
Abstract
Mfsd2a is the transporter for docosahexaenoic acid (DHA), an omega-3 fatty acid, across the blood brain barrier (BBB). Defects in Mfsd2a are linked to ailments from behavioral and motor dysfunctions to microcephaly. Mfsd2a transports long-chain unsaturated fatty-acids, including DHA and α-linolenic acid (ALA), that are attached to the zwitterionic lysophosphatidylcholine (LPC) headgroup. Even with the recently determined structures of Mfsd2a, the molecular details of how this transporter performs the energetically unfavorable task of translocating and flipping lysolipids across the lipid bilayer remains unclear. Here, we report five single-particle cryo-EM structures of Danio rerio Mfsd2a (drMfsd2a): in the inward-open conformation in the ligand-free state and displaying lipid-like densities modeled as ALA-LPC at four distinct positions. These Mfsd2a snapshots detail the flipping mechanism for lipid-LPC from outer to inner membrane leaflet and release for membrane integration on the cytoplasmic side. These results also map Mfsd2a mutants that disrupt lipid-LPC transport and are associated with disease.
Similar content being viewed by others

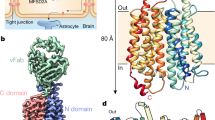

Introduction
Docosahexaenoic acid (DHA, 22:6) is an important component of cellular membranes where it forms structural and functional interactions with cholesterol and integral membrane proteins1 (Fig. 1a). DHA is highly enriched in the central nervous system (CNS), particularly at neuronal synapses2. Inadequate DHA results in several CNS-related ailments including learning and memory deficits, dyspraxia, and dyslexia3,4. Despite its essential roles in the CNS, the brain lacks robust mechanisms to biosynthesize DHA, which is estimated to have high turnover rates on the order of minutes5. Instead, the brain acquires DHA from plasma by transport across the blood brain barrier (BBB)6. Although many studies have gleaned important insights into the roles of tight junctions at the BBB7,8,9, recent work have revealed that DHA is delivered to the brain via selective transport through endothelial cells5,6.

a The preferred Mfsd2a DHA-LPC substrate and the ALA-LPC ligand observed in the drMfsd2a structures. b Overall architecture of drMfsd2a with the N-terminal domain, TM1-6, in blue and the C-terminal domain, TM7-12, in wheat. c Extracellular view of the substrate translocation pathway with the overlay of the four lysolipid positions observed in drMfsd2a. Dashed lines represent the proposed transport path of the C18 of the lipid tail through a cleft between the N- and C-domains. d Cytoplasmic view of the substrate translocation pathway with the overlay of the four lipid positions observed in drMfsd2a through a cleft between the N- and C-domains. Dashed lines represent the proposed transport path of the choline of the LPC headgroup. b–d Ligands illustrated as stick and sphere. e The ligand-free and the four elongated lipid-like densities fitted with ALA-LPC observed for drMfsd2a starting from the position closest to the extracellular towards the cytoplasmic side. Protein rendered as gray cartoon. Elongated densities shown as gray mesh. Lipid isomerism cannot be ascertained at the current resolution.
Phospholipids can move laterally in membranes but they cannot easily flip vertically between leaflets of the bilayer. Phospholipid flipping between membrane bilayers is energetically costly because this process entails the translocation of a charged headgroup across the hydrophobic region of the bilayer to realign with the opposite leaflet while maintaining the integrity of the membrane. Cells accomplish lipid flipping to establish membrane leaflet asymmetry by specialized transporters termed floppases10 and flippases11,12,13. These proteins couple lipid transport and flipping with energetically favorable processes such as ATP hydrolysis or shuttling ions down their electrochemical gradients11,12,13,14.
The major facilitator superfamily (MFS) is a large class of membrane proteins that span all kingdoms of life having diverse functions and substrate specificities15,16. MFS comprise over 25% of all transmembrane proteins and play key roles in cellular physiology. Although members of MFS share low sequence identity, they all share a similar twelve transmembrane helix (TM) topology and use the “rocker-switch” mechanism to transport cargos across membranes16,17. The MFS domain containing protein-2a (Mfsd2a) was initially discovered as an orphan receptor in 200818 and later revealed as the primary DHA flippase across the BBB6. Mechanistic studies revealed that Mfsd2a is a symporter for Na+ and is selective towards long chain, unsaturated fatty acids including DHA (22:6), α-linolenic acids (ALA, 18:3), and oleic acids (18:1) that are covalently tethered to the zwitterionic lysophosphatidylcholine (LPC) headgroup6,19 (Fig. 1a). Since most flippases so far are P4-ATP-dependent flippases, Mfsd2a is one of few known flippases outside this family6,11. The three other known MFS lipid transporters are Mfsd2b20, LplT21, and Spns222; however, they confer different substrate specificities and their transport mechanisms are yet to be defined. Importantly, Mfsd2a is required for the formation, development, and function of both the BBB and the CNS23,24,25. Defects in Mfsd2a disrupt normal brain development and function resulting in phenotypes including anxiety, learning deficits, ataxia, and severe microcephaly6,23,25,26,27. Consistent with these findings, it was recently demonstrated that Zika virus causes microcephaly by proteolytic degradation of Mfsd2a28.
Even with recently published structures of Mfsd2a19,29,30, the molecular details of how Mfsd2a performs lipid-LPC flipping and transport across the lipid bilayer remains obscure. Here, we present five structures of the Zebrafish (Danio rerio) isoform B Mfsd2a (drMfsd2a) in the inward-facing conformation determined by single-particle cryo-electron microscopy (cryo-EM), in the ligand-free state and displaying lipid-like densities at four distinct positions modeled as ALA-LPC (Fig. 1b–e). These four modeled ligand configurations allowed elucidation of (1) a clearer step-by-step transport and flipping mechanism of lipid-LPC, and (2) the release of the substrate to the cytoplasmic side of drMfsd2a for inner membrane leaflet integration (Fig. 1c–e). The details gleaned from these studies provide the molecular models of the proposed drMfsd2a lipid, phosphate, and choline binding sites, and how these components are orchestrated during the transport and flipping of the lysolipid. We also identify the interactions between the modeled lipid-LPC and drMfsd2a at four distinct positions, and map key residues that when mutated (1) disrupt lysolipid transport, and (2) are associated with known Mfsd2a-related diseases. Together this study adds to the repertoire of transport mechanisms by MFS by generating models for how members of this superfamily of proteins transport lipid cargos. Given its roles in brain function and development, our study lays the foundation for better understanding of neurological conditions and motor dysfunctions linked to defects in this family of transporters.
Results
Overall topology of drMfsd2a in complex with lipids
DrMfsd2a is a small protein at 59 kDa and structures of this size are exceedingly challenging to determine by single-particle cryo-EM31. As such, we generated and formed a complex between a FAB32 and drMfsd2a to facilitate cryo-EM structure determination (Supplementary Figs. 1–5, Supplementary Table 1). Further, alignment between the human and drMfsd2a sequences revealed a 76% similarity and 61% identity (Supplementary Fig. 6). Given that most MFS members share low sequence identities but a conserved fold15, sequence conservation between these two homologs suggests that the zebrafish protein can serve as a model for understanding human Mfsd2a.
Like other MFS members, drMfsd2a is comprised of twelve TMs that pack against one another with the substrate binding channel located at a cleft between the N- and C-terminal domains (Fig. 1b). The twelve TMs of drMfsd2a are, likewise, subdivided into two helical bundles called the N- (helices 1–6) and C-domains (helices 7–12) that are related by a twofold pseudosymmetry (Fig. 1b, Supplementary Fig. 6). This topology is consistent with other MFS members that have been structurally described, including the recent chicken19, mouse29, and human30 Mfsd2a structures. These three Mfsd2a structures were solved in the outward-facing, ligand-free conformation from mouse29, outward-occluded ligand-free state in complex with the placenta SYNC2 protein from human30, and the inward-facing complex bound with ALA-LPC from chicken19. These three structures are distinct from this study because none delineated the lipid flipping function of the transporter (Fig. 1d, e, Supplementary Fig. 7). Here, we report five structures of drMfsd2a in the inward-facing conformation in the ligand-free state and with bound lipid at four distinct locations in the substrate translocation tunnel (Fig. 1). We expressed and purified drMfsd2a from SF-9 insect cells and discovered that the protein copurified with endogenous phospholipids (Supplementary Fig. 8a–c). These results are consistent with past reports of Mfsd2a copurifying with native ALA-LPC from SF-9 insect cells19. Functional assays using reconstituted proteoliposome coupled with [14C]DHA-LPC uptake further confirmed that drMfsd2a is indeed active and able to bind lipids (Supplementary Fig. 9). We therefore modeled ALA-LPC into the four distinct lipid-like densities termed Lysolipid1A, 1B, 2B, 3C (numbers and letters refer to the lipid and LPC binding positions) (Fig. 1e, Supplementary Figs. 5, 10, 11).
Initial data processing yielded a 2.9 Å resolution map with three lipid-like densities representing three merged ligands, Lysolipid1A, 2B, 3C (Fig. 1e). However, our in vitro analysis indicates that there is only ~1 phospholipid per protein molecule (Supplementary Fig. 8c), suggesting that the lipids observed in this model is a merge of particles of substrates bound at three separate sites. Due to the small size of the phospholipid, traditional classification and 3D variability analysis30 were unable to separate individual states. Alternatively, we deployed 3D classification without alignment in cryoSPARC with five input references including Mfsd2a-FAB with (1) Lysolipid1A, 2B, 3C, (2) no ligand, (3) Lysolipid1A, (4) Lysolipid2B, and (5) Lysolipid3C bound (Supplementary Fig. 2). Maps with individual lipid-like densities were obtained after classification (Supplementary Figs. 2, 4, 5). To mitigate reference bias, reconstruction of each subclass was performed through ab-initio model building and non-uniform refinement with initial model lowpass-filtered to 30 Å, eventually yielding maps at resolutions from 3.3 to 3.4 Å (see methods). To gain more accurate assessment of how drMfsd2a coordinates these ligands, we plotted the intermolecular Lysolipid2C- and Lysolipid3C-drMfsd2a interactions using the model for the merged ligand map at 2.9 Å resolution (Table S2). A reference with a segment of TM1 truncated was included as a control (Supplementary Fig. 3). Recovery of TM1 segment density from classification and refinement indicates validity of the image processing scheme. We also obtained the lipid-like density for Lysolipid1B using an alternate strategy for particle picking, 2D classification, and 3D reconstruction (see methods).
The maps for drMfsd2a display four densities that cannot be attributed to the proteins chain (Fig. 1e, Supplementary Fig. 5). Closer examination revealed that the elongated densities are surrounded by hydrophobic residues on one side and charged residues at the other end (Figs. 2–3). As such, these elongated densities can each accommodate substrate with a lipid tail and a hydrophilic headgroup (Figs. 2–3, Supplementary Fig. 5). We considered several other ligands that could account for these densities including ALA-LPC, which is known to copurify with Mfsd2a when expressed and purified from SF-9 cells19, and DDM and DM which were used during protein purification. ALA-LPC, rather than DM and DDM, provide better fitting into the elongated densities (Supplementary Fig. 5) and resulted in lower B-factor and more stable binding energy calculations (Supplementary Fig. 10). For Lysolipid1A, the density for the headgroup is weaker than that observed for the other ALA-LPC sites (Supplementary Fig. 5a). This is likely because the LPC headgroup of Lysolipid1A is structurally dynamic in an open cavity between the ZA and ZB sites (Fig. 2a). Further, modeling detergent into the elongated density places the hydrophilic sugar rings of the detergent in the unfavorable hydrophobic cavity of Chamber1 (Supplementary Fig. 11a). This hydrophobic cavity was also observed for binding of the lipid tail of ALA-LPC in the chicken Mfsd2a structure20 and thus unlikely to bind the hydrophilic head of a detergent. It is therefore more likely that a lipid occupies this site (Supplementary Fig. 11b).

a Relative positions of Lysolipid1A-1B in Chamber1 and ZA-B. Lipid Chamber1 is outlined in cyan dotted lines. ZA-B are in dashed cyan and orange lines, respectively. The proposed Na+ binding site is highlighted by dashed purple lines. Protein rendered as surface. Lysolipids1A-1B are in stick and sphere. b Interactions between lipid tail and LPC of Lysolipid1A with residues in Chamber1 and ZA. c Interactions between lipid tail and LPC of Lysolipid1B with residues in Chamber1 and ZB. b, c Carbons of lipid chamber residues are in gray. Carbons of Z-site residues are in black. Black dotted lines represent H-bonding between 2.6 Å and 3.3 Å. Red dotted lines indicate salt-bridges with distances ≤4 Å. Blue half circles indicate choline coordinating residues within 3.5 Å. Red δ indicate residues with mutations that alter activity (Table 1). Water molecule shown as small red sphere.

a Relative positions of Lysolipid2B-3C in Chamber2-3 and ZB-C. Lipid Chambers2-3 are outlined in orange and yellow dotted lines, respectively. ZB-C are in dashed orange and yellow lines, respectively. Protein is rendered as surface. Lysolipids are in stick and sphere. b Interactions between lipid tail and LPC of Lysolipid2B with residues in Chamber2 and ZB. c Interactions between lipid tail and LPC of Lysolipid3C with residues in Chamber3 and Zc. b, c Lipid chamber residues are in gray. Z-site residues are in black. Black dotted lines represent H-bonding between 2.6 Å and 3.3 Å. Red dotted lines indicate salt-bridges with distances ≤4 Å. Red δ and ¥ indicate residues with mutations that alter activity and are associated with disease (see Table 1). Water molecule shown as small red sphere. d Substrate transport tunnel in closed conformation. The Lysolipid1A structure is shown with alternate conformations of the F396 and W400 from structures without lipid tail of ALA-LPC in Chamber2. e Substrate transport tunnel in open conformation with Lysolipid2B bound. d, e Protein rendered as cartoon. Gating residues are in stick. TM10 colored in wheat. f Lysolipid3C (yellow) at the cytoplasmic exit shown adjacent to the DDM belt with detergent molecules shown in stick and ball, and in tan transparent surface.
These results are consistent with the observation that ALA-LPC copurifies with the Mfsd2a when purified from SF-9 insect cells19. The placement of the four ALA-LPC in the elongated densities in drMfsd2a is unique from where this lysolipid was modelled in the structure of the chicken Mfsd2a19 (Fig. 1, Supplementary Fig. 7c). In drMfsd2a, the four lysolipids are positioned along the vertical axis of the substrate tunnel within a cleft between the N- and C-domains comprise of helices 1, 2, 4, 5, 7, 8, 9, 10, and 11 that open to the cytoplasmic side (Fig. 1b–e, Supplementary Fig. 6). Together, the four modeled ALA-LPC binding positions provide a unique opportunity to detail the transport and flippase mechanism of Mfsd2a as the substrate is shuttled through the transporter. Since Mfsd2a transports lipid-LPC from the outer to the inner leaflet, we propose the following order for the four ALA-LPC during substrate transport through Mfsd2a: Lysolipid1A, Lysolipid1B, Lysolipid2B, then Lysolipid3C (Fig. 1e).
Rocker-switch motion
To gain further insights into the differences between known major conformations of the rocker-switch, we mapped the interdomain interactions observed in the outward-facing mouse29, outward-occluded human30, and our inward-drMfsd2a structures (Supplementary Movie 1–2). The interdomain interactions in the mouse and drMfsd2a structures include hydrophobic, salt-bridge, and hydrogen bond contacts located on the (1) extracellular end, (2) cytoplasmic side, (3) between helices 5 and 8 and (4) across helices 2 and 11 of Mfsd2a (Supplementary Movie 1). Strikingly, the rocker-switch motion “rocks” along the vertical axes of TM5 and TM8, and TM2 and TM11 (Supplementary Movie 1–2). We also observe that the outward-facing mouse conformation29 is held in place by more hydrogen bonds and salt-bridges placed at the opposite, cytoplasmic side than observed for interactions at the extracellular end that locks drMfsd2a in the inward-facing state (Supplementary Movie 1).
Rotation of the LPC headgroup
During the transport cycle, as lipids traverse from the outer to the inner leaflet, the substrates must translocate and also invert. As such, it is expected that both the acyl tail and headgroup of lipid-LPC will rotate during lipid flipping and translocation through Mfsd2a. Tracing the relative positions between Lysolipid1A and Lysolipid1B illustrate a significant rotation of the LPC headgroup (Figs. 1d, e, 2, Supplementary Fig. 12a, b). Unlike previous studies that suggest a linear substrate tunnel, our drMfsd2a structures revealed three distinct compartments that each comprise a separate hydrophobic pocket and a charged cavity to house the lipid tail and LPC headgroup (Figs. 2–3). We term the three hydrophobic pockets Chambers1-3 and the three charged cavities the zwitterionic trapsA-C (ZA-C) (Figs. 2–3, Supplementary discussion). Lysolipid1A, with the lipid tail in Chamber1 and LPC in ZA, is the closest substrate to the extracellular side and, hence, likely the first of the four ligand states captured during the substrate transport in our drMfsd2a structures (Figs. 1c–e, 2a, b). Chamber1, also observed in the chicken Mfsd2a structure19, is more extensive here in drMfsd2a (Fig. 2a, b, Supplementary discussion). All the residues in Chamber1 are highly conserved (Supplementary Fig. 6) and mutations at F59, M181, L308, F312, I333, F396, and W400 reduce lysolipid transport by Mfsd2a19,29 (Table 1). Based on their placement in Chamber1 and ZA, the lipid tail and headgroup of Lysolipid1A are both pointed outwards, bent at an ~70° angle (Figs. 1e, 2a, b. Supplementary Fig. 12a). This suggest that lysolipid1A is an intermediate state with lipid tail already rotated to align with the inner membrane leaflet but a headgroup that is yet to be flipped.
Previous studies suggest that Mfsd2a transports lipid-LPC in a Na+ dependent manner6,33. It was proposed that the Mfsd2a symporter uses the flow of Na+ down its electrochemical gradient, from the outside to the inside of the cell, to drive the conformational changes for the transport and flipping of lysolipids. Molecular dynamics and functional analysis of the chicken Mfsd2a proposed a conserved and dynamic Na+ binding site annotated as D87, D91, E309, and K433 (drMfsd2a sequence numbers)19. Similar Na+ binding sites are also proposed in the mouse and human Mfsd2a structures29,30. These studies, however, do not provide clear structural details about how Na+ orchestrates lysolipid transport and flipping, thus require further investigation. In the Lysolipid1A structure, the LPC headgroup is placed adjacent to the previously proposed Na+ binding location6,19,33 (Fig. 2a). Although we are not able to confidently place Na+ at this resolution, the placement of the LPC of lysolipid1A into the elongated lipid-like density adjacent to the previously proposed Na+ site6,19,33 could provide important insights into the Na+-coupled transport mechanism by Mfsd2a. The colocalization of the headgroup and the cation suggest possible direct interaction of the Na+ with LPC, perhaps with the phosphate. In the lysolipid1A configuration, the zwitterion phosphate and choline moieties are rigidified by (1) salt-bridges to D87, R84 and K433, (2) hydrogen bonds to Y50, Q51, and T432, and (3) electrostatic interactions with Q149 and Q152 (Fig. 2b, Supplementary Fig. 12a). This LPC binding arrangement is supported by studies illustrating that Mfsd2a can only transport lysolipids with headgroups that comprise of (1) a negative charge, but not necessarily a phosphate, and (2) an amine moiety20,33. We term the Mfsd2a interactions with the Lysolipid1A LPC as zwitterion trapA (ZA). Consistent with these observations, Y50, Q51, D87, R84, T432, and K433 are highly conserved (Supplementary Fig. 6) and their mutations alters or prohibit Mfsd2a lysolipid transport19,29 (Table 1).
In the second elongated density fitted with Lysolipid1B, we observed a lipid tail that is also tethered in Chamber1 but the LPC headgroup is now translocated by 16.9 Å, rotated by ~70° inwards, bound by a second zwitterion trap, ZB (Figs. 1e, 2a, c, Supplementary Fig. 12b). The ZB site that coordinates the LPC of Lysolipid1B is comprised of (1) two salt-bridges by R180 and E421 to the phosphate and choline moieties and (2) is further rigidified by hydrogen bonding to M181, W400, and Y425 (Fig. 2c, Supplementary Fig. 12b). This LPC binding arrangement in ZB, again, supports the necessity of a headgroup that contains a negatively charged moiety and an amine group for lipid transport by Mfsd2a20,33. All residues that comprise ZB coordination sites are conserved (Supplementary Fig. 6) and mutations of Q51, M181, R180, W400, and E421 reduce Mfsd2a lipid-LPC transport activity19,29,33 (Table 1). Unlike the LPC headgroup, the translocation of C18 of the lipid tail of Lysolipid1B from Lysolipid1A is more modest and still adhered in Chamber1 (Figs. 1a-c, 2a-c). Due to the shift in Lysolipid1B, Chamber1 is modified for binding the lipid tail with fewer hydrophobic interactions (Fig. 2c, Supplementary discussion). Together, the trajectory from Lysolipid1A to Lysolipid1B describes a ~70° rotation and 16.9 Å translocation of the LPC from a head-out to a head-in orientation (Figs. 1d, e, 2, Supplementary Fig. 12a–b). In contrast to Lysolipid1A, which is only partially rotated, both the lipid tail and LPC headgroup of lysolipid1B have been flipped to align with the inner membrane leaflet.
Lipid translocation
After the rotation of the LPC headgroup, we propose that the next steps during lysolipid transport involve the translocation of the flipped lipid-LPC by Mfsd2a towards the cytoplasmic side (Fig. 3). In the Lysolipid2B configuration, the C18 of the ALA lipid tail is displaced 13.9 Å relative to Lysolipid1B (Fig. 1a, c, e) and is bound in Chamber2 located towards the cytoplasmic side of drMfsd2a (Fig. 3a, b). Chamber2 is shifted inward from Chamber1 and is comprised of highly conserved residues (Fig. 3b, Supplementary Fig. 6, Supplementary discussion) and mutation of M181, T188, and I341 disrupt or abolish Mfsd2a transport activity19,29 (Table 1). Unlike the large movement in the lipid tail, the LPC of Lysolipid2B is bound in a similar position to Lysolipid1B by ZB (Figs. 2a, 3a). The phosphate and choline of Lysolipid2B are also held in place by salt-bridges to R180 and E421 but is now shifted more towards the cytoplasmic side, forming hydrogen bonds with H156 (via a water), S160, and E184 (Fig. 3a, b, Supplementary Fig. 12c). Consistent with our findings, H156, S160, R180, E184, and E421 are conserved (Supplementary Fig. 6). Mutation of H156, S160, R180, E184, and E421 disrupt or abolish Mfsd2a transport activity19,26,29 (Table 1). It is noteworthy that mutation of S160 is associated with decreased DHA transport to the brain and results in severe microcephaly26.
Examination of Lysolipid3C revealed that the lipid tail is in a third hydrophobic chamber, Chamber3, located adjacent to the cytoplasmic opening of the drMfsd2a substrate tunnel (Fig. 3a, c). Comparison with Lysolipid2B indicates that the C18 of the lipid tail and choline of Lysolipid3C have translocated inwards by 8.1 and 4.9 Å, respectively, (Figs. 1c–e, 3a). The more modest sized Chamber3 is exposed at the opening of the cytoplasmic side of the drMfsd2a substrate tunnel (Figs. 1e, 3a, c, Supplementary discussion). All residues that comprise Chamber3 are highly conserved (Supplementary Fig. 6) and mutation of I341 and W400 limit or abolish Mfsd2a transport activity19 (Table 1). Similar to the fewer interactions that bind the lipid tail, the LPC of Lysolipid3C is coordinated by a single salt-bridge of the choline by E421 and two hydrogen bonds to D174 and Q345 (Fig. 3c, Supplementary Fig. 12d). Consistent with these observations, D174, Q345, and E421 are highly conserved (Supplementary Fig. 6) and mutation of the latter two decrease or prohibit Mfsd2a lysolipid transport activity19,33 (Table 1). After lipid rotation, the transition from Lysolipid1B to Lysolipid3C describes a 22 Å and 9.4 Å translocation of the C18 of the lipid tail and the choline of the headgroup from the center towards the cytoplasmic side of the drMfsd2a substrate tunnel (Fig. 1c–e).
Open and closed conformations of the Mfsd2a gates
It has been proposed that substrate “gating” of Mfsd2a is comprised of bulky hydrophobic residues that respond to the presence of and orchestrate the conformational changes required for transport of lysolipids19,34. Molecular dynamics simulations of the chicken Mfsd2a proposed “inner gates” that are located within the hydrophobic tunnel consisting of the conserved residues M181, F396, and W40019 (Supplementary Fig. 6). Indeed, we observe movements of F396 and W400 but not M181 that is dependent on the presence of a nearby acyl-chain (Fig. 3d, e). In the Mfsd2a substrate tunnel, F396 and W400 interact with lipid tails in Chamber1-2 and Chamber1-3, respectively (Figs. 2–3). In the structures with the lipid tail absent from Chamber2, F396 and W400 are flexible, occupying multiple conformations that block access to Chamber2 (Fig. 3d). In the presence of lipid tail of Lysolipid2B in the Chamber2, F396 and W400 occupy a single conformation that allow access to the substrate tunnel (Fig. 3e). Here, F396 and W400 are now also engaged in forming van der Waals interactions with the acyl-chain of the Lysolipid2B within Chamber2 (Fig. 3e). We propose that the “gating” interactions with these bulky hydrophobic residues facilitate the translocation of the lipid tail of lipid-LPC from Chamber1 through Chamber3. Consistent with the above postulate, functional analyses coupled with mutagenesis of F396 and W400 reduce Mfsd2a lipid transport19 (Table 1). Moreover, molecular dynamics simulations indicated similar findings where the presence of Na+ and substrate cargo stimulated the opening of the F396 and W400 gates19 to allow lipid tail access to Chamber2.
Lipid release and inner membrane integration
Based on its unique position, the following features observed in our studies suggest Lysolipid3C is poised for release from Mfsd2a for integration into the inner membrane leaflet. First, there are fewer contacts between the lipid tail and drMfsd2a of Chamber3 than Chamber1-2 (Figs. 2–3, Supplementary discussion). Second, there are also minimal contacts between LPC of Lysolipid3C and ZC (Fig. 3c, Supplementary Fig. 12d). Third, Lysolipid3C is placed in a groove at the opening of the drMfsd2a substrate tunnel that is exposed on the cytoplasmic side of the lipid bilayer (Figs. 1c–e, 3a, c, 3f). Fourth, when mapped to the adjacent DDM molecules, Lysolipid3C is closely aligned with the detergent belt that encircles the cytoplasmic side of drMfsd2a, likely resembling where the inner membrane leaflet would interact with the transporter (Fig. 3f). Given these rationales, we propose that Lysolipid3C is poised for release through the cytoplasmic opening of the substrate tunnel of Mfsd2a for integration into the inner membrane leaflet (Figs. 1e, 3f).
Proposed lipid flipping and Mfsd2a cycling
During the transport of the substrate, flippases perform the energetically unfavorable task of flipping the lipids from the outer to the inner membrane leaflet (Fig. 4). To our knowledge, a structural model fully detailing a lipid flipping mechanism has not been elucidated for flippases, particularly for the MFS system. The most well studied lipid flipping system is the P4-ATPase flippase. There are several nucleotide-dependent structures of P4-ATPases with alternate conformations of the soluble domains, however, with only one lipid bound on the extracellular side35. More recently, there is now a P4-ATPase flippase structure bound with the lipid already flipped with the headgroup towards the cytoplasmic side36. The missing gap is how the ATP-dependent structural changes of the soluble domains promote lipid rotation within the P4-ATPase transmembrane region. Further, P4-ATPase and Mfsd2a flippases are different structurally and one uses ATP and the other a Na+ gradient to drive lipid rotation and transport. As such and while it would provide important comparative analysis, these two flippase systems likely employ different mechanisms for lipid flipping and transport. Here, using the five structures determined, together with the previously determined structures of Mfsd2a, we propose that lipid flipping occurs in a series of finely orchestrated steps by which the lipid tail and LPC headgroup are interchangeably anchored by the transporter through interactions with Chambers1-3 and ZA-C that are separated into three distinct compartments as the phospholipid traverses through the substrate tunnel (Figs. 2–4).

a The ligand-bound, outward-facing mouse Mfsd2a structure docked with ALA-LPC is bound in a lateral position Chamber1 (see Supplementary discussion). The LPC headgroup is trapped by ZA in an outward pointing orientation. b The proposed and unknown occluded ligand-bound conformation. Mfsd2a rocks from an outward to inward-facing conformation. c–e The various conformations during flipping of the LPC headgroup. The lipid tail is held in position by Chamber1 while the LPC headgroup samples multiple configurations between ZA and ZB before it is moved to ZB. This movement flips the LPC headgroup from an outward to inward-pointing orientation. This process results in a substrate with the lipid tail pointing out and the LPC headgroup pointing in, a ligand configuration that is aligned with the inner membrane leaflet. f The translocation of the lysolipid to Chamber2 and ZB. The lipid tail is translocated from Chamber1 to Chamber2. The LPC headgroup is shifted inwards. This process translocates the entire lipid-LPC substrate closer to the cytoplasmic exit. g The release of the substrate to the cytoplasmic side. LPC headgroup is moved to ZC and the lipid tail is translocated to Chamber3. Chamber3 and ZC are located at the cytoplasmic exit. h The substrate-free structure of drMfsd2a in the inward-facing conformation. This structure represents the conformation after lysolipid release from Mfsd2a. i–j The conformations during resetting of the transporter. In the absence of the substrate, Mfsd2a resets to the outward-open conformation (j) by first transitioning through a ligand-free occluded state (i).
Since the lipid-LPC translocates from the outer to the inner membrane leaflet, the first anchoring event observed in this study is the transition between Lysolipid1A to Lysolipid1B where the lipid tail is tethered to Chamber1 while the headgroup is free to flip and sample the open cavity from ZA to ZB (Figs. 2, 4c–e, Supplementary Movie 3). Once the headgroup is attached to ZB, as seen for Lysolipid1B (Figs. 2c, 4e), the tail is released from Chamber1 and transferred to Chamber2 where Lysolipid2B is found (Figs. 3b, 4f). While the positions of the four lipid-LPC span the length of the drMfsd2a substrate tunnel (Fig. 1e), key steps are still missing. Namely, it is unclear whether the lipid tail or LPC headgroup anchors first to Chamber3 or Z3 (Figs. 3, 4f, g). Our structures do, however, demonstrate that the lysolipid exits Mfsd2a through an opening of the substrate tunnel on the cytoplasmic side (Figs. 1e, 3f, 4g, Supplementary Movie 3c).
In addition to the substrate, the roles of sodium in lipid transport and flipping remain elusive. It is still unclear whether sodium is involved directly in lipid flipping, however, the close proximity of the LPC of lysolipid1A to the proposed Na+ binding site suggests possible contacts between the cation and the headgroup (Fig. 2a). If true, it is tempting to suggest a model of the coupled movement of Na+ with lipid-LPC down the electrochemical gradient of the cation during translocation and flipping of the lysolipid through Mfsd2a (Figs. 2–3, 4c–f, Supplementary Movie 3). While the cryo-EM maps presented here do display a density at this site, it is unclear at this resolution whether there is indeed a sodium bound. As such, further studies are required to clarify the involvement of the sodium, or lack of, in the lipid flipping mechanism. Together, the lipid tail versus LPC anchoring model provides a mechanistic model where the lipid flips and translocate in a stepwise manner through three separate compartments until it emerges out at the apposing end and inserts into the inner membrane leaflet.
Discussion
In this study, five structures of drMfsd2a were described, including four unique lipid sites. The structures allow us to propose a stepwise lipid flipping mechanism of Mfsd2a. Extensive mutagenesis and functional analyses confirm the importance of key residues identified for lysolipid transport and flipping. While these snapshots allow us to propose a stepwise lipid flipping mechanism, several unanswered questions still remain. For example, is sodium necessary for lipid flipping? If yes, where is the sodium bound? Is this transport mechanism conserved for the various lipid-LPC that can be transported by Mfsd2a? When does acyl-chain inversion occur? Here, we have preliminary docking simulations that suggest that acyl-chain inversion happens before the rotation of the headgroup and before the transition of Mfsd2a from the outward to inward-facing conformation (Supplementary Fig. 13). As such, future studies should further investigate if and how the change in protein conformation, specifically the transition from outward to inward-facing positions, affect lipid flipping and transport. Along the same line, is the protein simply acting as a scaffold for lipid flipping or is the rocker-switch critical in ushering the lipid through this process? To provide a more complete understanding, future studies must delve more deeply into the proposed flippase and lipid transport for Mfsd2a and for other less common lipids that play critical roles in neuromodulation.
Methods
Protein expression and purification
Mfsd2a isoform B from zebrafish (Uniprot Q6DEJ6) was subcloned in the pFasbac1 vector (Invitrogen) with an N-terminal-10xHis tag. The Mfsd2a sequence was truncated at residue D22 of the N-terminus, D509 of the C-terminus, and with the N509Q, N214Q, N225Q mutations to remove post-translational glycosylation modifications. DH10bac was used for preparation of bacmids. Recombinant baculovirus was generated using the Spodoptera frugiperda Sf-9 system and transfection following the protocol provided for the Bac-to-bac Baculovirus Expression System. Mfsd2a-pFasbac1 construct was used to transfect into Sf-9 cells (ThermoFisher, 12659017) following a standard protocol to generate three generations of baculovirus. P3 generation of the Mfsd2a-pFasbac1 baculovirus was used to infect a batch culture of Sf-9 for protein expression.
Mfsd2a was overexpressed in Sf-9 insect cells, harvested 60 h after infection. The cell pellets were resuspended in lysis buffer containing 20 mM Tris (pH 8.0) and 150 mM NaCl and supplemented with EDTA-free protease inhibitor cocktail (Roche). The Sf-9 cells were lysed using 50 homogenizing cycles on ice. The lysate was clarified by centrifugation at 130,000 × g for 1 h. The pelleted membrane was resuspended in high-salt buffer containing 1.6 M NaCl and 20 mM Tris (pH 8.0) for washing and centrifuged again for 1 h at 130,000 × g to remove soluble debris. The pelleted membrane was rapidly frozen in liquid nitrogen and stored at –80 °C until further use.
To purify Mfsd2a, the membrane pellet was solubilized and membrane protein extracted in 2% n-dodecyl-β-d-maltopyranoside (DDM), 20 mM Tris (pH 8.0), 150 mM NaCl, 5% glycerol and 0.2% cholesteryl hemisuccinate Tris salt (CHS) for 4 h at 4 °C. After extraction, the insoluble debris were removed by centrifugation for 1 h at 130,000 × g. 20 mM imidazole (pH 8.2) was added to the supernatant and incubated with TALON beads for 16 h at 4 °C. The Mfsd2a-bound beads were washed with 6 column volumes of 20 mM imidazole, 20 mM Tris (pH 8.0), 500 mM NaCl and 0.1% DDM. The resin was then equilibrated in buffer composed of 20 mM Tris (pH 8.0), 150 mM NaCl, 0.4% decyl-β-d-maltoside (DM), and 0.02% DDM. At 4 °C, the 10 × His tag was removed by on-column TEV digestion overnight at an enzyme:protein molar ratio of 1:500. The cleaved Mfsd2a was collected in the flow-through and was flash frozen in liquid nitrogen and stored at −80 °C until further use.
FAB production and purification
Mouse IgG monoclonal antibodies against drMfsd2a were produced by the Monoclonal Antibody Core (D. Cawley). 330 µg of purified Mfsd2a in buffer containing 20 mM Tris (pH 8.0), 150 mM NaCl, 0.02% DDM and 0.002% CHS was used to immunize mice in three injections. 15 × 96-well plate fusions yielded 169 IgG-positive wells at a 1:30 dilution. Native and denatured Mfsd2a proteins were then used in ELISA to search for candidates that bound the conformational epitopes27, where Ni-NTA plates were used for Mfsd2a immobilization. Thirty-five of the 169 fusions showed significant preference for binding against well-folded Mfsd2a. Western blotting was performed to assess the binding affinity and specificity of the antibodies generated from hybridoma cell lines. Monoclonal antibody 11D3 was then purified from the hybridoma supernatants by 4-mercaptoethylpyridine (MEP) chromatography. FAB fragments were produced by papain digestion and purified in flow-through buffer containing 20 mM NaPi (pH 8.0) and 150 mM NaCl by protein A affinity chromatography.
Mfsd2a-FAB complex assembly
Purified 15A9 FAB fragment was incubated overnight at 4 °C with Mfsd2a at a 5:1 molar ratio. Mfsd2a–15A9 complex was injected into the size exclusion column Superdex200 to separate the unbound FAB. The size exclusion column step was also used to exchange the buffer of the drMfsd2a-FAB complex into 50 mM HEPES (pH 8.0), 150 mM NaCl and 0.02% DDM. SDS-PAGE analysis was used to pool peak fractions containing both Mfsd2a and FAB, indicating complex formation (Supplementary Fig. 1). The pooled fractions containing Mfsd2a-FAB were pooled and concentrated to 3 mg/ml using an Amicon spin concentrator with a 30 kDa cutoff.
Mfsd2a-FAB Cryo-EM grid preparation
400-mesh 1.2/1.3 Cu or 300-mesh UltrAuFoil 1.2/1.3 Au grids (Quantifoil) were made hydrophilic by glow discharging for two times 60 seconds with a current of 15 mA in a PELCO easiGlow system. The cryo grids were prepared using a Leica EM GP (Leica). The chamber was kept at 4 °C and 95% humidity (86–91% measured). Three microliters of sample at 3 mg/ml were applied to a glow-discharged holey grids, blotted for 6 s, and plunge frozen into liquid ethane and stored in liquid nitrogen.
Mfsd2a-FAB cryo-EM data collection
Cryo-EM grids were loaded into a 300 keV FEI Titan Krios cryo-electron microscope (ThermoFisher Scientific, formerly FEI) at HHMI Janelia Reasearch Campus, Janelia Krios 1, equipped with a Cs corrector, and Gatan energy filter and K3 camera (Gatan Inc.). Movies of 50 frames with 1 e−/Å2 per frame (50 e−/Å2 total dose) were automatically recorded at a nominal magnification of ×81,000, corresponding to a physical pixel size of 0.844 Å/px (superresolution pixel size 0.422 Å/px) in CDS mode at a dose rate of 9.5 e−/px/s (~7.5 e−/px/s on the camera through the sample) and a defocus range of −0.5 to −1.8 µm using SerialEM37. In total, 2653 and 8443 movies were collected in two separate imaging sessions, with the first dataset on a 400-mesh copper grid and the second on a 300-mesh UltrAuFoil gold grid (Table S1).
Mfsd2a-FAB cryo-EM data processing
The overall workflow of the image processing is illustrated in Supplementary Fig. 2. All preliminary steps were performed within RELION 3.138 unless specified. Movie alignment and dose weighting were performed with MotionCor239 in a 5 × 5 patch. Contrast transfer function (CTF) parameters were estimated with Gctf40. Good 2D classes generated from ~1000 manually picked particles served as templates for automatic particle picking in RELION, resulting in 623,644 and 3,033,688 particles in dataset 1 and dataset 2, respectively. Particle images extracted were 4× binned, resulting in a pixel size of 3.376 Å, and then subjected to several rounds of 2D classification and 3D classification, removing junk particles and particles without FAB bound. Particles were re-extracted, unbinned to a pixel size of 0.844 Å, and subjected to 3D auto-refinement with local angular searches (RELION additional arguments: --sigma_ang 1.667) followed by Bayesian polishing. Polished particles from 2 datasets were imported into cryoSPARC v3.3.241. For dataset 1, 146,914 particles were subjected to CTF refinement to correct for beam-tilt, spherical aberrations, and per-particle defocus parameters, followed by non-uniform refinement resulting in a map at 3.3 Å. For dataset 2, 346,394 particles were first cleaned up by heterogenous refinement, followed by CTF refinement and non-uniform refinement resulting in a map at 3.2 Å. Two datasets were merged and further classified by heterogenous refinement and refined to 2.9 Å with local resolution regions up to 2.4 Å using non-uniform refinement and local resolution filtering. Altogether, more than 60 maps (from cryoSPARC) between 2.9 and 4.13 Å were created using different strategies of processing the data using cisTEM42, RELION38, and cryoSPARC41, showing different degrees of details, especially around the ligand binding sites.
Densities for three ligands in the 2.9 Å map from 295,580 particles were clearly observed in the cavities of Mfsd2a which are unlikely to co-exist simultaneously. We hypothesized that the three ligand densities shown in that particular density map are a result of merged particle populations where subsets with different ligand positions have not been classified due to the small ligand size. To sort out subsets with different ligand positions, 3D variability analysis43 and 3D classification without alignment using a tight mask covering the ligand binding sites were used without obvious success. Alternatively, reference-based 3D classification without alignment was deployed. Individual ligand densities were segmented and extracted with UCSF Chimera44. Extracted ligand densities were then subtracted from the Mfsd2a-FAB map, generating references with (1) 3 ligands bound, (2) no ligand-bound, (3) only ligand 1 bound, (4) only ligand 2 bound, and (5) only ligand 3 bound, respectively. These maps served as references for 3D classification without alignment, with a tight mask covering ligand binding sites included. To avoid reference bias and overfitting, individual particle subsets obtained from 3D classification were subjected to ab-initio reconstruction, followed by non-uniform refinement, with initial model lowpass-filtered to 30 Å, resulting in maps at 3.3 Å, 3.4 Å, 3.3 Å, 3.4 Å, and 3.4 Å, with local resolutions to up to 2.8 Å (3 ligands, lower resolution), 2.8 Å (ligand-free), 2.8 Å ( A), 2.9 Å (2B), and 2.9 Å (3C), respectively (Supplementary Fig. 2, Supplementary Table 1). To ensure model bias was not inherited throughout the workflow after classification without alignment, a control experiment was performed. A Mfsd2a-FAB map and a map with TM1 segment subtracted were used for classification without alignment, followed by ab-initio reconstruction and non-uniform refinement (Supplementary Fig. 3). The results showed that the TM1 density was clearly observed in both classes and the maps after refinement of each subclass, demonstrating reference bias was mitigated throughout the workflow applied in the study. In addition, we imported the particles of the individual subclasses from cryoSPARC into RELION. 3D refinement of the individual subclass with an initial model lowpass-filtered to 40 Å yielded maps at 3.8–4.1 Å, which also showed individual ligand density. The map with the clearest Lysolipid1B configuration was generated from 413,435 particles using a combination of processing in cisTEM42 and cryoSPARC41 and has an average resolution of 4.1 Å (Table S1). The local resolution of all maps was additionally estimated using RELION’s own implementation where half maps and masks from cryoSPARC were imported as inputs (Table S1). Six of the >60 maps have been made available and have the following accession codes: EMD-27148, EMD-27149, EMD-27150, EMD-27151, EMD-27152, and EMD-27153.
Model building and refinement
The initial model fitting was achieved by manual rigid body rotation and translation and the “Fit Map” of the chicken Mfsd2a structure19 (PDB 7MJS) into the 2.9 Å electron density maps for drMfsd2a in UCSF Chimera44. The protein sequence of the structures was altered to match drMfsd2a and manually fitted into the maps in Coot45. Building of the FAB portion of the structure was achieve similar to drMfsd2a starting with the FAB model from the SLC38A9-FAB46 structure (PDB 6C08). Refinement of the drMfsd2a structure was accomplished by iterative cycles of manual flexible fitting and automatic refinement in Coot45 and Real Space Refine in Phenix47,48, respectively. The final structure validation was carried out in Real Space Refine in Phenix48 (Table S1). The fitted pdbs have been made available together with the EM maps with the following PDB IDs: 8D2S, 8D2T, 8D2U, 8D2V, 8D2W, and 8D2X. Figures and movies were generated in Pymol or UCSF Chimera44.
In vitro endogenous lipid analysis
Unsaturated free fatty-acids were detected and quantified using the Lipid Assay kit (abcam ab242305). Purified drMfsd2a was concentrated to 15 mg/mL and used for lipid quantification according to manufacturer’s instructions. Briefly, 15 μL of samples were dispensed in tubes and incubated at 90 °C for 30 min, then 4 °C for 5 min. 150 μL of 18 M sulfuric acid was added to each sample. Samples were then incubated for 10 min at 90 °C, then 5 min at 4 °C. One hundred microliters of each sample were transferred to a 96-well plate and mixed with 100 μL of vanillin and incubated at 37 °C for 15 min. Absorbance was measured at OD 560 nm. Data reported are mol-to-mol ratio of three technical replicates calculated using molecular weights 517.6 g/mol for lipids and 56.6 kDa for drMfsd2a. The standard curve for phospholipid quantification was generated using the standards from the Lipid Assay Kit.
Proteoliposome reconstitution
Chloroform-dissolved chicken egg phosphatidylcholine (egg-PC, Avanti Polar Lipids) was evaporated using dry nitrogen to yield a thin lipid film in a small glass vial and further desiccated under vacuum overnight. The lipids were hydrated in inside buffer (20 mM Tris (pH 8.0), 150 mM KCl, 5 mM NaCl) at 25 mg/mL by vortexing for 3 min and then aged at room temperature for 1 h. Liposomes were clarified by 5 rounds of freezing and thawing in liquid nitrogen and extruded through a 100 nm membrane filter (Millipore) with 21 passes. The liposomes were pre-incubated with 1% n-octyl-β-D-glucoside (β-OG) and 1 mM DDT for 1 h at 4 °C before protein reconstitution. Purified Mfsd2a protein was incorporated at 1:100 (w/w) ratio into destabilized liposomes for 1 h in a 4 °C rotator. Five percent glycerol-supplemented protein buffer was used in lieu of Mfsd2a protein in liposome-only control groups. The detergents were removed by incubation overnight with 250 mg per reaction of Bio-Beads, and the proteoliposomes were further incubated with 50 mg per reaction of fresh Bio-Beads for an additional hour. The proteoliposomes and liposomes-only controls were collected using ultracentrifuge at 100,000 × g for 30 min at 4 °C and then resuspended in outside buffer (20 mM Tris (pH 8.0), 150 mM NaCl, 5 mM KCl) to a final lipid concentration of 32 μg/μL.
Proteoliposome [14C]DHA-LPC uptake assay
Transport reactions were initiated by adding 0.1 μCi [14C]DHA-LPC (American Radiolabeled Chemicals, Inc) to 30 μL aliquots of proteoliposomes to a final concentration of 60 μM [14C]DHA-LPC. Assay of Liposome-only controls were carried out in parallel to experimental groups as negative controls. All buffers were chilled, and assays were performed at 30 °C in a metal bath.
For the steady-state uptake assay, after 10 min incubation, proteoliposomes were filtered, washed by 5 mL of ice-cold outside buffer, and collected on 0.22 μm nitrocellulose membrane filters (Millipore) which had been pre-wet with washing buffer (PBS pH 8.0). After washing, each filter was dried by vacuum for exactly 1 min and transferred into a glass vial with 10 mL scintillation fluid for counting. Non-specific adsorptions of [14C]DHA-LPC by liposomes-only controls were shown as control groups. The experiment was repeated three times and figures show the results with S.E.M error bar.
Computational analysis of ALA-LPC versus DDM binding
For binding energy comparisons between the lysolipid and DDM modeled in each of the liganded states, pdb coordinates were prepared in MOE (Molecular Operating Environment) with the default simulation forcefield Amber10:EHT with R-Field solvation. QuickPrep-Protonate3D is used to prepare the PDB coordinates for adding hydrogens and assigning protonation states. Atom constraints were applied to the coordinates to fix the modeled positions in the electron density maps. Ligand properties were calculated after energy minimization for the chosen ligands.
Molecular docking for ALA-LPC in the outward-open conformation of mouse Mfsd2a
The molecular docking of ALA-LPC in the outward-open conformation of mouse Mfsd2a31 (PDB 7N98) was calculated with the Chimera Autodock Vina tool47. The docking was performed within the defined bounding box in the extracellular access of the transmembrane domain, which included a volume of ~30,000 A3 as the search space. Ten docking poses were generated with exhaustiveness of search set at 8, maximum energy difference (kcal/mol) set at 2. The resulting poses yielded scores ranging from 6.2 to −5.7 kcal/mol. The best one was chosen following biochemical rationales.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon request. Cryo-EM maps have been deposited in the Electron Microscopy Data Base (EMDB) under accession codes EMD-27148 (Merged Lysolipid1A,2B,3C), EMD-27149 (Ligand-free), EMD-27150 (Lysolipid1A), EMD-27151 (Lysolipid1B), EMD-27152 (Lysolipid2B), and EMD-27153 (Lysolipid3C). The atomic coordinates have been deposited in the Protein Data Bank (PDB) under accession codes 8D2S (Merged Lysolipid1A,2B,3C), 8D2T (Ligand-free), 8D2U (Lysolipid1A), 8D2V (Lysolipid1B), 8D2W (Lysolipid2B), and 8D2X (Lysolipid3C).
References
Stillwell, W. & Wassall, S. R. Docosahexaenoic acid: membrane properties of a unique fatty acid. Chem. Phys. Lipids 126, 1–27 (2003).
Sastry, P. S. Lipids of nervous tissue: composition and metabolism. Prog. Lipid Res. 24, 69–176 (1985).
Boucher, O. et al. Neurophysiologic and neurobehavioral evidence of beneficial effects of prenatal omega-3 fatty acid intake on memory function at school age. Am. J. Clin. Nutr. 93, 1025–1037 (2011).
Stordy, B. J. Dark adaptation, motor skills, docosahexaenoic acid, and dyslexia. Am. J. Clin. Nutr. 71, 323–326 (2000).
Lacombe, R. J. S., Chouinard-Watkins, R. & Bazinet, R. P. Brain docosahexaenoic acid uptake and metabolism. Mol. Aspects Med. 64, 109–134 (2018).
Nguyen, L. N. et al. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509, 503–506 (2014).
Zihni, C., Mills, C., Matter, K. & Balda, M. S. Tight junctions: from simple barriers to multifunctional molecular gates. Nat. Publ. Gr. https://doi.org/10.1038/nrm.2016.80 (2016).
Lochhead, J. J., Yang, J., Ronaldson, P. T. & Davis, T. P. Structure, function, and regulation of the blood-brain barrier tight junction in central nervous system disorders. Front. Physiol. 11, 914 (2020).
Greene, C. & Campbell, M. Tight junction modulation of the blood brain barrier: CNS delivery of small molecules. Tissue Barriers 4, 1–10 (2016).
Prescher, M. et al. Evidence for a credit-card-swipe mechanism in the human PC floppase ABCB4. Structure 29, 1144–1155.e5 (2021).
Nintemann, S. J., Palmgren, M. & López-Marqués, R. L. Catch you on the flip side: a critical review of flippase mutant phenotypes. Trends Plant Sci. 24, 468–478 (2019).
Andersen, J. P., Vestergaard, A. L., Mikkelsen, S. A. & Mogensen, L. S. P4-ATPases as phospholipid flippases—structure, function, and enigmas. Front. Physiol. 7, 1–23 (2016).
Hankins, H. M. et al. Role of flippases, scramblases, and transfer proteins in phosphatidylserine subcellular distribution. Traffic 16, 35–47 (2015).
Zhang, T. et al. TMEM41B and VMP1 are phospholipid scramblases. Autophagy 17, 2048–2050 (2021).
Saier, M. H. et al. The major facilitator superfamily. J. Mol. Microbiol. Biotechnol. 1, 257–279 (1999).
Quistgaard, E. M., Löw, C., Guettou, F. & Nordlund, P. Understanding transport by the major facilitator superfamily (MFS): Structures pave the way. Nat. Rev. Mol. Cell Biol. 17, 123–132 (2016).
Law, C. J., Maloney, P. C. & Wang, D. Ins and outs of major facilitator superfamily antiporters. Ann. Rev. MicroBiol. 62, 289–305 (2008).
Angers, M., Uldry, M., Kong, D., Gimble, J. M. & Jetten, A. M. Mfsd2a encodes a 14 novel major facilitator superfamily domain-containing protein highly induced in brown 15 adipose tissue during fasting and adaptive thermogenesis. Biochem. J. 23, 1–7 (2008).
Cater, R. J. et al. Structural basis of omega-3 fatty acid transport across the blood–brain barrier. Nature 595, 315–319 (2021).
Vu, T. M. et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature 550, 524–528 (2017).
Harvat, E. M. et al. Lysophospholipid flipping across the Escherichia coli inner membrane catalyzed by a transporter (LplT) belonging to the major facilitator superfamily. J. Biol. Chem. 280, 12028–12034 (2005).
Mendoza, A. The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep. https://doi.org/10.1016/j.celrep.2012.09.021.The (2012).
Chan, J. P. et al. The lysolipid transporter Mfsd2a regulates lipogenesis in the developing brain. PLoS Biol. 16, 1–30 (2018).
Huang, B. & Li, X. The role of Mfsd2a in nervous system diseases. Front. Neurosci. 15, 1–9 (2021).
Ben-Zvi, A. et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509, 507–511 (2014).
Guemez-Gamboa, A. et al. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat. Genet. 47, 809–813 (2015).
Harel, T. et al. Homozygous mutation in MFSD2A, encoding a lysolipid transporter for docosahexanoic acid, is associated with microcephaly and hypomyelination. Neurogenetics 19, 227–235 (2018).
Zhou, J. et al. Zika virus degrades the w -3 fatty acid transporter Mfsd2a in brain microvascular endothelial cells and impairs lipid homeostasis. Sci. Adv. 5, eaax7142 (2019).
Wood, C. A. P. et al. Structure and mechanism of blood–brain-barrier lipid transporter MFSD2A. Nature 596, 444–448 (2021).
Martinez-Molledo, M., Nji, E. & Reyes, N. Structural insights into the lysophospholipid brain uptake mechanism and its inhibition by syncytin-2. Nat. Struct. Mol. Biol. 29, 604–612 (2022).
Nygaard, R., Kim, J. & Mancia, F. Cryo-electron microscopy analysis of small membrane proteins. Curr. Opin. Struct. Biol. 64, 26–33 (2020).
Kobayashi, N. et al. MFSD2B is a sphingosine 1-phosphate transporter in erythroid cells. Sci. Rep. 8, 1–11 (2018).
Quek, D. Q. Y., Nguyen, L. N., Fan, H. & Silver, D. L. Structural insights into the transport mechanism of the human sodium-dependent lysophosphatidylcholine transporter MFSD2A. J. Biol. Chem. 291, 9383–9394 (2016).
Drew, D., North, R. A., Nagarathinam, K. & Tanabe, M. Structures and general transport mechanisms by the major facilitator superfamily (MFS). Chem. Rev. 121, 5289–5335 (2021).
Hiraizumi, M., Yamashita, K., Nishizawa, T. & Nureki, O. Cryo-EM structures capture the transport cycle of the P4-ATPase flippase. Science 365, 1149–1155 (2019).
Bai, L. et al. Transport mechanism of P4 ATPase phosphatidylcholine flippases. Elife 9, 1–20 (2020).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, 1–22 (2018).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. CryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Grant, G., Rohou, A. & Grigorieff, N. cisTEM, user-friendly software for single- particle image processing. Cancer Res. 36, 1883–1885 (1976).
Punjani, A. & Fleet, D. J. 3D variability analysis: resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 213, 107702 (2021).
Pettersen, E. F. et al. UCSF Chimera−a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics research papers. Acta Crystallogr. Sect. D https://doi.org/10.1107/S0907444904019158 (2004).
Lei, H., Ma, J., Martinez, S. S. & Gonen, T. Crystal structure of arginine-bound lysosomal transporter SLC38A9 in the cytosol-open state. Nat. Struct. Mol. Biol. 25, 522–527 (2018).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221 (2010).
Afonine, P. V. et al. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. Sect. D Struct. Biol. 74, 531–544 (2018).
Acknowledgements
We thank Rui Yan at the HHMI Janelia research campus for her help with data collection. We also thank the Janelia Research Campus cryo-EM center for the use of their electron microscope. The Gonen laboratory is supported by funds from the Howard Hughes Medical Institute and by the National Institutes of Health P41-GM136508. This research was supported by the Division of Intramural Research of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH (grant NICHD intramural project ZIA HD008998). This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Author information
Authors and Affiliations
Contributions
C.N., H.T.L., and M.J.G. performed cloning, protein expression, purification, antibody generation and complex formation, and initial screening by cryo-EM; C.N., H.T.L., M.J.G., and D.M. performed cryo-EM screening and data collection; C.N., H.T.L., L.T.F.L., M.J.G., and D.M. performed single-particle reconstructions, worked on model building and refinement. M.J.G. performed lipid detection experiments. C.N. and X.M. performed drMfsd2a proteoliposome reconstitution and in vitro uptake assays. C.N. and H.T.L. performed energetic calculations. All authors participated in manuscript preparation, figure preparation, and interpretation of the data and analyses. H.T.L. and T.G. conceived the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nguyen, C., Lei, HT., Lai, L.T.F. et al. Lipid flipping in the omega-3 fatty-acid transporter. Nat Commun 14, 2571 (2023). https://doi.org/10.1038/s41467-023-37702-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37702-7
This article is cited by
-
Structural basis of Sphingosine-1-phosphate transport via human SPNS2
Cell Research (2023)
-
Molecular basis of Spns2-facilitated sphingosine-1-phosphate transport
Cell Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
