared to interlayer excitons, where the separation is limited by the interlayer distance in vertical heterostructures. The CT binding energy decreases with the increasing dipole (cf. the Supplementary material), similar to the behaviour of interlayer excitons with increasing interlayer spacing16. Only for free-standing lateral heterostructures, we predict that CT excitons have dipoles de−h ≈ 1 nm comparable with vertical heterostructures, resulting in comparable binding energies. In contrast, for substrates with an increasing dielectric constant, we find significantly enhanced in-plane dipole moments, e.g. de−h ≈ 5 nm for the SiO2 substrate or de−h ≈ 9.6 nm for hBN-encapsulated samples. The increase in de−h leads to a decrease in the CT binding energy as well as of the radiative recombination rate by one order of magnitude compared to the free-standing case.
The behaviour of XCT in Fig. 3a results from the non-trivial interplay of \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) and \({X}_{{{{{{{{\rm{Mo}}}}}}}}}^{{\rm {b}}}\). The CT exciton binding energy \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\) decreases from about 200 meV in the free-standing case (ε = 1) to just a few meV in the presence of high-dielectric substrates (Fig. 3b). As a consequence, bound and unbound CT energies almost coincide for large ε (red and purple line in Fig. 3a). Furthermore, they shift well below the intralayer MoSe2 energy XMo. In the limiting case of very large ε, the separation between XCT and XMo tends toward the value of the band offset due to the negligible excitonic binding energies, (cf. Fig. 1). In the free-standing limit, we find XCT ≈ XMo, despite the large CT exciton binding energy. This occurs since for ε → 1 also the unbound CT energy is shifted up relative to XMo (purple line in Fig. 3a) and cancels out the change in \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}}\), such that \({X}_{{{{{{{{\rm{CT}}}}}}}}}={E}_{{{{{{{{\rm{CT}}}}}}}}}^{0}-{X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}} \, \approx \, {X}_{{{{{{{{\rm{Mo}}}}}}}}}\). In this regime, the bound CT excitons are thermally stable thanks to binding energies of a few hundred meV (Fig. 3b), but they are located only slightly below XMo (Fig. 3a). Thus, they are weakly populated and not visible in PL spectra, cf. the supplementary material. It is, however, in the intermediate range of 2 < ε < 5 that one finds the optimal situation where we have a considerably large CT binding energy and at the same time the CT exciton is located well below the MoSe2 exciton. For a SiO2-air environment (ε ≈ 2.4), we predict the CT exciton to be ~35 meV below XMo with a binding energy of \({X}_{{{{{{{{\rm{CT}}}}}}}}}^{{\rm {b}}} \, \approx \,\)63 meV. The energy separation between CT and intralayer MoSe2 excitons increases significantly in hBN-encapsulated heterostructures (ε ≈ 4.5), but this comes at the price of a smaller binding energy of X\({}_{\rm {CT}}^{{\rm {b}}} \, \approx\) 20 meV.
In a nutshell, high-dielectric substrates lead to bound CT excitons that are located much below the intralayer exciton and thus carry a large occupation, however, they are weakly bound and hence thermally unstable. The optimal case is reached for ε ≈ 2−5 where we find CT excitons with a considerably large binding energy and still a sufficient occupation.
Optical spectra
Now, we investigate whether bound CT excitons can be observed in photoluminescence spectra. First, we calculate a PL spectrum of an hBN-encapsulated MoSe2–WSe2 lateral heterostructures (with the band offset ΔEv = 0.215 eV and the interface width w = 2.4 nm) and then we perform cryogenic PL measurements. The starting point of our calculation is a focused laser excitation spot with an FWHM of 700 nm as in a typical experiment36. In a homogeneously excited system, the PL can be expressed by the Elliott formula describing the emission of bright exciton states37. In our case, this Elliott formula has to be extended to take into account the spatially confined laser excitation and excitonic states. To this purpose, we assume a Gaussian excitonic distribution N(x0, Rx) localized around \({x}_{0}\equiv {R}_{x}^{0}\) with a spatial width Δx in agreement to the FWHM of the laser pulse. Here, Rx is the exciton center-of-mass position. In the case without a junction, the spectral distribution is governed by the Boltzmann distribution. In the presence of a junction, however, the wavefunction of each state \(\left|{\psi }_{n}\right\rangle\) plays an important role and determines the relative occupation of the state via a weight coefficient cn(Rx), i.e. Nn(Rx) = N(x0, Rx)cn(Rx) (cf. “Methods” section for more details). This makes sure that we have a local thermal distribution. We generalize the Elliott formula for the PL intensity In(E) of the state \(\left|{\psi }_{n}\right\rangle\) taking into account that the laser pulse excites a spatially inhomogeneous exciton distribution. Thus, the spatially dependent PL reads after an optical excitation centered at x0
i.e. we sum over all emitting states \(\left|{\psi }_{n}\right\rangle\) and weight the emission by the coefficient cn(Rx). Note that we limit our study to momentum-direct radiative recombination since phonon sidebands are expected only in the WSe2 but not in the MoSe2 monolayer (as here momentum-dark excitons are not the energetically lowest states, cf. the Supplementary material)38,39,40. As a result, we also do not expect efficient indirect recombination of CT excitons as here the electron is located in the MoSe2 layer (Fig. 1b). Furthermore, funneling effects41 and exciton thermalization/charge transfer effects42,43 are beyond the scope of this work.
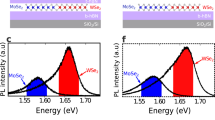
Now, we evaluate Eq. (3) and calculate spatially and spectrally dependent PL spectra at different temperatures for the hBN-encapsulated MoSe2–WSe2 lateral heterostructures. We tune the laser pulse position x0 and fix the junction characteristics to values of ΔEv = 0.215 eV and w = 2.4 nm in accordance with predicted and measured values14,18. At moderate and high temperatures far away from the junction, we reproduce the regular monolayer PL spectrum and find the XMo and XW excitons on the right-hand and the left-hand side, respectively (cf. Fig. 4a, b). When exciting at the interface, both features are still visible reflecting the large spatial width of the laser pulse (FWHM of 700 nm) that excites both sides of the heterojunction. Interestingly, when decreasing the temperature, a low-energy resonance appears approximately 90 meV below XMo (cf. Fig. 4c, d). This can be clearly ascribed to the position of the CT exciton XCT (Fig. 2d). At low temperatures, CT excitons can result in a strong PL despite their low oscillator strength due to their large occupation as energetically lowest states. The PL emitted from CT excitons is particularly strong compared to XW, as the bright exciton XW in the WSe2 layer is higher in energy than XMo, and is thus only weakly populated. For the same reason, we find that XMo is more intense than XW at low temperatures (Fig. 4c). Importantly, the new low-energy peak XCT is visible only in the presence of a narrow junction, (i.e. w = 2.4 nm), while it disappears for larger junction widths, as shown by the dashed orange line in Fig. 4d. This can be explained by the smaller spectral separation of CT excitons from the monolayer resonance at broader junctions (Fig. 2d), resulting in a smaller occupation of the CT state. In addition, the CT exciton binding energy also considerably drops, and the electron–hole separation drastically increases (Fig. 2e). As a direct consequence, the radiative decay rate γ0, which is given by the wavefunction overlap of electrons and holes (cf. “Methods” section), decreases by 4 orders of magnitude when moving from w = 2.4 nm to w = 12 nm.

Photoluminescence (PL) spectra of hBN-encapsulated MoSe2–WSe2 lateral heterostructures with the band offset ΔEv = 0.215 eV and the interface width w = 2.4 nm studied at a 300 K, b 150 K, and c 30 K. We excite the material with a laser excitation spot with an FWHM of 700 nm. d Cuts of the PL spectrum at the interface at 30 K. We also show the comparison to the larger interface width of 12 nm (dashed orange line). e, f Experimental PL spectrum at the junction and at MoSe2 monolayer region, with two different interfaces considered. We find both in experiment and theory a low-energy resonance that we assign to a bound CT exciton.
To test the theoretical prediction we perform spatially dependent cryogenic PL measurements on the very same sample system, i.e. hBN-encapsulated MoSe2-WSe2. This sample set has shown high structural quality at the junction in electron microscopy and clear exciton transport from WSe2 to MoSe2 through the junction14. This allows us to show a direct comparison between theory and experiment (cf. Fig. 4d–f). In Fig. 4e, f we present the spectra from two different junctions. We find in the experiment a clear PL emission of about 80–100 meV below the XMo resonance at several junctions. The emission in the CT-exciton energy range is absent far away from the junction (cf. thin bright blue line in Fig. 4e, f). This is in excellent agreement with the theoretical prediction (Fig. 4d) and is a strong indication of the direct emission from CT excitons. To further support this assignment, we have performed power-dependent studies, cf. the Supplementary material. The integrated intensity of the low-energy peak increases linearly with the excitation power, contrary to the saturating behaviour expected from defects44,45. In addition, we also observe a blue-shift of the peak with increasing excitation powers, similar to the behaviour of interlayer excitons, which blueshift due to dipole–dipole repulsion46. The observed shift could thus further confirm the dipolar origin of the low-energy peak.
By investigating samples in PL at cryogenic temperatures, the chances for the observation of the CT exciton are optimized also by the hBN encapsulation which, besides reducing the disorder (resulting in linewidth of less than 10 meV for XMo), leads to large energy separations between XCT and XMo excitons, as explained in the dielectric engineering part of the manuscript (Fig. 3). We emphasize that both the narrow linewidth and the large-energy separation between XCT and XMo are needed to observe CT excitons. The broader nature of the CT exciton in the experiment is likely to be related to sample imperfections or strain. A moderate red-shift of 20–30 meV of XW close to the junction14 suggests the presence of strain that could result in an inhomogeneous broadening of XCT, together with dielectric disorder and impurities. Furthermore, we observe trionic features resulting in multiple peaks around the energy of the MoSe2 exciton that have not been taken into account in the theory. Note that PL emission at the calculated CT exciton energy has been observed in several junctions (cf. Fig. 4e, f). From a material perspective, it is likely that the junction width w might vary for junctions grown on the same substrate. As a result, CT exciton formation does not necessarily occur at all junctions, due to the strong dependence on w as shown in our calculations (Fig. 2f).
Discussion
We have presented a joint theory–experiment study investigating the bound charge-transfer excitons at the interface of lateral two-dimensional heterostructures. We find in theory and experiment first signatures for the appearance of bound charge transfer excitons in cryogenic photoluminesce spectra of hBN-encapsulated lateral MoSe2–WSe2 heterostructures. We perform interface and dielectric engineering in our calculations and reveal critical conditions for the observation of charge transfer excitons including narrow junction widths (in the range of a few nm), relatively large band offsets (above 100 meV), and an intermediate dielectric screening (ε ≈ 2−5). Our study provides novel insights into the characteristics of bound charge transfer excitons and will trigger future experimental and theoretical studies in the growing research field of lateral heterostructures. The latter also has a large technological potential as ultrathin junctions present quasi-one-dimensional channels with a strongly suppressed scattering with phonons and thus significantly enhanced exciton mobility47. Additionally, the large intrinsic dipole of CT excitons is expected to lead to an efficient dipole–dipole repulsion that together with the 1D confinement could lead to excitonic highways as recently proposed13. A further key for technological application is exciton dissociation, i.e. the conversion of light absorption into electrical currents. Due to the huge excitonic binding energies of hundreds of meV, the charge separation is largely ineffective in TMD monolayers. In contrast, weakly bound CT excitons in lateral heterostructures will efficiently dissociate and thus facilitate charge separation. In our work, we show how to engineer lateral TMD heterostructures to obtain stable and highly dipolar CT excitons that have a high potential to boost exciton transport and exciton dissociation—both highly relevant for optoelectronic applications.
Methods
Microscopic modeling
To microscopically model charge transfer excitons in lateral TMD heterostructures, we solve the Schrödinger equation including the strong Coulomb interaction in TMD monolayers and the space-dependent dispersion relations induced by the junction (Fig. 1b). The Coulomb interaction VC(r) is described introducing a generalized Keldysh potential30,31,48 for charges in a thin-film surrounded by a dielectric environment that is spatially homogeneous along the plane in terms of thickness and dielectric constant30,31,48. The in-plane variation of energy is described via spatially dependent single-particle energies \({E}_{{\rm {c/v}}}^{0}({{{{{{{\bf{r}}}}}}}})\) of electrons and holes, respectively. In particular, we take \({E}_{{\rm {c/v}}}^{0}({{{{{{{\bf{r}}}}}}}})={{\Delta }}{E}_{{\rm {c/v}}}/2(1-\tanh (4x/w))+{E}_{{{{{{{{\rm{Mo}}}}}}}}}^{0}(1\pm 1)/2\), which recovers the situation in Fig. 1b15. The Schrödinger equation can be separated into equations for the relative and the center-of-mass motion (Eqs. (1) and (2)). We focus on electrons and holes located at the K valley in MoSe2 and WSe2, respectively, as all other CT electron–hole pairs are energetically higher, cf. the Supplementary material. While lateral heterostructures involving TMDs with different chalcogen atoms have a lattice mismatch, in MoSe2–WSe2 we can assume a strain-free interface11,19. Finally, we solve the coupled Eqs. (1) and (2) with space-independent WSe2 electron masses49 to obtain the eigenenergies and eigenfunctions, which in turn allow to determine the radiative recombination rate and the dipole d\({}_{{\rm {e-h}}}\equiv \left|{{{{{{{{\bf{d}}}}}}}}}_{{\rm {e-h}}}\right |=\left|\int\,{{\rm {d}}R}_{x}d{{{{{{{\bf{r}}}}}}}}{{{{{{{\bf{r}}}}}}}}|{{\Psi }}({R}_{x},{{{{{{{\bf{r}}}}}}}}){|}^{2}\right|\). Note that finite dipoles are present only for CT states and only across the interface, i.e. de−h = d\({}_{{\rm {e-h}}}^{\, x}\) and d\({}_{{\rm {e-h}}}^{\, y}\) = 0, where d\({}_{{\rm {e-h}}}^{\, x/y}\) describes the component across and along the interface of de−h, respectively.
To model the spatially dependent PL, we must take into account the junction in lateral heterostructures yielding the PL formula in Eq. (3). The appearing coefficients cn(Rx) can be obtained starting from the total center-of-mass excitonic distribution \(N({{{{{{{\bf{R}}}}}}}})\propto {\sum }_{n{n}^{{\prime} }}\langle {\hat{X}}_{{n}^{{\prime} }}^{{{{\dagger}}} }{\hat{X}}_{n}\rangle {\psi }_{{n}^{{\prime} }}^{*}({{{{{{{\bf{R}}}}}}}}){\psi }_{n}({{{{{{{\bf{R}}}}}}}})\), where \({\hat{X}}_{n}^{{{{\dagger}}} },\, {\hat{X}}_{n}\) are the creation/annihilation operators of an exciton in the state n and where \(\langle {\hat{X}}_{{n}^{{\prime} }}^{{{{\dagger}}} }\,{\hat{X}}_{n}\rangle\) is the single-exciton density matrix. In the equilibrium of homogeneous low-density excitations, we find \(\langle {\hat{X}}_{{n}^{{\prime} }}^{{{{\dagger}}} }\,{\hat{X}}_{n}\rangle \equiv c{e}^{-{E}_{n}/{k}_{{\rm {B}}}T}{\delta }_{n{n}^{{\prime} }}\) with c being the normalization constant reflecting the local density.
Applying such equilibrium condition to the general definition of N(R) yields
with \({c}_{n}({{{{{{{\bf{R}}}}}}}})={e}^{-\frac{{E}_{n}}{{k}_{{\rm {B}}}T}}|{\psi }_{n}({{{{{{{\bf{R}}}}}}}}){|}^{2}{\big[{\sum }_{{n}^{{\prime} }}{e}^{-\frac{{E}_{{n}^{{\prime} }}}{{k}_{{\rm {B}}}T}}|{\psi }_{{n}^{{\prime} }}({{{{{{{\bf{R}}}}}}}}){|}^{2}\big]}^{-1}\) providing the local occupation of state \(\left|n\right\rangle\). In the monolayer limit one has ∣ψn(R)∣2 = 1/A with A being the area of the sample. Hence, the coefficients cn(R) become the normalized spatially independent Boltzmann distribution. A highly non-trivial dynamics is expected at the interface, where the charge transfer42,43 into bound CT states is likely to lead to local features similar to those of phonon-induced carrier-capture50,51. In this work, we focus on stationary PL after exciton thermalization has occurred.
The space-independent PL of \({I}_{n}(E)={\tilde{\gamma }}_{n}\frac{{\tilde{\gamma }}_{n}+{{{\Gamma }}}_{n}}{{(E-{E}_{n})}^{2}+{({\tilde{\gamma }}_{n}+{{{\Gamma }}}_{n})}^{2}}\) entering Eq. (3) describes the emission spectrum after radiative recombination of the state \(\left|n\right\rangle\) according to the excitonic Elliott formula37,38. Phonon-assisted mechanisms are not included as they are expected to be strong only on the WSe2 side of the junction but negligible for both MoSe2 and CT excitons in the junction. The oscillator strength \({\tilde{\gamma }}_{n}={\gamma }_{n}\,|{\psi }_{{Q}_{x}=0}{|}^{2}\) is given by the product of the radiative rate γn and the Qx = 0 component of the squared wavefunction in center-of-mass momentum space \({\psi }_{{Q}_{x}}\) in view of the conservation of momentum after recombination into photons52. The radiative rate γn can be extracted from the monolayer case48 as \({\gamma }_{n}=\tilde{M}|\phi ({{{{{{{\bf{r}}}}}}}}=0){|}^{2}/{E}_{n}\) with \(\tilde{M}\) depending on the material and the substrate (via optical dipole moment or refractive index), while En and \(|\phi ({{{{{{{\bf{r}}}}}}}}=0){|}^{2}=\int\,{{\rm {d}}R}_{x}|\psi ({R}_{x}){|}^{2}|{\phi }^{{R}_{x}}({{{{{{{\bf{r}}}}}}}}=0){|}^{2}\) are obtained from the solution of Eqs. (1) and (2), i.e. in particular including effects from the junction. While \({\tilde{\gamma }}_{n}\) determines the oscillator strength, i.e. the height of the resonances, Γn describes the impact of exciton–phonon scattering on the shape of the resonances. As a full microscopic calculation of the latter including the junction is beyond the scope of this work, we estimate Γn with the values obtained in the low-density limit for MoSe2 and WS2 monolayers53.
Sample fabrication and photoluminescence measurements
Our MoSe2–WSe2 lateral monolayer heterojunction is grown by chemical vapor deposition (CVD) synthesis that we reported recently20. For the hBN encapsulation we follow the water-assisted transfer method to pick up as-grown, chemical vapor deposition (CVD) lateral heterostructures using polydimethylsiloxane (PDMS) and deterministically transfer and encapsulate them in hBN54,55. Photoluminescence spectra are collected at T = 4 K in a closed-loop liquid helium (LHe) system. A 633 nm HeNe laser is used as an excitation source with a spot size diameter of ≈ 1 μm and 6 μW power, while in the Supplementary material we present results gradually increasing the power up to 25 μW.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding authors on reasonable request.
Code availability
The codes used to generate the data are available from the corresponding authors on reasonable request.
References
Wang, G. et al. Colloquium: excitons in atomically thin transition metal dichalcogenides. Rev. Mod. Phys. 90, 021001 (2018).
Mueller, T. & Malic, E. Exciton physics and device application of two-dimensional transition metal dichalcogenide semiconductors. npj 2D Mater. Appl. 2, 29 (2018).
Rivera, P. et al. Observation of long-lived interlayer excitons in monolayer MoSe2–WSe2 heterostructures. Nat. Commun. 6, 6242 (2015).
Unuchek, D. et al. Room-temperature electrical control of exciton flux in a van der Waals heterostructure. Nature 560, 340–344 (2018).
Sun, Z. et al. Excitonic transport driven by repulsive dipolar interaction in a van der Waals heterostructure. Nat. Photonics 16, 79–85 (2022).
Duan, X. et al. Lateral epitaxial growth of two-dimensional layered semiconductor heterojunctions. Nat. Nanotechnol. 9, 1024–1030 (2014).
Gong, Y. et al. Vertical and in-plane heterostructures from WS2/MoS2 monolayers. Nat. Mater. 13, 1135–1142 (2014).
Huang, C. et al. Lateral heterojunctions within monolayer MoSe2–WSe2 semiconductors. Nat. Mater. 13, 1096–1101 (2014).
Li, Ming-Yang et al. Epitaxial growth of a monolayer WSe2–MoS2 lateral p–n junction with an atomically sharp interface. Science 349, 524–528 (2015).
Heo, H. et al. 2d materials: rotation-misfit-free heteroepitaxial stacking and stitching growth of hexagonal transition-metal dichalcogenide monolayers by nucleation kinetics controls. Adv. Mater. 27, 3839–3839 (2015).
Zhang, C. et al. Strain distributions and their influence on electronic structures of WSe2–MoS2 laterally strained heterojunctions. Nat. Nanotech. 13, 152–158 (2018).
Sahoo, P. K., Memaran, S., Xin, Y., Balicas, L. & Gutiérrez, H. R. One-pot growth of two-dimensional lateral heterostructures via sequential edge-epitaxy. Nature 553, 63–67 (2018).
Yuan, L. et al. Non-equilibrium first-order exciton Mott transition at monolayer lateral heterojunctions visualized by ultrafast microscopy. arXiv preprint arXiv:2111.07887 (2021).
Beret, D. et al. Exciton spectroscopy and unidirectional transport in MoSe2–WSe2 lateral heterostructures encapsulated in hexagonal boron nitride. npj 2D Mater. Appl. 6, 84 (2022).
Lau, Ka. Wai, Calvin, Gong, Z., Yu, H. & Yao, W. Interface excitons at lateral heterojunctions in monolayer semiconductors. Phys. Rev. B 98, 115427 (2018).
Latini, S., Winther, K. T., Olsen, T. & Thygesen, K. S. Interlayer excitons and band alignment in MoS2/hBN/WSe2 van der Waals heterostructures. Nano Lett. 17, 938–945 (2017).
Ovesen, S. et al. Interlayer exciton dynamics in van der Waals heterostructures. Commun. Phys. 2, 23 (2019).
Guo, Y. & Robertson, J. Band engineering in transition metal dichalcogenides: Stacked versus lateral heterostructures. Appl. Phys. Lett. 108, 233104 (2016).
Xie, S. et al. Coherent, atomically thin transition-metal dichalcogenide superlattices with engineered strain. Science 359, 1131–1136 (2018).
Najafidehaghani, E. et al. 1D p–n junction electronic and optoelectronic devices from transition metal dichalcogenide lateral heterostructures grown by one-pot chemical vapor deposition synthesis. Adv. Funct. Mater. 31, 2101086 (2021).
Ichinose, N. et al. Two-dimensional atomic-scale ultrathin lateral heterostructures. arXiv preprint arXiv:2208.12696 (2022).
Kang, J., Tongay, S., Zhou, J., Li, J. & Wu, J. Band offsets and heterostructures of two-dimensional semiconductors. Appl. Phys. Lett. 102, 012111 (2013).
Chu, Y.-H. et al. Atomic scale depletion region at one dimensional MoSe2–WSe2 heterointerface. Appl. Phys. Lett. 113, 241601 (2018).
Herbig, C. et al. Local electronic properties of coherent single-layer WS2/WSe2 lateral heterostructures. Nano Lett. 21, 2363–2369 (2021).
Pospischil, A., Furchi, M. M. & Mueller, T. Solar-energy conversion and light emission in an atomic monolayer p–n diode. Nat. Nanotechnol. 9, 257–261 (2014).
Baugher, B. W. H., Churchill, H. O. H., Yang, Y. & Jarillo-Herrero, P. Optoelectronic devices based on electrically tunable p–n diodes in a monolayer dichalcogenide. Nat. Nanotechnol. 9, 262–267 (2014).
Ross, J. S. et al. Electrically tunable excitonic light-emitting diodes based on monolayer WSe2 p–n junctions. Nat. Nanotechnol. 9, 268–272 (2014).
Thureja, D. et al. Electrically tunable quantum confinement of neutral excitons. Nature 606, 298–304 (2022).
Zipfel, J. et al. Spatial extent of the excited exciton states in WS2 monolayers from diamagnetic shifts. Phys. Rev. B 98, 075438 (2018).
Rytova, N. S. The screened potential of a point charge in a thin film. Proc. MSU. Phys. Astron. 30, 3 (1967).
Keldysh, L. V. Coulomb interaction in thin semiconductor and semimetal films. JETPL 29, 658 (1979).
Rhodes, D., Chae, SangHoon, Ribeiro-Palau, R. & Hone, J. Disorder in van der Waals heterostructures of 2D materials. Nat. Mater. 18, 541 (2019).
Pielić, B. et al. Electronic structure of quasi-freestanding WS2/MoS2 heterostructures. ACS Appl. Mater. Interfaces 13, 50552–50563 (2021).
Shimasaki, M. et al. Directional exciton-energy transport in a lateral heteromonolayer of WSe2–MoSe2. ACS Nano 16, 8205–8212 (2022).
Perea-Causín, Raül, Brem, S. & Malic, E. Phonon-assisted exciton dissociation in transition metal dichalcogenides. Nanoscale 13, 1884–1892 (2021).
Shree, S., Paradisanos, I., Marie, X., Robert, C. & Urbaszek, B. Guide to optical spectroscopy of layered semiconductors. Nat. Rev. Phys. 3, 39–54 (2021).
Koch, S. W., Kira, M., Khitrova, G. & Gibbs, H. M. Semiconductor excitons in new light. Nat. Mater. 5, 523–531 (2006).
Brem, S. et al. Phonon-assisted photoluminescence from indirect excitons in monolayers of transition-metal dichalcogenides. Nano Lett. 20, 2849–2856 (2020).
Lu, Z. et al. Magnetic field mixing and splitting of bright and dark excitons in monolayer MoSe2. 2D Mater. 7, 015017 (2019).
Robert, C. et al. Measurement of the spin-forbidden dark excitons in MoS2 and MoSe2 monolayers. Nat. Commun. 11, 1–8 (2020).
Rosati, R. et al. Dark exciton anti-funneling in atomically thin semiconductors. Nat. Commun. 12, 7221 (2021).
Schmitt, D. et al. Formation of moiré interlayer excitons in space and time. Nature 608, 499–503 (2022).
Meneghini, G., Brem, S. & Malic, E. Ultrafast phonon-driven charge transfer in van der Waals heterostructures. Nat. Sci. 2, e20220014 (2022).
Wu, Z. & Ni, Z. Spectroscopic investigation of defects in two-dimensional materials. Nanophotonics 6, 1219–1237 (2017).
Hernández López, P. et al. Strain control of hybridization between dark and localized excitons in a 2D semiconductor. Nat. Commun. 13, 7691 (2022).
Erkensten, D., Brem, S., Perea-Causín, R. & Malic, E. Microscopic origin of anomalous interlayer exciton transport in van der Waals heterostructures. Phys. Rev. Mater. 6, 094006 (2022).
Dirnberger, F. et al. Quasi-1D exciton channels in strain-engineered 2D materials. Sci. Adv. 7, eabj3066 (2021).
Brem, S. et al. Intrinsic lifetime of higher excitonic states in tungsten diselenide monolayers. Nanoscale 11, 12381–12387 (2019).
Kormányos, A. et al. k ⋅ p theory for two-dimensional transition metal dichalcogenide semiconductors. 2D Mater. 2, 022001 (2015).
Glanemann, M., Axt, V. M. & Kuhn, T. Transport of a wave packet through nanostructures: quantum kinetics of carrier capture processes. Phys. Rev. B 72, 045354 (2005).
Reiter, D., Glanemann, M., Axt, V. M. & Kuhn, T. Spatiotemporal dynamics in optically excited quantum wire-dot systems: capture, escape, and wave-front dynamics. Phys. Rev. B 75, 205327 (2007).
Feierabend, M., Khatibi, Z., Berghäuser, G. & Malic, E. Dark exciton based strain sensing in tungsten-based transition metal dichalcogenides. Phys. Rev. B 99, 195454 (2019).
Selig, M. et al. Excitonic linewidth and coherence lifetime in monolayer transition metal dichalcogenides. Nat. Commun. 7, 13279 (2016).
Jia, H. et al. Large-scale arrays of single-and few-layer MoS2 nanomechanical resonators. Nanoscale 8, 10677–10685 (2016).
Paradisanos, I. et al. Controlling interlayer excitons in MoS2 layers grown by chemical vapor deposition. Nat. Commun. 11, 1–7 (2020).
Acknowledgements
We acknowledge funding from the Deutsche Forschungsgemeinschaft (DFG) via SFB 1083 and project 512604469 as well as from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 881603 (Graphene Flagship). Toulouse acknowledges partial funding from ANR IXTASE, Growth of hexagonal boron nitride crystals was supported by JSPS KAKENHI (Grants Nos. 19H05790, 20H00354, and 21H05233). Jena group financial support of the Deutsche Forschungsgemeinschaft (DFG) through CRC 1375 NOA (Project B2), SPP2244 (Project TU149/13-1), DFG grant TU149/16-1.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
R.R. and E.M. developed the theoretical model for CT-exciton engineering and optics. I.P., L.L., P.R., and B.U. devised and performed the PL measurements. Z.G., A.G., and A.T. grew the CVD samples. T.T. and K.W. grew the hBN bulk. All authors contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Carino Ferrante, Junyi Liu and Alessandro Surrente for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rosati, R., Paradisanos, I., Huang, L. et al. Interface engineering of charge-transfer excitons in 2D lateral heterostructures. Nat Commun 14, 2438 (2023). https://doi.org/10.1038/s41467-023-37889-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37889-9
This article is cited by
-
Kapitza-resistance-like exciton dynamics in atomically flat MoSe2-WSe2 lateral heterojunction
Nature Communications (2023)
-
Exciton transport in atomically thin semiconductors
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
