
Abstract
Epigenetic markers are potential biomarkers for diabetes and related complications. Using a prospective cohort from the Hong Kong Diabetes Register, we perform two independent epigenome-wide association studies to identify methylation markers associated with baseline estimated glomerular filtration rate (eGFR) and subsequent decline in kidney function (eGFR slope), respectively, in 1,271 type 2 diabetes subjects. Here we show 40 (30 previously unidentified) and eight (all previously unidentified) CpG sites individually reach epigenome-wide significance for baseline eGFR and eGFR slope, respectively. We also develop a multisite analysis method, which selects 64 and 37 CpG sites for baseline eGFR and eGFR slope, respectively. These models are validated in an independent cohort of Native Americans with type 2 diabetes. Our identified CpG sites are near genes enriched for functional roles in kidney diseases, and some show association with renal damage. This study highlights the potential of methylation markers in risk stratification of kidney disease among type 2 diabetes individuals.
Similar content being viewed by others
Introduction
There is a global epidemic of type 2 diabetes. The increasing prevalence of young-onset diabetes has contributed to the increasing burden of end-stage kidney disease (ESKD) due to the associated long disease duration1,2. Given the preventable nature of diabetic kidney disease (DKD), there is a need to identify individuals at risk of progression of DKD and ESKD for early intensive interventions. Several treatments have recently been proven to retard the progression of DKD, including sodium glucose transporter 2 (SGLT2) inhibitors3 and selective mineralocorticoid receptor antagonist such as Finerenone4. These expanding treatment options for DKD have increased the urgency to develop new models that can stratify those at high risk of kidney dysfunction.
There have been numerous efforts to identify biomarkers that can guide the stratification of DKD, including the use of genetic and other types of biomarkers. Whilst genome-wide association studies (GWAS) have had considerable success in identifying genetic markers for type 2 diabetes and other complex diseases, the progress in identifying loci associated with DKD had been less impressive5,6. Epigenetic markers, including methylation changes and miRNA, may be able to capture the interaction between environmental factors and the genome, and may provide new biomarkers for diabetes-related complications7. Methylation markers, in particular, have been postulated to mediate the effects of metabolic memory8, and are promising biomarkers for diabetic complications. Some previous studies have investigated DNA methylation changes associated with DKD based on human blood9,10, human kidney tubules11,12, or mouse samples13. These studies involved control samples from healthy individuals or individuals with diabetes.
In this study, we examine whether methylation at CpG sites, measured in peripheral blood, may be associated with renal function, and whether this information can be used to predict deterioration in kidney function in type 2 diabetes for prognostication purpose.
Results
Genome-wide DNA methylation trends are associated with baseline kidney function
We studied a cohort of 1271 patients with type 2 diabetes from the Hong Kong Diabetes Register (HKDR). Among the patients, 19.7% had DKD at baseline, defined as eGFR<60 ml/min/1.73 m2 (Supplementary Table 1 and Supplementary Fig. 1). During a median follow-up period of 14.6 (Q1–Q3: 8.3–19.4) years, 33% developed ESKD. During the follow-up period, the included subjects had a median number of eGFR measurements of 29 (Q1–Q3: 15-46), and the median eGFR slope during follow-up was −2.27% (Q1–Q3: −9.11 to −0.65) change of eGFR per year.
We profiled the DNA methylome of whole-blood samples of the patients using the Illumina Infinium HumanMethylation450K BeadChip (“Methods”). For the DNA methylation data produced, after filtering and normalization, 434,908 CpG sites and 1268 samples were retained. Data reproducibility was confirmed by replicate samples (Supplementary Results and Supplementary Fig. 2). The top principal components (PCs) of our DNA methylation data were strongly indicative of sex, age, and smoking status (Supplementary Methods, Supplementary Results, and Supplementary Fig. 3), which are consistent with previous studies14,15,16,17,18,19 and further confirm the quality of our data.
DNA methylation was associated with renal function, with the models for baseline eGFR achieving a high mean area under the receiver–operator characteristic (AUROC) of 0.76 (Supplementary Fig. 4a). This association was not due to confounding factors caused by sex, age, or smoking status (Supplementary Results and Supplementary Fig. 4b, c). In contrast, most of the other clinical variables were not strongly associated with DNA methylation (Supplementary Fig. 5).
Methylation levels of individual CpG sites are associated with baseline renal function and renal function decline
To discover individual CpG sites associated with kidney function, we performed an epigenome-wide association study (EWAS) of baseline eGFR. Since recent studies have reported that CpG methylation levels are predictive of the decline of eGFR over time11,20, we also set eGFR slope as an additional target trait. We included sex, age, smoking status, duration of diabetes, hemoglobin A1c, blood pressure, batch of experiment, and cell-type composition estimations21 as covariates.
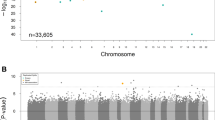
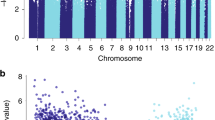
For baseline eGFR, 40 CpG sites reached epigenome-wide significance (Bonferroni-corrected P value below 0.05) and 386 CpG sites were statistically significant at FDR = 0.05 (Fig. 1a–c, Table 1, and Supplementary Data 1). The most significant CpG site was cg17944885 (Bonferroni-corrected P = 6.11 × 10−15), located between ZNF788 and ZNF20 on chromosome 19. The DNA methylation level of this CpG site had also been associated with kidney function in various populations22,23,24,25 (Supplementary Fig. 6 and Supplementary Data 1 and 2). Interestingly, two of the sites with a Bonferroni-correctedP value below 0.05 (cg04983687, cg01676795) and one other significant site at FDR = 0.05 (cg22460173) in our cohort had also been reported as significant in a recent multiethnic meta-analysis22, but they had not been reported to have a significant association with kidney function in earlier studies of cohorts that involved a single ethnic group10,22.

The methylation level of each CpG site was tested for its association with baseline eGFR (a–c) and eGFR slope (d–f). The results of all the 434,908 CpG sites analyzed in this study are shown using Manhattan plots (a, d), quantile–quantile (QQ) plots (b, e), and volcano plots (c, f). P values were computed using two-sided Student’s t test. In the Manhattan plots, CpG sites with a Bonferroni-corrected P value <0.05 are shown in red. The horizontal red lines show the cutoff above which all sites are significant at FDR = 0.05. In the QQ plots, the diagonal straight line is the expectation under the null hypothesis. λ is the inflation factor. In the volcano plots, CpG sites with a Bonferroni-corrected P value <0.05 are shown in red.
For eGFR slope, eight CpG sites had a Bonferroni-correctedP value below 0.05 and 74 CpG sites were significant at FDR = 0.05 (Fig. 1d–f, Table 1, and Supplementary Data 1). The most significant CpG site was cg10272901 (Bonferroni-corrected P = 3.41 × 10−5), located in a CpG island on chromosome 21. None of these 74 sites was reported to be associated with eGFR slope in previous studies, conducted mainly in the general population rather than population with diabetes (Supplementary Data 1 and 2). When we performed reciprocal lookup of the previously reported top sites, we found several sites reported by Gluck et al., based on data from multiple populations11, to have marginally significant P values in our data (Supplementary Fig. 6). These included cg15826891 (P = 5.29 × 10−5 in our data), which is located within the MIR100HGnon-coding gene locus on chromosome 11, and cg02950701 (P = 1.26 × 10−4 in our data), which is located within the protein-coding gene CCNY locus on chromosome 10.
A multisite approach to identifying sets of CpG sites indicative of renal function
The single-site approach described above, though commonly used in the literature, has two important limitations. First, some CpG sites that are not strongly associated with kidney function by themselves could complement other sites to explain residual kidney function differences. These “auxiliary” sites cannot be identified by the single-site approach. Second, some significant CpG sites identified by the single-site approach could be strongly correlated with each other (Supplementary Fig. 7), due to genomic spatial dependency or other reasons, leading to redundancy and diversion of attention to non-functional sites.
To tackle these limitations, we developed a multisite approach that considered all CpG sites at the same time and selected a subset of them to create the best model to infer baseline eGFR or eGFR slope (“Methods”). Considering both the model performance and complexity of the models, our procedure automatically determined the feature selection thresholds (“Methods” and Supplementary Results). According to left-out testing data not involved in this procedure, at these selected thresholds, the Pearson correlation between the measured baseline eGFR values and the values inferred by the models was 0.704, and that of eGFR slope was 0.386 (Supplementary Fig. 8a, d).
The multisite models capture relationships between DNA methylation and renal function in multiple populations
After confirming the validity of our procedure, we then implemented it to rebuild the models using the whole set of samples. In these “final” models, 64 and 37 CpG sites were included for predicting baseline eGFR and eGFR slope, respectively (Tables 2 and 3 and Supplementary Data 3).
For baseline eGFR and eGFR slope, the actual values and the values inferred by our final models had Pearson correlations of 0.806 and 0.635, respectively (Table 4 and Fig. 2a, b). The performance of the models was better with the covariates than without (Table 4 and Fig. 2c, d), and they were substantially better than models constructed from the same number of random CpG sites (Supplementary Fig. 9) and several alternative models (Supplementary Results).

Scatter plots of inferred baseline eGFR and eGFR slope against their corresponding actual measurements using selected CpG sites based on the models constructed from the primary cohort and applied to the primary cohort (a–d) or the Native American cohort (trained using CpG sites available to both cohorts) (e–h). In each panel, the black lines mark the best fit lines of linear regression. Source data are provided as a Source Data file.
In our final models, some of the CpG sites included were also significantly associated with kidney function in the single-site analysis, such as the most significant sites cg17944885 for baseline eGFR and cg10272901 for eGFR slope. Other sites demonstrated significant associations only in the multisite models, showing that they carried additional information for inferring the target traits. Intriguingly, the most significant site cg17944885 for baseline eGFR was also included in the multisite model for eGFR slope, although it was not significant for eGFR slope in the single-site analysis. One of the selected sites for the baseline eGFR model, cg13408344, was previously associated with baseline eGFR22.
To evaluate whether the selected sites could successfully classify people with or without kidney disease, we constructed regularized logistic regression models using the above choices of CpG sites for baseline eGFR and eGFR slope. All the models performed well in these classification tasks, achieving a mean AUROC of 0.89 for baseline eGFR and 0.81 for eGFR slope (Supplementary Table 2), demonstrating the ability of these sites to recognize people with potential renal dysfunction.
In the final models, since all samples were used in training the model, there were no left-out samples for evaluating the model performance in an unbiased fashion. Therefore, we further tested our models using genome-wide methylation measurements of blood samples from an independent cohort of 326 Native American subjects with type 2 diabetes. The results (Table 4, Supplementary Table 3, Fig. 2e–h, and Supplementary Fig. 10) show that our models also achieved good performance for predicting baseline eGFR and eGFR decline in type 2 diabetes in this independent cohort despite differences in ethnicity.
Proximal genes of the selected sites in the single-site and multisite analyses have potential kidney functions
We next evaluated the functional significance of the genes proximal to (within 1 kb) the sites identified in our single-site and multisite analyses by checking whether they have been reported as potentially related to kidney function. We collected these potential kidney function-related genes from a number of previous studies that identified the genes using various types of data, including DNA methylation data of blood samples from people with or without kidney disease9,26,27,28, bulk RNA expression data of human kidneys29,30,31, single-cell RNA sequencing data of mouse kidneys32,33, and GWAS prioritized genes34,35.
Of the 348 CpG sites (which corresponded to 358 genes) identified by our single-site and multisite analyses as associated with baseline eGFR and proximal to (within 1 kb) annotated genes, 228 (which corresponded to 215 genes) of them (65.5%) were reported in at least one of these previous studies (Fig. 3 and Supplementary Data 4), which corresponded to a 1.25-fold enrichment as compared to the set of all human genes (P = 3.78 × 10−6, hypergeometric test).

Each row corresponds to a CpG site and all genes within 1 kb from it. The “Single-site” and “Multi-site” columns show whether a site is significant at FDR = 0·05 in our single-site analysis and whether it is included in the final multisite model, respectively. The “DNAm” and “DEGs” columns show whether at least one of the nearby genes is differentially methylated or differentially expressed in samples with and without kidney function decline in one or more previous methylation9,25,26,27,28 or gene expression studies29,31,32, respectively. The “eQTL” column shows whether at least one of the nearby genes is associated with an expression quantitative trait locus identified in human kidney samples in a previous study30. The “MarkerGenes” column shows whether at least one of the nearby genes is a cell-type-specific marker of a major kidney cell type as identified previously33. The “GWAS” column shows whether at least one of the nearby genes is prioritized by GWAS results in two recent studies34,35. Only CpG sites where the nearby genes have at least 3 and 1 functional supports, respectively, for baseline eGFR and eGFR slope, are shown.
Noticeably, the CpG site cg24707889, located in the upstream region of the ITGB2 gene, was identified in the multisite model but not recognized as significant at FDR = 0.05 in the single-site analysis. The association between ITGB2 and kidney function was supported by various data such as blood DNA methylation27, RNA expression and expression quantitative trait loci (eQTLs) in human kidney samples30,31, and single-cell RNA expression in mouse kidneys32,33. The ITGB2 gene encodes integrin subunit beta 2 (also known as archetypal innate immune receptor CD11b/CD18), which plays an important role in immune response, and defects in this gene may cause leukocyte adhesion deficiency. A recent study reported that inhibition of CD11b/CD18 prevented long-term fibrotic ESKD from acute kidney injury (AKI) in cynomolgus monkeys36.
Interestingly, our analysis identified several previously unidentified CpG sites associated with baseline eGFR with nearby genes having differential expression between samples from people with and without kidney disease, such as RFTN1 and CTSB (“Discussion”).
For eGFR slope, 51 of the 76 CpG sites (67.1%) (which corresponded to 52 of 89 genes) were reported as potentially related to kidney function in previous studies (Fig. 3 and Supplementary Data 4), which corresponded to a 1.21-fold enrichment as compared to the set of all human genes (P = 0.03, hypergeometric test).
One CpG site, cg19693031, which was selected by our multisite model but not recognized as significant at FDR = 0.05 in the single-site analysis, is located in the 3’-UTR (untranslated region) of the TXNIP gene. TXNIP encodes a thioredoxin-interacting protein, which was implicated in the pathogenesis of DKD. CpG sites within this gene were differentially methylated between patients with type 1 diabetes with and without complications26. TXNIP expression is related to DKD31, structural abnormalities such as cortical interstitial fractional volume (VvInt)29, an index of tubule-interstitial damage, as well as folic acid nephropathy (FAN)32. Previous studies suggested that hyperglycemia might contribute to DKD by increasing the level of inflammatory factors via upregulating the expression of TXNIP through histone modifications, such as increase in H3K9ac, H3K4me3, and H3K4me1, and decrease in H3K27me3 at TXNIP promoter region, whereas the contributory roles of DNA methylation required further elucidation37,38. Another CpG site, cg13591783, identified in both our single-site and multisite analyses for eGFR slope, is located within the ANXA1 gene. ANXA1 encodes annexin A1, which is a membrane-localized protein that binds phospholipids, inhibits phospholipase A2, and has anti-inflammatory activity. ANXA1 was differentially expressed in kidney tubules between human samples of DKD and control samples31 and correlated with VvInt29 in patients with DKD. In addition, annexin A1 was a potential therapeutic target in diabetes and the treatment of microvascular disease such as DKD39,40.
To further confirm that the CpG sites identified in our single-site and multisite analyses are statistically near genes related to kidney function, we sampled many sets of random genes proximal to (within 1 kb) CpG sites profiled by the Infinium HumanMethylation450 BeadChip with the same sizes as the actual numbers of genes proximal to the CpG sites we identified in our analyses. For these random sets of genes, the numbers of them related to kidney function were significantly smaller than our actual sets of genes identified (Supplementary Fig. 11).
Taken together, among the genes near the CpG sites associated with baseline eGFR or eGFR slope in our single-site and multisite analyses, many of them had been reported to be related to normal kidney function or kidney diseases. These results were based on various types of data, including data curated from human kidney samples, which provides strong support for the functional relevance of our reported CpG sites obtained from blood samples.
To further validate the relevance of our selected CpG sites in the kidney, we selected seven CpG sites that were associated with baseline eGFR in our single-site and multisite analyses, namely cg21573651, cg17944885, cg06449934, cg02304370, cg21919729, cg04610187, and cg18593194 (“Methods”). For two of these seven CpG sites (cg21573651 and cg04610187) their methylation levels in kidney samples were significantly different between kidney disease patients and control groups (Supplementary Fig. 12a, d). Their methylation levels in kidney samples also had significant correlations with eGFR and fibrosis (Supplementary Fig. 12b, c, e, f). These results further supported that the CpG sites we identified from blood samples had functional significance in the kidney. In a different cohort of 84 individuals with type 2 diabetes from the Native American population, two out of the seven CpG sites identified (cg02304370 and cg18593194) showed a suggestive association between methylation measured in peripheral blood with global glomerular sclerosis on morphometric variables of kidney biopsy samples in the same individuals (Supplementary Table 4), again highlighting the potential link between methylation level in blood and kidney pathology.
eGFR slope inferred by the multisite model can predict future renal failure
There are existing risk equations using clinical variables to predict kidney-related outcomes such as ESKD41,42,43,44,45. To see if our multisite model for eGFR slope can also predict future ESKD cases, we used it to predict the 5-year eGFR value of each patient and then determined the corresponding 5-year ESKD status based on it (defined as calculated eGFR<15 ml/min/1.73 m2). To avoid over-fitting, we inferred the eGFR slope and predicted the ESKD risk using a cross-validation procedure, in which the multisite model was built on the training samples and the inference was made on the left-out validation samples. The performance of predictions was then evaluated by considering all these left-out predictions together. For the benchmarking purpose, we also predicted 5-year ESKD status of all patients using three clinical risk equations. These included the Joint Asia Diabetes Evaluation (JADE) model developed in Chinese patients with type 2 diabetes41,46, an equation based on data from the United Kingdom Prospective Diabetes Study (UKPDS)44, and a simple equation that ranks patients by the negative values of baseline eGFR where a lower baseline eGFR value predicted a higher risk of developing ESKD in 5 years.
When considering all patients, our multisite model with covariates achieved an AUROC of 0.94 and an AUPR (area under the precision-recall curve) of 0.73 (Fig. 4). When excluding patients with baseline eGFR<30 ml/min/1.73 m2, who had very high risk of developing ESKD in 5 years, our model with covariates achieved an AUROC of 0.88 and an AUPR of 0.36 (Fig. 4). In both cases, the performance of our model, even without clinical covariates, was comparable to the performance of the clinical equations, and the inclusion of clinical covariates further enhanced the performance of the models.

AUROC and AUPR of the risk scores from simple negative value of baseline eGFR, JADE risk model, UKPDS-OM2, and our multisite models with or without covariates. The risk scores of the JADE model and UKPDS-OM2 were calculated with the risk equations in the original paper. The risk scores of the multisite models were calculated using the inferred eGFR slope with 5-fold cross-validation. Source data are provided as a Source Data file.
In an independent nested case–control cohort of 181 Native Americans with type 2 diabetes, of which 80 developed ESKD during follow-up, baseline methylation scores for baseline eGFR or eGFR slope were both associated with incident ESKD (Supplementary Table 5). The association was rendered non-significant after inclusion of baseline eGFR into the model, highlighting that the ability of the methylation changes to predict incident ESKD was mediated by methylation changes associated with baseline eGFR.
Discussion
In this study of methylation profiles from a cohort of patients with type 2 diabetes, our major findings are as follows: (1) DNA methylation level was associated with renal function in type 2 diabetes; (2) methylation levels of previously unidentified CpG sites were associated with baseline eGFR; (3) a set of eight previously unidentified CpG sites was associated with the rate of eGFR decline; (4) it is possible to construct prediction models using methylation data for baseline eGFR and decline in eGFR with replication in independent cohorts with type 2 diabetes; (5) proximal genes of the previously unidentified CpG sites and those included in the prediction models were implicated in pathways related to the pathogenesis of kidney diseases; and (6) the prediction models constructed can achieve comparable prediction to models incorporating clinical risk factors. Our study provides insights on the potential of incorporating methylation biomarkers to facilitate risk stratification in type 2 diabetes.
Our results extend earlier work by ourselves and others in highlighting the potential link between kidney function and methylation profile. In particular, the top sites identified in our study, cg17944885, near ZNF20, corresponded to a CpG site which had been reported in several EWAS for kidney function22,23,24,25. Furthermore, CpG sites identified in other studies with methylation levels associated with kidney function in the general population also demonstrated nominal association in our analysis of methylation changes. These results suggested that methylation changes associated with kidney function in the general population may also be applicable to a population with type 2 diabetes. The earlier EWAS were mainly conducted in European populations with subsequent replication in multiethnic cohorts. Together with our results, we might conclude that methylation profiles are not ethnic-specific, as in the case of genetic loci identified from GWAS. Several of our findings were also reported in two recent meta-analysis of EWAS10,22, although many of them had not been identified in the earlier individual cohort studies. While this may reflect improved statistical power from the recent larger meta-analysis, a trans-ethnic meta-analysis may be a more powerful strategy for discovering sites that are relevant for different ethnic populations.
In general, there was greater consistency for findings related to methylation changes associated with baseline eGFR compared to decline in kidney function. This is not surprising, given that multiple factors such as blood pressure, lipids, and glycaemia as well as medications might modulate renal and vascular pathology to influence progression of kidney function. Despite the strong association, it is difficult to disentangle the causal relationship between methylation changes and kidney function at baseline. The strong association between baseline eGFR and methylation changes might be consequences of the altered metabolic milieu related to kidney dysfunction. On the other hand, methylation changes predictive of kidney function decline, with minimal overlap with sites associated with baseline eGFR, are more likely to be of use as prognostic biomarkers.
In general, our EWAS results of baseline eGFR were most consistent with those reported by Chu et al. in the ARIC and FHS cohorts23 and Breeze et al. in multiple studies and ethnicities22. A number of their top sites also had significant P values in our data, even though none of these previous studies was conducted on Chinese-specific cohorts or cohorts consisting only of patients with type 2 diabetes (Supplementary Fig. 6, Supplementary Data 1, and Supplementary Data 2). Other than cg17944885, 13 significant CpG sites at FDR = 0.05 in our cohort, including cg25364972, cg02304370, cg12065228, cg21745599, cg16292343, cg05554494, cg22386583, cg09299075, cg13924998, cg07814567, cg03919650, cg19942083, and cg26099045, were also reported as significant signals in either ARIC or FHS cohort, and one significant CpG site in our data, cg23597162, was identified in both the ARIC and FHS cohorts23.
Both our single-site and multisite analyses identified cg00506299 as being associated with baseline eGFR. This site is located within the RFTN1 gene, the methylation level of which has not been previously associated with kidney function. However, RFTN1 was found differentially expressed between DKD and controls31 and correlated with VvInt29 in patients with DKD. In FAN mouse kidneys, Rftn1 was differentially expressed as compared to kidneys from healthy mice32. As another example, cg21919729, located within the CTSB gene and identified by our single-site analysis, did not have its methylation reported to be associated with kidney disease. However, its expression was correlated with VvInt29 in patients with DKD. Its mouse homologous gene Ctsb was also differentially expressed in proximal tubule (PT) cells between FAN mice and healthy controls32. CTSB encodes cathepsin B, a member of the C1 family of peptidases, which produces a lysosomal cysteine protease with both endopeptidase and exopeptidase activity that may play a role in protein turnover. Cathepsin B is involved in inflammation, apoptosis and autophagy during ESKD, chronic kidney disease, and AKI47.
Interestingly, the majority of the most significant CpG sites reside in the gene body, highlighting the increasingly recognized role of gene body and non-promoter methylation as important mechanisms of gene regulation in metabolic diseases48,49. Among the 74 CpG sites associated with rate of decline in eGFR in people with diabetes in our study, none was reported in previous studies of the general population, which demonstrates the utility of undertaking discovery efforts specifically in people with diabetes to advance precision medicine in diabetes.
From our functional evaluation using other datasets, these CpG sites identified to be associated with diabetic kidney disease point towards other genes implicated in kidney function and kidney diseases, highlighting the potential to use methylation markers in peripheral blood to obtain important biological insights in organ-specific diseases, such as examples from previously noted inter-individual variation in methylation across blood and brain50. The fact that methylation level of some of the CpG sites in blood, or in kidneys, show correlation to fibrosis and glomerulosclerosis, further highlight the potential of identifying organ-specific pathology using methylome from peripheral blood.
Although we identified several methylation sites strongly associated with kidney function and decline in kidney function which reached stringent threshold of statistical significance after considering the number of statistical tests, the construction of a prediction model did not necessarily include all of these individually significant CpG sites. This might be because of strong correlation among individual CpG sites, due to spatial dependency or other reasons, leading to redundancy.
In the future, it will be useful to directly test the functional significance of the CpG sites identified in this study in kidney tissues or cell/organoid models. One possible way is to use CRISPR-Cas9-based genome editing to turn a CpG site into a non-CpG sequence, to test the effect of losing CpG methylation at the site. Another possible way is to use CRISPR-based epigenome editing, such as dCas9 fused with a DNA methyltransferase to test the effect of gaining DNA methylation, or dCas9 fused with the ten-eleven translocation (TET) methylcytosine dioxygenase to test the effect of losing DNA methylation.
The prediction model with the best performance included a combination of CpG sites, many of which were not individually strongly associated with eGFR or eGFR decline. The difference in performance between prediction models incorporating multiple sites versus ones including only top individual CpG sites is analogous to the recent development of genome-wide polygenic risk scores51. The latter tend to have better performance and utility than the traditional approach of developing polygenic risk scores based on only GWAS-significant hits52. Given a large number of methylation datasets currently available, our approach may be applicable for developing other prediction models based on epigenome-wide methylation data, an approach taken by the pioneering work of epigenetic clocks53. The prediction model based on our multisite methylation signature also had comparable performance with established risk equations using clinical parameters to predict adverse renal outcome, whereas our data provided additional insights on biological pathways. Our results also suggest that the methylation signature could capture most of the information provided by clinical risk factors, and that inclusion of clinical risk factors did not substantially improve prediction. In contrast, most studies that add genetic variables to clinical markers only marginally improved the prediction of diabetes-related complications54. Thus, our results highlight the potential utility of incorporating methylation changes to risk models to improve risk stratification.
Our study has several strengths, including methylation profiling of a moderately large number of subjects with type 2 diabetes with long duration of follow-up for kidney outcomes and assessment of kidney function decline. Most subjects were free of DKD at baseline. We acknowledge several limitations. The discovery was undertaken in a cohort of Chinese patients at comparatively high risk of DKD progression, though the model developed was applied to a group of patients with diabetes in a different clinical setting. In particular, our methylation signature was based on methylation changes in circulating leukocytes as opposed to methylation changes within the kidney. Nevertheless, peripheral blood is a readily accessible tissue for risk stratification in clinical practice, and numerous studies have demonstrated the ability to identify biomarkers in disease-relevant pathways using methylation changes in leukocytes55.
Our results highlight the potential utility of using methylation levels in blood samples to predict eGFR or change in eGFR in different populations. We have also identified previously unidentified methylation markers associated with kidney function and decline in kidney function and kidney pathology. Our study highlights the potential of using methylation markers in the risk stratification of renal disease among individuals with type 2 diabetes.
Methods
Participant recruitment and clinical variable measurements
We included subjects from HKDR, which was established at the Prince of Wales Hospital, the teaching hospital of the Chinese University of Hong Kong. The HKDR consecutively enrolled patients who were referred to the Diabetes Mellitus and Endocrine Centre for comprehensive assessment of complications and metabolic control, including patients referred from specialty clinics, community clinics and general practitioners46. Subjects with diabetes were evaluated as part of a structured assessment for diabetes complications according to a modified European DiabCare protocol. All patients in the HKDR underwent clinical assessments and laboratory investigations after 8-hour overnight fast, including eye, feet, urine, and blood examinations. Eye examination included visual acuity and fundoscopy through dilated pupils or retinal photography. Retinopathy was defined by typical changes due to diabetes, laser scars, or a history of vitrectomy. Foot examination was performed using Doppler ultrasound scan and monofilament and graduated tuning fork. Fasting blood was sampled for measurement of plasma glucose, HbA1c, lipid profile (total cholesterol, high-density lipoprotein [HDL] cholesterol, triglycerides and calculated low-density lipoprotein [LDL] cholesterol), and random spot urinary sample was used to assess albumin to creatinine ratio (ACR). The CKD-EPI equation56 was used to estimate glomerular filtration rate.
Ethical approval was obtained from the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee. Written informed consent was obtained from all subjects at the time of enrollment for the collection of clinical information and biosamples for archival and research purposes.
Between 1995 and December 31, 2007, a consecutive cohort consisting of 10,129 patients with diabetes was assessed, with follow-up. Clinical outcomes were defined using hospital discharge diagnoses based on the International Classification of Diseases, Ninth Revision (ICD-9). The Hong Kong Hospital Authority Central Computer System records admissions to all public hospitals, which provides about 95% of inpatient bed-days in Hong Kong. All hospitalization records were retrieved from this system using a unique identifier number. Results of follow-up investigations, including eGFR were likewise retrieved for each subject from the electronic health record from the Central Computer System57. For the current analysis, we created a nested case–control cohort based on incident ESKD or incident cardiovascular disease (defined according to the censor date of June 30th, 2017, around the time when the EWAS was initiated and when the case–control status was defined), whereby each subject free of DKD at follow-up was matched with a case of incident ESKD with a similar age at baseline. ESKD was defined by the codes of dialysis (procedure codes 39.95 or 54.98), kidney transplant (procedure code 55.6 or diagnosis codes 996.81 or V42.0), or eGFR<15 ml/min/1.73 m2. All subjects were selected based on being free of known cardiovascular events at baseline. In addition to baseline kidney function data, we retrieved follow-up laboratory data through June 30th, 2017, in order to calculate the eGFR slope during follow-up for each individual, up to the censor date, eGFR<15 ml/min/1.73 m2, or death, whichever event occurred sooner.
DNA methylation data production and processing
Whole blood was taken at the baseline assessment visit in a fasting state. Genomic DNA from leukocytes was extracted using traditional phenol-chloroform methods and quantified using Picogreen. Bisulfite conversion was performed using EZGold Methylation kit (Zymo), as per standard protocol. After DNA extraction and bisulfite treatment, DNA methylation in each sample was measured using the Illumina Infinium HumanMethylation450K BeadChip, which covered around 485,000 CpG sites across the genome.
The RnBeads package (version 1.6.1)58 was used to preprocess the raw data. First, 10,119 sites were removed due to overlapping with single nucleotide polymorphisms (SNPs). Probes and samples with a large fraction of unreliable measurements, defined as those with detection P values larger than 0.05, were also removed. Furthermore, probes in contexts other than CpG sites and probes on sex chromosomes were removed, as was done in some previous studies11,20. Background correction was conducted using the “noob” method in the methylumi package (version 2.20.0)59 and the signal intensities were normalized using the SWAN method60 in the minfi package (version 1.20.2)61. After these filtering and normalization steps, 453,128 probes and 1268 samples remained, each quantified by a beta value. In all downstream analyses, we excluded probes with missing methylation values in any sample, resulting in the final number of 434,908 probes. In the whole study, genomic coordinates were based on the reference human genome hg19.
Modeling the clinical variables using top DNA methylation principal components
Dimensionality reduction of the methylation data was performed using PCA. The top PCs were taken as features of each sample to model each of the clinical variables in a classification setting. Specifically, for each clinical variable, we mapped their values to binary class labels using the criteria listed in Supplementary Table 6. When considering each clinical variable, samples with missing values were omitted. We then constructed logistic regression models with L2 regularization using the Python scikit-learn package (version 0.20.3)62 following a 10-fold cross-validation procedure. In this procedure, the whole set of samples was randomly divided into 10 subsets, and each time 9 subsets were used to construct a model while the remaining subset was used to evaluate the model performance, quantified by AUROC. The ten sets of results were then reported separately, together with their mean values. We also tried two other modeling methods, namely support vector classifier with a radial-basis kernel and random forest, and obtained largely comparable results as the logistic regression models (Supplementary Table 7). This same procedure was also used when we modeled eGFR using sex, age, and smoking status alone and with the top PCs.
Cell-type composition estimation
To adjust for cell heterogeneity of whole-blood samples, cell-type compositions were estimated using a reference-based approach21. Using raw methylation data as input, we generated estimated cell counts for CD4 + T cells, CD8 + T cells, NK cells, B cells, monocytes, and granulocytes, using the estimateCellCounts function implemented in the minfi package (version 1.28.4)61.
Single-site epigenome-wide association study (EWAS)
Baseline eGFR was calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation56. The eGFR slope of each individual was determined by fitting a linear mixed model63 and expressed as the percentage change of eGFR per year:
where \({{log }}\left({eGF}{R}_{{ij}}\right)\) is the log-transformed eGFR of i-th individual at jth measurement, \({t}_{{ij}}\) is the time for measuring eGFRij, \({\beta }_{0}\) and \({\beta }_{1}\) are coefficients for the fixed effects while \({b}_{0i}\) and \({b}_{1i}\) are coefficients for the random effects that are specific to the ith individual, and \({\epsilon }_{{ij}}\) is the random noise. After fitting the model, the individual-specific slope is given by the following:
which is expressed as the percentage change of eGFR per year.
For each CpG site, a linear model was constructed using either baseline eGFR or eGFR slope as the dependent variable and the methylation level as the independent variable. Sex, age, smoking status, duration of diabetes, hemoglobin A1c, blood pressure, experiment batch and the cell-type composition estimations (Supplementary Methods) were included as additional independent variables for models that involved covariates. The P value of each CpG site was calculated based on the null hypothesis that it had a zero coefficient in its linear model using two-sided Student’s t test. The Bonferroni procedure was used to perform multiple hypothesis testing correction of the raw P values. In addition, the Benjamin–Hochberg procedure was used to identify significant sites at a given false discovery rate (FDR).
Using M values in EWAS
Apart from using beta values to quantify methylation levels, we also used M values (where M = log2 beta/(1-beta)) which yielded similar results. The Pearson correlations of association P values of CpG sites with baseline eGFR and eGFR slope were 0.967 and 0.956, respectively. The corresponding Spearman correlations were 0.928 and 0.927 for baseline eGFR and eGFR slope, respectively.
Multisite models
We also developed a multisite approach that considered all CpG sites at the same time and selected a subset of them to create the best model to infer baseline eGFR or eGFR slope. Briefly, we used LASSO (least absolute shrinkage and selection operator) to construct regression models, which aims at fitting linear models with only a small number of CpG sites having a nonzero coefficient. Performance of each model was evaluated using cross-validation, while the final set of CpG sites (and the corresponding value of the L1 regularization hyperparameter) was selected using a nested procedure that involves the Bayesian Information Criterion (BIC) to balance between model complexity and performance. The constructed models were finally evaluated using left-out testing sets not involved in either training the models or tuning the hyper-parameters.
In detail, we used a multi-step procedure with nested cross-validation to perform model learning, hyperparameter tuning, and unbiased model evaluations (Supplementary Fig. 13). As a data pre-processing step, the methylation levels of each CpG site and the values of each covariate were individually standardized to have zero mean and unit variance.
In our multi-step procedure, we first randomly split the 1268 samples into training (90%) and testing (10%) sets. Using the samples in the training set, we used the tenfold cross-validation procedure to construct linear regression models with LASSO. The value of the regularization parameter \(\alpha\) was chosen using grid search based on a nested fivefold cross-validation within each training fold. The value of \(\alpha\) chosen (denoted as \({\alpha }^{*}\)) for each of the 10 outer training folds was determined using the following criterion:
Where \({R}_{\alpha }^{2}\) is the \({R}^{2}\) of the LASSO model using parameter \(\alpha\), \({{max }}({R}^{2})\) and \({SD}({R}^{2})\) are the maximum and standard deviation of \({R}^{2}\), respectively, among all the models with different values of \(\alpha\) in the set \(D\) considered during the grid search. This criterion aims at finding the largest value of \(\alpha\) that still gives a model performance close to the one with maximal \({R}^{2}\). The goal of choosing a large value of \(\alpha\) is to ensure that only a small set of the most important CpG sites is selected from each model. Using this selected value of \(\alpha\), a model was trained with all the samples in the outer training fold. The model was then applied to the samples in the outer testing fold to compute the performance measures. After applying these to all the ten outer training folds, ten sets of performance measures were produced. This whole procedure was further repeated ten times with different random splits of data into ten folds each time, leading to a total of 100 models and correspondingly 100 sets of performance measures.
To produce a single model based on these 100 sets of results, we assigned a weight to each CpG site based on the number of times that it was included in the models and the performance of these models, using the following formula:
where \({w}_{k}\) is the weight of the kth CpG site, \({\rho }_{{ij}}\) is the Pearson correlation between prediction and actual values in the ith outer testing fold for the jth repeat, and \({S}_{{ij}}\) is the set of CpG sites selected by the ith outer training fold for the jth repeat with a nonzero coefficient. Based on this formula, a CpG site would generally get a higher weight if it has a nonzero coefficient in more models and/or in models that have better performance in terms of Pearson correlation.
All the CpG sites were then sorted in descending order according to their weights. A second series of linear regression models with LASSO were then constructed using different numbers of CpG sites with the largest weights as features with all samples in the original training set for training. The final number of CpG sites to use, \({n}^{*}\), was determined using the following formula that involves the Bayesian Information Criterion (BIC):
where \({BI} {{C_n}}\) is the BIC of the model involving the nhighest-weight CpG sites as features, and \({{\min }}\left({BIC}\right)\) and \({SD}({BIC})\) are the minimum and standard deviation of BIC among all the models with different numbers of CpG sites, respectively. This formula aims at maximizing the number of CpG sites while having a model with a BIC close to the one with the minimal BIC. This time, the number of CpG sites was maximized because the highest-weight CpG sites should already be the most important ones, and including more of them in the model could ensure its robustness. The performance of the model that involved the n highest-weight CpG sites was then evaluated objectively using the original testing set, which was not involved in any training and parameter tuning steps described above.
Finally, all 1268 samples were used together to train a final model for baseline eGFR and another model for eGFR slope, both using the same procedure described above to determine the number of CpG sites. With the selected CpG sites, we trained another version of these two models without including the covariates. Since these final models involved all 1268 samples in model training and parameter tuning, there were no left-out samples in the primary cohort that could independently evaluate their performance.
Validation of the models in a cohort of Native Americans
Our multisite models were tested in an independent Native American cohort, which contained 326 participants with type 2 diabetes. Baseline eGFR, eGFRs during a mean follow-up of 9.5 years and other clinical variables were measured for each participant. The raw eGFR slope for each subject was calculated using linear regression across all available eGFR measures. The mean baseline eGFR is 106.7 ± 15.1 ml/min/1.73 m2. DNA methylation was measured by Illumina Infinium HumanMethylation450K BeadChip and processed as described before20. In brief, Minfi61 was used to preprocess the data and perform quality control. RnBeads58 was then used for beta-mixture quantile normalization.
To use this replication cohort to evaluate the performance of models constructed from the primary cohort, we took the intersection of CpG sites which passed quality control in both cohorts. All samples in the primary cohort were used to learn the baseline eGFR and eGFR slope models based on the subset of CpG sites found in both cohorts, using the same procedure as described above. These models were then applied to the Native American cohort to compare the predicted baseline eGFR or eGFR slope values and their corresponding actual measurements (Fig. 2e–h and Table 4). To check whether our original model is directly applicable to other cohorts, we also applied the original model in the Native American cohort without re-training and performed the same evaluation. All CpG sites not available in the Native American cohort had their methylation levels set to 0 in this case (Supplementary Table 3 and Supplementary Fig. 10). All analyses were performed using R.
Functional significance of our CpG sites’ methylation levels in kidney samples
Seven CpG sites were selected to check their methylation levels in kidney samples using a published dataset with methylation data from 506 human kidneys64. In this dataset, the samples belong to five groups based on the donors’ disease status, namely Con (normal kidneys, 113 samples), CKD (eGFR <60, 101 samples), DKD (having both CKD and diabetes, 63 samples), DM (having diabetes but not CKD, 97 samples), and HTN (having hypertension but not CKD, 132 samples).
Among the seven CpG sites selected for lookup, one (cg21573651) was associated with both baseline eGFR and eGFR slope in the single-site analysis. The other six CpG sites (cg17944885, cg06449934, cg02304370, cg21919729, cg04610187 and cg18593194) were associated with baseline eGFR and were the top six sites among the 36 CpG sites identified in both single-site and multisite analyses.
Morphometric analysis in kidney biopsies of patients with type 2 diabetes
To evaluate links between blood methylation and structural changes in the kidney, we utilized data from analyses of the morphometric variables from the kidney biopsies from 84 individuals who had biopsies done and methylation in peripheral blood has been measured using the Illumina Infinium HumanMethylation450 array in the same individuals20,65. Results are reported as partial correlation and P value for each of the nine key variables. The parameters evaluated included: FPW—podocyte foot process width (nm) (higher is worse), GBM—glomerular basement membrane width (nm) (higher is worse), GS—global glomerular sclerosis (%) (higher is worse), VG—mean glomerular volume (×106 µm3) (higher is worse), non-Podo—mean non-podocyte number per glomerulus (N) (lower is worse), Fen—percent fenestrated endothelium (%) (higher is worse), SV (glomerular filtration surface density) (µ2/µ3) (lower is worse), VvInt—cortical interstitial fractional volume (%) (higher is worse), VvMes—mesangial fractional volume (%) (higher is worse). These morphometric parameters were selected because they were previously associated with loss of kidney function in this cohort29,66.
Risk equations comparison
To calculate the eGFR of each subject five years after the baseline measurements using the eGFR slope determined by Eqs. (1) and (2), the following formula is used:
where \({\left({eGFR}\right)}_{i0}\) and \({\left({eGFR}\right)}_{i5}\) are the eGFR of i-th individual at baseline and five years after the baseline, respectively. We defined subject i to have ESKD in five years after the baseline if \({\left({eGFR}\right)}_{i5}\) < 15 ml/min/1.73 m2.
For each patient, the actual ESKD status was determined using the above method based on his/her actual eGFR slope obtained by making use of all his/her eGFR measurements during the follow-up period. Similarly, the ESKD status predicted by our model was produced using the above method based on the predicted eGFR slope, the multisite model of which was constructed using DNA methylation. This was achieved by a fivefold cross-validation procedure, in which every time 4/5 of the patients were used to train the multisite model, which was applied to the remaining 1/5 of the patients to predict their 5-year ESKD status. The risk scores of the risk equations for renal outcomes by JADE risk model41,46 and UKPDS-OM244 were calculated following the descriptions in the original publications.
An independent nested case–control cohort of 181 individuals with type 2 diabetes, of which 80 developed ESKD during follow-up20, were included to examine association between blood methylation level and progression to ESKD.
Inclusion and ethics statement
In this research study, local researchers were included throughout the research process, including study design, study implementation, data ownership, intellectual property, and authorship of the publication. This study is locally relevant and has been determined in collaboration with local partners. Roles and responsibilities were agreed among collaborators ahead of the research. This study was approved by a local ethics review committee. Local and regional research relevant to this study has been taken into account in citations.
The original discovery analysis in Hong Kong Diabetes Register is supported by ethics approval from the Joint Chinese University of Hong Kong-New Territories East Cluster Clinical Research Ethics Committee. Written informed consent was obtained from all participants.
The validation of the models in a cohort of Native Americans was conducted by the NIDDK Intramural Research Program, and these protocols were approved by the NIDDK/NIH Institutional Review Board. Written informed consent was obtained from all participants.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Individual-level data are protected and are not available because of ethical restriction, as they were not consented for sharing on a public platform. Summary methylation data are available for analysis by qualified researchers who fulfill the criteria for access by providing a copy of the research proposal and analysis plan, proof of ethics approval for the planned methylation analysis, and institutional endorsement of server data security. Readers and colleagues who are interested to obtain further information about the study can contact the Hong Kong Institute of Diabetes and Obesity, The Chinese University of Hong Kong, Hong Kong at hkido@cuhk.edu.hk. The summary statistics of significant CpG sites and the multisite models generated in this study are provided in the Supplementary Information. We have also created a web-based tool using Shiny app so that readers can use the tool to calculate eGFR and eGFR slope based on methylation data, or perform lookup of association between CpG methylation and eGFR. The tool can be accessed at http://hkdbrmlab.shinyapps.io/DKD_EWAS/. Contact person for the Hong Kong Diabetes Register: Professor Ronald Ma, rcwma@cuhk.edu.hk. Contact person for the Native American cohorts: Dr Robert Hanson, rhanson@phx.niddk.nih.gov. Source data are provided with this paper.
Code availability
Source code for single-site and multisite analyses can be accessed from our GitHub repository, https://github.com/kellyliyichen/eGFR_450k and Zenodo67, under GPL-3.0 license.
References
Nanditha, A. et al. Diabetes in Asia and the Pacific: implications for the global epidemic. Diabetes Care 39, 472–485 (2016).
Wu, H. et al. Trends in kidney failure and kidney replacement therapy in people with diabetes in Hong Kong, 2002-2015: a retrospective cohort study. Lancet Reg. Heal. West. Pac. 11, 100165 (2021).
Wheeler, D. C. et al. Effects of dapagliflozin on major adverse kidney and cardiovascular events in patients with diabetic and non-diabetic chronic kidney disease: a prespecified analysis from the DAPA-CKD trial. Lancet Diabetes Endocrinol. 9, 22–31 (2021).
Bakris, G. L. et al. Effect of finerenone on chronic kidney disease outcomes in type 2 diabetes. N. Engl. J. Med. 383, 2219–2229 (2020).
Ma, R. C. W. & Cooper, M. E. Genetics of diabetic kidney disease-from the worst of nightmares to the light of dawn? J. Am. Soc. Nephrol. 28, 389–393 (2017).
van Zuydam, N. R. et al. Genome-wide association study of diabetic kidney disease in subjects with type 2 diabetes. Diabetes 67, 1414–1427 (2018).
Fan, B., Luk, A. O. Y., Chan, J. C. N. & Ma, R. C. W. MicroRNA and diabetic complications: a clinical perspective. Antioxid. Redox Signal. 29, 1041–1063 (2018).
Aranyi, T. & Susztak, K. Cytosine methylation studies in patients with diabetic kidney disease. Curr. Diabetes Rep. 19, 91 (2019).
Smyth, L. J. et al. Assessment of differentially methylated loci in individuals with end-stage kidney disease attributed to diabetic kidney disease: an exploratory study. Clin. Epigenet. 13, 99 (2021).
Schlosser, P. et al. Meta-analyses identify DNA methylation associated with kidney function and damage. Nat. Commun. 12, 7174 (2021).
Gluck, C. et al. Kidney cytosine methylation changes improve renal function decline estimation in patients with diabetic kidney disease. Nat. Commun. 10, 2461 (2019).
Park, J. et al. Functional methylome analysis of human diabetic kidney disease. JCI Insight 4, 11 (2019).
Nguyen, L. T. et al. Blood DNA methylation predicts diabetic kidney disease progression in high fat diet-fed mice. Nutrients 14, 785 (2022).
McCarthy, N. S. et al. Meta-analysis of human methylation data for evidence of sex-specific autosomal patterns. BMC Genom. 15, 981 (2014).
Yousefi, P. et al. Sex differences in DNA methylation assessed by 450 K BeadChip in newborns. BMC Genom. 16, 911 (2015).
Field, A. E. et al. DNA methylation clocks in aging: categories, causes, and consequences. Mol. Cell 71, 882–895 (2018).
Li, X., Li, W. & Xu, Y. Human age prediction based on DNA methylation using a gradient boosting regressor. Genes 9, 424 (2018).
Dugué, P.-A. et al. Smoking and blood DNA methylation: an epigenome-wide association study and assessment of reversibility. Epigenetics 15, 358–368 (2020).
Lee, K. W. K. & Pausova, Z. Cigarette smoking and DNA methylation. Front. Genet. 4, 132 (2013).
Qiu, C. et al. Cytosine methylation predicts renal function decline in American Indians. Kidney Int. 93, 1417–1431 (2018).
Houseman, E. A. et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinforma. 13, 86 (2012).
Breeze, C. E. et al. Epigenome-wide association study of kidney function identifies trans-ethnic and ethnic-specific loci. Genome Med. 13, 74 (2021).
Chu, A. Y. et al. Epigenome-wide association studies identify DNA methylation associated with kidney function. Nat. Commun. 8, 1286 (2017).
Chen, J. et al. Epigenetic associations with estimated glomerular filtration rate among men with human immunodeficiency virus infection. Clin. Infect. Dis. 70, 667–673 (2020).
Sheng, X. et al. Systematic integrated analysis of genetic and epigenetic variation in diabetic kidney disease. Proc. Natl Acad. Sci. USA 117, 29013–29024 (2020).
Chen, Z. et al. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the DCCT/EDIC type 1 diabetes cohort. Proc. Natl Acad. Sci. USA 113, E3002–E3011 (2016).
Smyth, L. J., Patterson, C. C., Swan, E. J., Maxwell, A. P. & McKnight, A. J. DNA methylation associated with diabetic kidney disease in blood-derived DNA. Front. Cell Dev. Biol. 8, 561907 (2020).
Wing, M. R. et al. DNA methylation profile associated with rapid decline in kidney function: findings from the CRIC study. Nephrol. Dial. Transpl. 29, 864–872 (2014).
Nair, V. et al. A molecular morphometric approach to diabetic kidney disease can link structure to function and outcome. Kidney Int. 93, 439–449 (2018).
Qiu, C. et al. Renal compartment-specific genetic variation analyses identify new pathways in chronic kidney disease. Nat. Med. 24, 1721–1731 (2018).
Woroniecka, K. I. et al. Transcriptome analysis of human diabetic kidney disease. Diabetes 60, 2354–2369 (2011).
Dhillon, P. et al. The nuclear receptor ESRRA protects from kidney disease by coupling metabolism and differentiation. Cell Metab. 33, 379–394.e8 (2021).
Park, J. et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360, 758–763 (2018).
Liu, H. et al. Epigenomic and transcriptomic analyses define core cell types, genes and targetable mechanisms for kidney disease. Nat. Genet. 54, 950–962 (2022).
Stanzick, K. J. et al. Discovery and prioritization of variants and genes for kidney function in >1.2 million individuals. Nat. Commun. 12, 4350 (2021).
Dehnadi, A. et al. Prophylactic orthosteric inhibition of leukocyte integrin CD11b/CD18 prevents long-term fibrotic kidney failure in cynomolgus monkeys. Nat. Commun. 8, 13899 (2017).
De Marinis, Y. et al. Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int. 89, 342–353 (2016).
Gu, C., Liu, S., Wang, H. & Dou, H. Role of the thioredoxin interacting protein in diabetic nephropathy and the mechanism of regulating NOD‑like receptor protein 3 inflammatory corpuscle. Int. J. Mol. Med. 43, 2440–2450 (2019).
Purvis, G. S. D., Solito, E. & Thiemermann, C. Annexin-A1: therapeutic potential in microvascular disease. Front. Immunol. 10, 938 (2019).
Wu, L. et al. The attenuation of diabetic nephropathy by annexin A1 via regulation of lipid metabolism through the AMPK/PPARα/CPT1b pathway. Diabetes 70, 2192–2203 (2021).
Yang, X. L. et al. Modified end-stage renal disease risk score for Chinese type 2 diabetic patients–the Hong Kong Diabetes Registry. Diabetologia 50, 1348–1350 (2007).
Wan, E. Y. F. et al. Prediction of new onset of end stage renal disease in Chinese patients with type 2 diabetes mellitus—a population-based retrospective cohort study. BMC Nephrol. 18, 257 (2017).
Basu, S., Sussman, J. B., Berkowitz, S. A., Hayward, R. A. & Yudkin, J. S. Development and validation of risk equations for complications of type 2 diabetes (RECODe) using individual participant data from randomised trials. lancet Diabetes Endocrinol. 5, 788–798 (2017).
Hayes, A. J., Leal, J., Gray, A. M., Holman, R. R. & Clarke, P. M. UKPDS outcomes model 2: a new version of a model to simulate lifetime health outcomes of patients with type 2 diabetes mellitus using data from the 30 year United Kingdom Prospective Diabetes Study: UKPDS 82. Diabetologia 56, 1925–1933 (2013).
Jardine, M. J. et al. Prediction of kidney-related outcomes in patients with type 2 diabetes. Am. J. Kidney Dis. 60, 770–778 (2012).
Chan, J. C. N. et al. From Hong Kong diabetes register to JADE program to RAMP-DM for data-driven actions. Diabetes Care 42, 2022–2031 (2019).
Cocchiaro, P. et al. The multifaceted role of the lysosomal protease cathepsins in kidney disease. Front. Cell Dev. Biol. 5, 114 (2017).
Lou, S. et al. Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol. 15, 408 (2014).
Li, L., Gao, Y., Wu, Q., Cheng, A. S. L. & Yip, K. Y. New guidelines for DNA methylome studies regarding 5-hydroxymethylcytosine for understanding transcriptional regulation. Genome Res. 29, 543–553 (2019).
Davies, M. N. et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 13, R43 (2012).
Khera, A. V. et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 50, 1219–1224 (2018).
Tam, C. H. T. et al. Development of genome-wide polygenic risk scores for lipid traits and clinical applications for dyslipidemia, subclinical atherosclerosis, and diabetes cardiovascular complications among East Asians. Genome Med. 13, 29 (2021).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Morieri, M. L. et al. Genetic tools for coronary risk assessment in type 2 diabetes: a cohort study from the ACCORD clinical trial. Diabetes Care 41, 2404–2413 (2018).
Chambers, J. C. et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol. 3, 526–534 (2015).
Levey, A. S. et al. A new equation to estimate glomerular filtration rate. Ann. Intern. Med. 150, 604–612 (2009).
Jiang, G. et al. Progression of diabetic kidney disease and trajectory of kidney function decline in Chinese patients with Type 2 diabetes. Kidney Int. 95, 178–187 (2019).
Assenov, Y. et al. Comprehensive analysis of DNA methylation data with RnBeads. Nat. Methods 11, 1138–1140 (2014).
Buhule, O. D. et al. Stratified randomization controls better for batch effects in 450K methylation analysis: a cautionary tale. Front. Genet. 5, 354 (2014).
Maksimovic, J., Gordon, L. & Oshlack, A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 13, R44 (2012).
Aryee, M. J. et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Asar, Ö., Ritchie, J., Kalra, P. A. & Diggle, P. J. Joint modelling of repeated measurement and time-to-event data: an introductory tutorial. Int. J. Epidemiol. 44, 334–344 (2015).
Liu, J., Morgan, M., Hutchison, K. & Calhoun, V. D. A study of the influence of sex on genome wide methylation. PLoS ONE 5, e10028 (2010).
Weil, E. J. et al. Effect of losartan on prevention and progression of early diabetic nephropathy in American Indians with type 2 diabetes. Diabetes 62, 3224–3231 (2013).
Fufaa, G. D. et al. Structural predictors of loss of renal function in American Indians with type 2 diabetes. Clin. J. Am. Soc. Nephrol. 11, 254–261 (2016).
Li, K. Y. & Yip, K. Y. DNA methylation markers for kidney function and progression of diabetic kidney disease, eGFR_450k. https://doi.org/10.5281/zenodo.7713732 (2023).
Acknowledgements
We thank Qin Cao for the helpful discussions. This project was supported by the Research Grants Council Theme-based Research Scheme T12-402/13N and Research Impact Fund R4012-18 (to R.C.W.M.). R.C.W.M. also acknowledges support from a Croucher Foundation Senior Medical Research Fellowship, and the Chinese University of Hong Kong Focused Innovation Scheme, Outstanding Fellowship, and the Faculty Postdoctoral Fellowship Scheme. K.Y.Y. was additionally supported by Hong Kong Research Grants Council Collaborative Research Funds C4015-20E, C4045-18W, C4057-18E, and C7044-19G and General Research Funds 14107420 and 14203119, the Hong Kong Epigenomics Project (EpiHK), and the Chinese University of Hong Kong Young Researcher Award, Outstanding Fellowship, and Project Impact Enhancement Fund. K.Y.Y. is currently supported by the National Institute of Health P30 CA030199-41, U54 AG079758-01, and R21 AG075483-01S1. K.S. has been supported by the National Institute of Health NIH R01 DK087635, DK076077, and DK105821. R.L.H. and R.G.N. are supported by the Intramural Research Program of the United States National Institute of Diabetes and Digestive and Kidney Diseases.
Author information
Authors and Affiliations
Consortia
Contributions
J.C.N.C., K.Y.Y., and R.C.W.M. contributed substantially to the conception and design of the study. C.K.P.L., W.Y.S., H.M.L., C.C.S., J.C.N.C., and R.C.W.M. contributed to the sample collection and production of DNA methylation data. K.Y.L., C.H.T.T., C.H., G.J., M.S., H.Y.L., K.Y.Y., J.C.N.C., and R.C.W.M. performed data analysis and interpretation. H.L., S.D., R.L.H., R.G.N., and K.S. contributed to the replication studies. K.Y.L. and C.H. created the website for data access. K.Y.L., J.C.N.C., K.Y.Y., and R.C.W.M. wrote the manuscript. K.Y.L., K.Y.Y., and R.C.W.M. had full access to all the data in the study. K.Y.Y. and R.C.W.M. are the guarantors of this work and, as such, take responsibility for the integrity of the data and the accuracy of the data analysis. This work was part of the TRANSCEND Consortium project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
J.C.N.C. has received research grants and/or honoraria for consultancy and/or giving lectures from AstraZeneca, Bayer, Boehringer Ingelheim, Celltrion, Eli-Lilly, Hua Medicine, Lee Powder, Merck Serono, Merck Sharp & Dohme, Pfizer, Servier, Sanofi and Viatris, holds patents for using biomakers to predict risks of diabetes and its complications and is a co-founder of GemVCare, a biotechnology company partially supported by the Hong Kong Government startup fund. RCWM has received research grants for clinical trials from AstraZeneca, Bayer, MSD, Novo Nordisk, Sanofi, Tricida Inc., and honoraria for consultancy or lectures from AstraZeneca, Bayer, and Boehringer Ingelheim, all used to support diabetes research at the Chinese University of Hong Kong. RCWM is a co-founder of GemVCare, a technology start-up initiated with support from the Hong Kong Government Innovation and Technology Commission and its Technology Start-up Support Scheme for Universities (TSSSU). K.Y.L., J.C.N.C., K.Y.Y., and R.C.W.M. submitted a patent related to this study. The remaining authors declare no other competing interests.
Peer review
Peer review information
Nature Communications thanks Pascal Schlosser and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, K.Y., Tam, C.H.T., Liu, H. et al. DNA methylation markers for kidney function and progression of diabetic kidney disease. Nat Commun 14, 2543 (2023). https://doi.org/10.1038/s41467-023-37837-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37837-7
This article is cited by
-
Tangshen formula improves diabetic nephropathy in STZ-induced diabetes rats fed with hyper-methionine by regulating the methylation status of kidney
Clinical Epigenetics (2024)
-
Metabolic memory: mechanisms and diseases
Signal Transduction and Targeted Therapy (2024)
-
Targeted identification of risk and treatment of diabetic kidney disease
Nature Reviews Nephrology (2024)
-
Lessons and Applications of Omics Research in Diabetes Epidemiology
Current Diabetes Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
