
Abstract
Recently, layered kagome metals AV3Sb5 (A = K, Rb, and Cs) have emerged as a fertile platform for exploring frustrated geometry, correlations, and topology. Here, using first-principles and mean-field calculations, we demonstrate that AV3Sb5 can crystallize in a mono-layered form, revealing a range of properties that render the system unique. Most importantly, the two-dimensional monolayer preserves intrinsically different symmetries from the three-dimensional layered bulk, enforced by stoichiometry. Consequently, the van Hove singularities, logarithmic divergences of the electronic density of states, are enriched, leading to a variety of competing instabilities such as doublets of charge density waves and s- and d-wave superconductivity. We show that the competition between orders can be fine-tuned in the monolayer via electron-filling of the van Hove singularities. Thus, our results suggest the monolayer kagome metal AV3Sb5 as a promising platform for designer quantum phases.
Similar content being viewed by others


Introduction
The kagome lattice refers to a two-dimensional (2D) planar crystal composed of corner-sharing triangles. Unique electronic structures emerge owing to the geometrical frustration of the lattice, featuring a flat band, a pair of Dirac points, and saddle-point van Hove singularities (VHSs). A prominent example of candidate kagome metals is the recently discovered vanadium-based kagome metals AV3Sb5 (A = K, Rb, and Cs)1,2. A cascade of correlated electronic states have been observed in AV3Sb5, associated with charge density waves (CDWs)2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17 and superconductivity2,14,15,16,17,18,19,20,21,22,23. These phases are reported to be accompanied by concomitant unexpected properties such as giant anomalous Hall effects24,25 without long-ranged magnetic ordering26, potential Majorana zero modes14, and edge supercurrent18. The VHSs in conjugation with the Coulomb interaction is suggested as an impetus of the unconventional properties27,28.
Although many outstanding materials have been found to comprise a kagome lattice in a layered form29,30,31, the kagome lattice in genuine two dimensions is rare in nature. This scarcity leads to prior explorations of the kagome materials based on the assumption that a three-dimensional (3D) layered system can be regarded as decoupled kagome layers. Similarly, in the case of the vanadium-based kagome metals, current experiments are mainly focused on the 3D layered structures1,2,2,3,4,5,11,12,13,14,15,16,18,19,21,22,23,24,25,26, while their theoretical analysis largely relies on an effective kagome model in two dimensions6,7,8,9,20. The dimensionality has been tacitly assumed as an irrelevant parameter, but this assumption has been generically refuted in layered systems32,33. A few research groups have made pioneering efforts to tackle this issue by successfully exfoliating thin films of AV3Sb534,35,36. However, the importance of dimensionality in this family of kagome metals has remained elusive to date.
In this work, we theoretically demonstrate that the AV3Sb5 monolayer is different from the 3D layered bulk by performing density-functional theory (DFT) and mean-field theory (MFT) calculations. At the crux of our results is the absence of a dimensional crossover between the monolayer and the layered bulk. We argue that symmetry-lowering is inevitable in the monolayer, enforced by the stoichiometry of AV3Sb5. The reduced symmetries give rise to significant changes in the formation of VHSs. Notably, unconventional VHSs appear, referred to as type-II VHSs. As a consequence, enhanced electronic instabilities are observed, leading to the emergence of competing orders such as CDW doublets, time-reversal breaking CDWs, s- and d-wave superconductivity. Our calculations predict that the AV3Sb5 monolayer can be thermodynamically stable. The stable AV3Sb5 monolayer becomes a unique platform to study the intriguing interplay between the VHSs and competing order parameters because any monolayer systems are under significantly enhanced fluctuations, as manifested in the celebrated Mermin-Wagner theorem37,38,39. In connection with future experiments, we also calculate the anomalous Hall conductivity that can probe the correlated orders. Possible experimental schemes are discussed to tune the electron-filling based on mechanical and chemical treatment.
Results
Crystal structure and symmetry
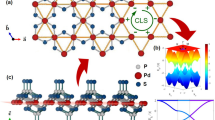
We begin by elucidating the similarities and differences between the crystal structures of the bulk and monolayer AV3Sb5 (A = K, Rb, Cs). As delineated in Fig. 1a and b, both systems comprise multiple sub-layers. Most importantly, a 2D kagome sub-layer is formed from V atoms, coexisting with Sb sub-layers. While these are similar in both systems, differences arise from the alkali atoms A. In the monolayer (bulk) system, alkali metals energetically favor to form rectangular (triangular) sub-layers shown in Fig. 1b(a) (see Supplementary Note 1 for the detailed analysis of the energetics using first-principles calculations). The different formation of alkali atoms is traceable to the stoichiometry of AV3Sb5. The kagome layer of the monolayer takes all the electrons donated from the alkali atoms, while those in the bulk system are shared between the adjacent two sub-layers. Therefore, to preserve the stoichiometry, the number of neighboring alkali atoms is halved by doubling the unit cell, such that they form rectangular sub-layers.

a, b Atomic structures of the bulk and monolayer AV3Sb5 (A = K, Rb, Cs). The monolayer structure preserves \(\sqrt{3}\times 1\) translational and D2h point group symmetries. The layered bulk preserves 1 × 1 translational and D6h point group symmetries. Black solid lines indicate the primitive unit cells and blue dashed lines indicate the y-axis. In b, dashed open circles represent vacant alkali sites. c Schematic illustration of the VHS points in momentum space rearranged by symmetry lowering. The type-I (type-II) VHS points are marked by blue (red) circles. In the middle and right panels, the reduced (pristine) BZ of the \(\sqrt{3}\times 1\) (1 × 1) unit cell is indicated by solid (dashed) line. The bottom panels delineate the energy level of the VHS points.
The rectangular sub-layer with the doubled unit cell breaks translational and rotational symmetries of bulk AV3Sb5. The translational symmetry \({{{{{{{{\mathscr{T}}}}}}}}}_{1\times 1}\) is reduced to \({{{{{{{{\mathscr{T}}}}}}}}}_{\sqrt{3}\times 1}\), and correspondingly, a three-fold rotational symmetry C3z is lifted. This reduces the D6h symmetry of the bulk to D2h in the monolayer. We stress that the lowered symmetry of the monolayer AV3Sb5 is the fundamental symmetry, which can be stable even at room temperature protected by energy barriers from the stoichiometry enforcement, while the same symmetry is only reachable at low temperature from a cascade of phase transitions in bulk AV3Sb54,15. We also note that any incommensurate CDW (IC-CDW) orders are prohibited at non-zero temperatures in monolayer systems unless either long-range interactions or substrate effects play a significant role. Thus, a dimensional crossover is unlikely to occur from the bulk to the monolayer AV3Sb5, and unique properties arise as a result.
Rearrangement of VHSs
Most importantly, the lowered symmetry of the monolayer AV3Sb5 leads to the rearrangement of the VHS in energy-momentum space. The mechanism of the rearrangement is illustrated in Fig. 1c, in which we trace the k-points that host the VHS. Hereafter, we refer to these momenta as the VHS points. The pristine BZ with the \({{{{{{{{\mathscr{T}}}}}}}}}_{1\times 1}\) translational symmetry initially hosts three inequivalent VHS points at Mi (i = 1, 2, 3), as in the case of the bulk AV3Sb5 (left panel in Fig. 1c). Upon the zone folding by lowering \({{{{{{{{\mathscr{T}}}}}}}}}_{1\times 1}\) to \({{{{{{{{\mathscr{T}}}}}}}}}_{\sqrt{3}\times 1}\), the M1 VHS point is folded to Γ and the M1 and M2 VHS points are merged to the M point of the reduced BZ (middle panel in Fig. 1c). The symmetry-lowering further hybridizes the two states at M (M2 and M3), such that it annihilates the VHS at M and creates four new VHS points off M, marked as Pi (i = 1, 2, 3, 4) in the right panel of Fig. 1c.
The rearrangement of VHS points is observed in our first-principles calculations. Figure 2a, b show exemplary DFT bands of KV3Sb5 with archetypal kagome bands distilled by our tight-binding theory (see Methods for the details of the TB model). In Fig. 2b, the divergence of the density of states (DOS) is clearly observed at E = −6( + 9) meV. A close inspection reveals that the diverging DOS at E = −6 meV arises from off high-symmetry momenta at Pi = M + ( ± 0.054, ± 0.021) Å−1 (Fig. 2d), while the divergence at E = 9 meV arises from the exact high-symmetry Γ point (Fig. 2c). In this respect, the VHS points at E = 9 meV (E = − 6 meV) belong to the type-I (type-II) class, where the type-I (II) refers to a class of VHSs that originates from (off) time-reversal invariant momenta40,41,42. Our calculations further reveal that the type-II VHSs generically occur in the kagome metals regardless of A = K, Rb, and Cs (see Supplementary Note 2 for the DFT results of the RbV3Sb5 and CsV3Sb5 monolayers).

a DFT and TB band structures of the KV3Sb5 monolayer. The DFT (TB) bands are colored by black (red). The kagome bands are reproduced by using our TB model (see Methods for the details of the TB model). A solid rectangle (dashed hexagon) in the inset depicts the reduced (pristine) BZ of the \(\sqrt{3}\times 1\) (1 × 1) unit cell. b (Left panel) Magnified view of the blue box in a. (Right panel) Density of states (DOS) of KV3Sb5. The type-I and type-II VHSs are indicated by blue and red arrows, respectively. c, d Energy contours of the kagome bands calculated from the TB bands. Solid (dashed) white lines show the reduced (pristine) BZ. The contour curves in c, d correspond to E = 9 and − 6 meV, respectively. The saddle points reside at the crossing points of the contour curves in c, d. The magnified view of the red box in d illustrates the position of type-II VHS points off M, marked by Pi (i = 1, 2, 3, 4).
The emergence of the type-II VHS is one of the key features of the monolayer AV3Sb5. Few remarks are as follows. First, the appearance of the type-II VHS off time-reversal invariant momenta is understood as a generic result attributed to the band hybridization allowed by the symmetry lowering. Since the two VHSs at M2 and M3 before the symmetry lowering are at the exact same energy level, they can be mixed together and contribute to generating the type-II VHSs (See Fig. 1c). On the other hand, the VHS at M1 still remains a type-I VHS because no states are available to hybridize with. We find that the number of the generated type-II VHS points (in this case, four) is not universal and depends on the microscopic details of the system (see details in Supplementary Note 3.2). Second, the type-II VHS points Pi (i = 1, 2, 3, 4) (Fig. 2d) consist of a mixed contribution from both the B and C sublattices (Supplementary Note 3, Fig. S5), referred to as a mixed-type flavor12,20,43. This is in contrast to the type-I VHS point at Γ (Fig. 2c), which is purely contributed from the A sublattice, referred to as a pure-type flavor. Third, the increased number of VHS points quantitatively changes the characteristic of the diverging DOS at − 6 meV. Namely, the peak at − 6 meV is significantly enhanced than that of the type-I VHS at 9 meV (Fig. 2b), contributed from the quartet VHS points at Pi (i = 1, 2,3, 4). Such quantitative changes are of immediate impact on the electronic properties, such as instabilities driven by the type-II VHS, as we will show below.
Instability of the monolayer
The pristine monolayer harbors intrinsic instability captured in the electronic susceptibility. Figure 3a–d show the real part of the bare static charge susceptibility χ(q) at various fillings (see Methods). We find that the presence of both type-I and type-II VHSs leads to diversified instability sensitively depending on electron filling. For example, at the type-II VHS filling (Fig. 3a), a significant contribution arises from the q vectors that connect the van Hove saddle points. The peaks at q1 and q2 in χ(q) correspond to the nesting vectors mediating distinct Pi points. The presence of these q vectors that are incommensurate to the reciprocal lattice vector necessitates the consideration of an incommensurate order parameter, such as IC-CDW phases. By contrast, when the chemical potential increases, the peaks quickly merge and give rise to the significant enhancement of χ(q) arises at the M point (Fig. 3b–d). This significant peak at q = M, as well as at q = Γ in the folded BZ, corresponds to the commensurate 2 × 2 CDW vectors. Notably, the large values of χ(q) at M survive for a wide range of chemical potentials. These results suggest a significant role of 2 × 2 CDW orders in stabilizing the monolayer. Moreover, the sublattice character of χ(q) reveals that sublattice interference is significant in the monolayer (See Supplementary Note 4 for the calculations of sublattice-resolved susceptibility). This result warrants the consideration of longer-range interactions, which promotes unconventional orders as in the bulk cases6,20,43,44.

a–c Electronic susceptibility χ(q) is calculated in 2D q-space for a the type-II VHS filling (μ = − 6 meV), b neutral filling (μ = 0 meV), and c type-I VHS filling (μ = 9 meV). The right panels show a magnified view of the boxed areas of the left panel near the high-symmetric Γ and M points, respectively. q1 and q2 in a represent the nesting vectors that connect the type-II VHS points (black circles). d Line profiles of χ(q) along the high-symmetry lines of the BZ for various chemical potentials. The corresponding chemical potential (in meV) is shown in each profile. The momenta correspond to q1 and q2 vectors in a are indicated by arrows with vertical lines. e Phonon bands of the pristine KV3Sb5 monolayer as a function of electronic temperature. The electronic temperature effect is introduced by tuning the smearing factor σ (from 0.1 to 0.3 eV) of the Fermi-Dirac distribution function.
The DFT phonon energy spectra also capture the instability of the pristine monolayer. We find that significant phonon softening arises at M and Γ, associated with the displacement of the V atoms. Figure 3e shows a representative example of the DFT phonon spectra for monolayer KV3Sb5. The Γ (M) point corresponds to the one (other two) of the 2 × 2 3Q CDW vectors in the folded BZ. Thus, these negative branches are a clear indicative of the structural instability associated with the 2 × 2 distortion of V atoms, similar to the bulk counterparts17,45,46,47. We also find that these 2 × 2 CDW instabilities are generically present in monolayer AV3Sb5 for A = K, Rb, and Cs (see Supplementary Fig. S3). We note that the monolayer hosts the broadened softened phonon modes in the momentum space compared to the bulk17, which signals various CDW instabilities other than 2 × 2. Nonetheless, we confirm that the 2 × 2 CDW instability is a leading instability by performing energy profile analysis for the various imaginary phonon modes (see Supplementary Note 5.1). Thus, it is imperative to consider the 2 × 2 3Q CDWs in the monolayer, such as the star of David (SD) and inverse star of David (ISD) distortions in the monolayer.
To investigate the role of the electronic instability in 2 × 2 CDW instabilities, we additionally calculated the phonon spectra as a function of electronic temperature, smearing factor σ, as shown in Fig. 3e. The softened phonon at M is lifted as we destroy the Fermi surface by increasing the electronic temperature. These calculations demonstrate that the electronic instability associated with the VHS plays a role in the formation of structural distortion. Since the DFT phonon spectra include the effects of electron-phonon coupling, our results suggest that either electronic or phononic contributions cannot be excluded in forming structural instability. In addition, the occurrence of the 2 × 2 instability in the phonon spectra is plausible in view of the electronic susceptibility calculations. Our DFT calculations are performed off the type-II VHS filling, in which large susceptibility values arise near the M point. While the reduced symmetry featured in the monolayer leads to the rearrangement of the VHSs into type-I and type-II VHSs at different fillings and correspondingly two types of distinct CDW orders—2 × 2-commensurate CDWs and IC-CDWs, we believe that the commensurate orders are more likely to occur in the monolayer, especially at non-zero temperature. The pristine monolayer AV3Sb5 is under strong fluctuations as dictated in the celebrated Mermin-Wagner theorem, prohibiting any IC-CDW orders at non-zero temperature. Thus, hereafter we mainly focus on commensurate order parameters in this work and discuss the possibility of incommensurate orders at zero temperature in Supplementary Note 11.
Competing orders
The rearranged VHSs manifest their properties in competing orders of correlated electronic states. In monolayer AV3Sb5, we employ the standard mean-field theory with the constructed TB model and uncover phase diagrams with CDWs and superconductivity (SC). The onsite and nearest-neighbor Coulomb interactions are introduced,
where U (V) describes the on-site (nearest-neighbor) density-density type interaction and R, α, and σ represent the lattice site, sublattice, and spin, respectively. We consider two classes of order parameters, CDWs and SC, which can significantly reduce the energy by gapping out the Fermi surface with diverging DOSs. CDWs and SC are of particular interest as they have been observed in the bulk AV3Sb5 in a variety of forms, including SD, ISD, and time-reversal symmetry breaking (TRSB) CDWs6,7,10,17.
Remarkably, any CDW order in the monolayer AV3Sb5 forms a doublet, as illustrated in Fig. 4c. The doublet formation of CDWs is one of the key characteristics of the monolayer AV3Sb5, originating from the reduced symmetry. The lowered \({{{{{{{{\mathscr{T}}}}}}}}}_{\sqrt{3}\times 1}\)-translational symmetry of AV3Sb5 monolayer plays a crucial role to double the CDW channels of the higher \({{{{{{{{\mathscr{T}}}}}}}}}_{1\times 1}\)-symmetry, and the number of multiplets is solely determined by their quotient group, \({{{{{{{{\mathscr{T}}}}}}}}}_{1\times 1}/{{{{{{{{\mathscr{T}}}}}}}}}_{\sqrt{3}\times 1}={{\mathbb{Z}}}_{2}\). For example, the two SD-CDW phases, dubbed SD-1 and SD-2, are the \({{\mathbb{Z}}}_{2}\) members, distinguished by the alkali chains hosted on and off the center of SD, respectively, as illustrated in Fig. 4c.

a, b Phase diagrams at the chemical potentials aμ1 = − 6 meV and bμ2 = 35 meV in U-V space, where U and V are the onsite and the nearest neighbor density-density type interactions. Different CDW and SC orders are indicated by different colors. The gap functions of s-wave and d-wave superconducting states correspond to \({A}_{g}^{(1)}\) and \({B}_{1g}^{(1)}\) provided in Table 2. The superconducting phases only exist at zero temperatures, and their phase boundaries are denoted by dashed lines to contrast the boundaries of commensurate CDWs denoted by the solid lines. X denotes a phase unidentified within our mean-field scheme. c Schematic illustration of six distinct CDW configurations. (c1-c2) Inverse star of David (ISD)-1,2, (c3-c4) star of David (SD)-1,2, (c5-c6) time-reversal symmetry breaking (TRSB)-1,2. In (c1-c4), solid and dashed blue lines indicate distinct bonding strengths. In (c5-c6), red arrows on the bonds indicate the direction of current bond orders. The oriented circles represent magnetic fluxes threading the hexagons and triangles where their magnitudes are proportional to the radius (see Supplementary Note 6 for the detailed descriptions). The corresponding CDW and SC order parameters are detailed in Tables 1 and 2.
The corresponding phenomenological Landau theory of the doublet CDWs becomes exotic. Introducing a bosonic real two-component spinor, \({\Psi }_{{{{{{{{\rm{SD}}}}}}}}}^{T}\equiv ({\psi }_{{{{{{{{\rm{SD-1}}}}}}}}},\,{\psi }_{{{{{{{{\rm{SD-2}}}}}}}}})\) with order parameters of SD-1 (ψSD−1) and SD-2 (ψSD−2), the Landau functional for the SD-CDW phases is given by
with phenomenological constants rSD and sSD. Here, the Pauli matrix \({\hat{\rho }}_{z}\) describes the spinor space, and higher order terms are omitted for simplicity. The sSD-term describes a free-energy difference between SD-1 and SD-2, which is nonzero when the \({{{{{{{{\mathscr{T}}}}}}}}}_{1\times 1}\)-translational symmetry is broken. Depending on sSD, the system energetically favors one of the doublet CDWs, enriching phase diagrams of the monolayer AV3Sb5.
Our mean-field analysis indeed finds enriched phase diagrams of the AV3Sb5 monolayer. We consider six configurations of CDWs (see Fig. 4c) and nine spin-singlet channels of SCs (see Table 2). In Fig. 4a, b, we illustrate representative mean-field phase diagrams of KV3Sb5 in U-V space obtained at two different chemical potentials μ1 = − 6 meV and μ2 = 35 meV, where the zero chemical potential is set to the neutral filling. We emphasize that our mean-field analysis of AV3Sb5 monolayer is reliable at zero and very low temperatures since any monolayer systems suffer from significant thermal fluctuations. Thus, the phase diagrams of Fig. 4 need to be understood as the ones in the limit of lowering temperature down to zero, T → 0+. In what follows, we point out key observations made from the phase diagrams.
First, five distinct CDW orders can be accessible by fine-tuning the chemical potential μ. For example, in the vicinity of type-II VHS at μ1 = −6 meV (Fig. 4a), SD-2 and ISD-2 dominantly occur with sizable regions of ISD-1 in the energy ranges of −0.6 eV < U < 0.4 eV and −20 meV < V < 40 meV. Similarly, a TRSB-2 CDW phase is uncovered under the condition μ ≥ 30 meV (Fig. 4b) in a wide range of U and V values. Moreover, the SD-1 phase appears near the type-I VHS at μ3 = 9 meV (see Supplementary Note 9). A small variation of chemical potential −6 meV < μ < 40 meV can tune the types and flavors of the VHSs, which should enable an on-demand onset of a variety of CDW phases, ranging from ISD-1/2, SD-1/2, to TRSB-2.
Second, competition between CDWs and SC is generically observed. A conventional s-wave SC phase is observed near μ1 = − 6 meV, which competes with ISD-1/2 and SD-2 at negative U and positive V as shown in Fig. 4a, b. Similarly, an unconventional d-wave SC phase is observed near μ2 = 35 meV. This competes with ISD-1 at positive U and negative V interactions as shown in Fig. 4b. Note that the chemical potential for the d-wave SC is quite higher than the energy of the type-II VHS μ1 and closer to the edge of the high energy band, which indicates that the VHS itself is not the unique reason to stabilize SC or CDW phases. The interplay between the VHS, filling, and interaction strengths should be considered together.
The final observation from our mean-field study is the nontrivial topology of the correlated CDW gaps. Notably, a non-zero Chern number \({{{{{{{\mathscr{C}}}}}}}}\) is induced in the energy spectra when gapped by the two time-reversal symmetry broken CDW phases TRSB-1 and TRSB-2. For example, the lowest unoccupied and highest occupied energy spectra of TRSB-1 (TRSB-2) host the Chern number \({{{{{{{\mathscr{C}}}}}}}}=-2\) and \({{{{{{{\mathscr{C}}}}}}}}=-1\) (\({{{{{{{\mathscr{C}}}}}}}}=-3\) and \({{{{{{{\mathscr{C}}}}}}}}=2\)), respectively. As shown in Fig. 5a, the different Chern numbers between the two phases arise due to the concurrent sign-change of the Berry curvature at high-symmetry momenta M1, K1, and K2. We note that the monolayer in the time-reversal symmetry broken CDW phase hosts the Fermi pockets that carry the Berry curvature, referred to as the Fermi Chern pockets48 (see Supplementary Fig. S13). However, unlike the bulk case48, asymmetric distribution of the Berry curvature occurs near the M1 and M2 in the monolayer due to the symmetry lowering. This results in distinct experimental observables, such as anomalous Hall conductivity σxy (see detailed comparison between bulk and monolayer in Supplementary Note 6). In Fig. 5b, we calculate σxy of the TRSB phases in the monolayer, which demonstrates the enriched structure of the anomalous Hall conductivities, distinct between TRSB-1 and TRSB-2. A sign change of σxy is also found as a function of chemical potential near the Fermi energy E = 0. We believe this nontrivial behavior featured in σxy can be readily observed in the Hall current measurements, leading to the experimental discovery of the TRSB-CDW phases.

a TB band structures of the TRSB-1 and TRSB-2 CDW phases. The Berry curvature Ωz(k) are overlaid at the corresponding k-points in the bands. The Chern numbers \({{{{{{{\mathscr{C}}}}}}}}\) induced in the bands are shown in green color. b Anomalous Hall conductivity σxy as a function of energy E for the TRSB-1 and TRSB-2 CDW phases. c DFT calculations of the chemical potential μ as a function of an uniform biaxial strain in AV3Sb5 for A = K, Rb, and Cs. d DFT calculations of the chemical potential μ as a function of doping concentration x for A1+xV3Sb5. The detailed DFT band structures under the biaxial strain and alkali doping are provided in Supplementary Note 12.
The exotic orders should be accessible in monolayer AV3Sb5 in a controlled fashion. Our mean-field phase diagrams (Fig. 4a, b) show that the occurrence of a specific electronic order is highly contingent upon the correct filling of electrons, which can be fine-tuned via mechanical and chemical means. For example, by applying a uniform biaxial strain in a range of ± 2% variations, μ can be tuned from 50 meV to −100 meV (Fig. 5c). We also find that U and V values are functions of applied strains by performing the ab initio calculations with constrained random-phase approximation49. This indicates that different phases in the mean-field phase diagrams are accessible using strains (See Supplementary Note 10 for the detailed discussion and computational methods). Moreover, the light doping of alkali atoms A (see Fig. 5d) or substituting Sb with Sn50 can be a fine knob to adjust the chemical potential. Owing to the 2D geometry of the AV3Sb5 monolayer, we believe that there exist more opportunities (such as ionic gating) to tailor the competing orders hosted therein.
Discussion
We have so far investigated unique features of the AV3Sb5 monolayer. Our system is unlike the bulk, hosted in a distinct symmetry class. The contrast is even more evident in the phase diagrams that we calculated with the bulk D6h and the monolayer D2h symmetries, respectively (see Supplementary Fig. S19 for the D6h phase diagrams). The lowered D2h symmetry in the monolayer features a tendency to foster the CDW orders. This is in line with the previous experiments, in which a CDW order is observed to suppress SC as the thickness of the AV3Sb5 film decreases34,35.
Our study arguably suggests that monolayer AV3Sb5 should be an exciting platform for studying intriguing 2D physics, such as the interplay between thermally suppressed IC-CDW orders and the type-II VHSs. At zero temperature, an IC-CDW associated with the type-II VHSs appears in the (U ≠ 0, V = 0) phase space. We investigate the two different fillings (μ = − 5, − 7 meV) near type-II VHSs and find that the IC-CDW phase is accessible with a negative U in the fine-tuned range of chemical potential (see Supplementary Fig. S21). Thus, not only the electron filling but also interaction strengths are important to stabilize the IC-CDW phases. At any non-zero temperature, the quasi-long range IC-CDW order can exist, and uncovering the possibility of topological phase transitions such as Kostelitz-Thouless transitions is certainly an important issue in both theoretical and experimental regards. Therefore, we believe that the AV3Sb5 monolayer offers timely new physics in the kagome metal community, calling for future sophisticated theoretical and experimental studies.
Our results also provide further insights into the unconventional CDW orders of the bulk systems. Notably, our DFT phonon and electronic susceptibility calculations have shown that the 2 × 2 instability of the bulk systems remains a robust feature of the V-based kagome metals against the weakening of the VHS nesting by symmetry-lowering. We have attributed its rationale to significant electronic instability remaining at q = M for a wide range of electron filling. Similar mechanisms may explain the robustness of the CDW orders in the bulk systems under a weakened nesting condition due to, for example, out of the exact VHS filling51. In this regard, further studies in the monolayer should be complementary in pinning down the origin of the CDW orders. The observation of CDW without acoustic phonon anomaly and evolution of CDW amplitude modes may be important to resolve the CDW mechanism of the monolayer as discussed in the bulk case52,53.
We conclude our discussion by arguing that the AV3Sb5 monolayer should be possible to synthesize based on the following facts. First, the cohesive energy generically indicates the thermodynamic stability of the monolayer systems. The cohesive energy is calculated as 3.8 eV/atom for all three alkali atoms (see details in Supplementary Note 1). This value is comparable to the bulk value of ~ 3.9 eV/atom, supporting the thermodynamical stability of the monolayer. In Fig. 6a, we plot the cohesive energy difference of the ISD-1 and SD-2 phases ΔEcoh(d) = Ecoh(d = 0) − Ecoh(d) as a function of the distortion of V atoms d. The CDW phases form a local minimum in the configuration space with an energy barrier of around 4 meV/atom. Second, our DFT phonon calculations indicate that the monolayer should be dynamically stable. Exemplary phonon bands of the ISD-1 phase are shown in Fig. 6b, which are clean of imaginary frequencies, showing the dynamical stability of the monolayer structure. Finally, the exfoliation energies of the monolayer are calculated as 42, 45, and 45 meV/Å2 for A = K, Rb, and Cs, respectively. These values are amount to existing two-dimensional materials, such as graphene ( ~ 21 meV/Å2)54, hBN ( ~ 28 meV/Å2)54, and Ca2N ( ~ 68 meV/Å2)55. We note that the recent experiments have successfully exfoliated thin layers of AV3Sb5 up to five layers using the taping methods35. Current developments of the chemical solution reaction method could be an appropriate technique to weaken the interlayer interaction of the bulk system and separate the monolayer56.

a Cohesive energy difference (per atom) as a function of the CDW distortion d of the KV3Sb5 monolayer. The cohesive energy difference is defined as ΔEcoh(d) = Ecoh(d = 0) − Ecoh(d), where the positive (negative) value of the distortion d represents the shift of the V atom in the ISD-1 (SD-2) phase with respect to the pristine structure. b Phonon bands of monolayer KV3Sb5 in the ISD-1 phase. In the inset, the solid and dashed lines represent the BZs for the 2 × 2 and \(\sqrt{3}\times 1\) structures, respectively. For Rb and Cs, see Supplementary Fig. S4.
Methods
Tight-binding model
We construct a TB model for the monolayer with D2h symmetry in the \(\sqrt{3}\times 1\) unit cell. Introducing the six-component spinor, \({\Psi }_{{{{{{{{\boldsymbol{k}}}}}}}}}^{T}=({A}_{{{{{{{{\boldsymbol{k}}}}}}}}}^{T},\,{B}_{{{{{{{{\boldsymbol{k}}}}}}}}}^{T},\,{C}_{{{{{{{{\boldsymbol{k}}}}}}}}}^{T})\) with \({\alpha }_{{{{{{{{\boldsymbol{k}}}}}}}}}^{T}=({\alpha }_{1,{{{{{{{\boldsymbol{k}}}}}}}}},\,{\alpha }_{2,{{{{{{{\boldsymbol{k}}}}}}}}})\), the Hamiltonian becomes \({H}_{0}={\sum }_{{{{{{{{\boldsymbol{k}}}}}}}}}{\Psi }_{{{{{{{{\boldsymbol{k}}}}}}}}}^{{{{\dagger}}} }{{{{{{{{\mathscr{H}}}}}}}}}_{0}({{{{{{{\boldsymbol{k}}}}}}}}){\Psi }_{{{{{{{{\boldsymbol{k}}}}}}}}}\). The indices for sublattice α ∈ {A, B, C} and site i ∈ {1, 2} are used. The Bloch Hamiltonian \({{{{{{{{\mathscr{H}}}}}}}}}_{0}({{{{{{{\boldsymbol{k}}}}}}}})\) is given by
where \(({c}_{i},\,{s}_{i})\equiv (\cos {{{{{{{\boldsymbol{k}}}}}}}}\cdot {{{{{{{{\boldsymbol{r}}}}}}}}}_{i},\sin {{{{{{{\boldsymbol{k}}}}}}}}\cdot {{{{{{{{\boldsymbol{r}}}}}}}}}_{i})\) with \({{{{{{{{\boldsymbol{r}}}}}}}}}_{1}=\frac{1}{2}(\sqrt{3},\,1),\,{{{{{{{{\boldsymbol{r}}}}}}}}}_{2}=\frac{1}{2}(\sqrt{3},-1),\,{{{{{{{{\boldsymbol{r}}}}}}}}}_{3}=(0,\,1),\,{{{{{{{{\boldsymbol{r}}}}}}}}}_{4}={{{{{{{{\boldsymbol{r}}}}}}}}}_{2}-{{{{{{{{\boldsymbol{r}}}}}}}}}_{3}\), r5 = r1 + r3, and r6 = r1 + r2. Here, t, t2, ϵ, σi and λi are nearest-neighbor hopping, next nearest-neighbor hopping, onsite energy, 2 × 2 Pauli matrices and 3 × 3 Gell-Mann matrices, respectively. The sublattice information and the definition of the Gell-Mann matrices are provided in Supplementary Fig. S5a and Supplementary Note 7, respectively. The last two terms give rise to onsite energy difference 2δϵ between two A sublattices and staggered hopping ± δt between the B and C sublattices, respectively. These two terms lead to the symmetry-lowering from \({{{{{{{{\mathscr{T}}}}}}}}}_{1\times 1}{D}_{6h}\) to \({{{{{{{{\mathscr{T}}}}}}}}}_{\sqrt{3}\times 1}\,{D}_{2h}\). With parameters (ϵ, t, t2, δϵ, δt) = (0.01, 0.42, 0.03, − 0.033, 0.01), our TB model well reproduces the DFT bands as well as the irreducible representations of the VHS bands at Γ and M (Ag) for KV3Sb5 (see Fig. 2a, b). For Rb and Cs, we also find good agreement between the TB and DFT (see Supplementary Note 2). The TB band structures of the TRSB-1/2 phases in Fig. 5a are obtained by using parameters (ϵ, t, t2, δϵ, δt, Φ) = (0.035, 0.42, 0.03, − 0.033, 0.01, 0.07) where Φ is a CDW magnitude of the TRSB-1/2 phases (see Supplementary Note 8 for detailed order parameters of the TRSB-1/2 phases).
Electronic susceptibility
We calculate the real part of the bare electronic susceptibility in the constant matrix approximation χ(q) given by
where nF(ϵ) is the Fermi-Dirac distribution and (μ, ν) is band index. For the calculation of χ(q) in Fig. 3, TB bands with 200 × 348 k-points and β = 1000 are used.
Mean-field theory
We perform the conventional zero-temperature mean-field method to investigate the interaction effects. Six distinct CDW configurations and nine spin-singlet SC orders are chosen to uncover CDW and SC instabilities in our analysis (see Fig. 4c and Table 1, 2). We introduce the CDW and SC order parameters (Φ, Δ) that break translational and U(1) symmetry. In the 2 × 2 unit cell, the mean-field Hamiltonians for CDW and SC orders are written as,
and
with the twelve-component spinor, \({\tilde{\Psi }}_{{{{{{{{\boldsymbol{k}}}}}}}}}^{T}=({\tilde{A}}_{{{{{{{{\boldsymbol{k}}}}}}}}}^{T},\,{\tilde{B}}_{{{{{{{{\boldsymbol{k}}}}}}}}}^{T},\,{\tilde{C}}_{{{{{{{{\boldsymbol{k}}}}}}}}}^{T})\) and \({\tilde{\alpha }}_{{{{{{{{\boldsymbol{k}}}}}}}}}^{T}=({\alpha }_{1,{{{{{{{\boldsymbol{k}}}}}}}}},\,{\alpha }_{2,{{{{{{{\boldsymbol{k}}}}}}}}},\,{\alpha }_{3,{{{{{{{\boldsymbol{k}}}}}}}}},\,{\alpha }_{4,{{{{{{{\boldsymbol{k}}}}}}}}})\). The indices i ∈ {1, 2, 3, 4} and σ ∈ {↑, ↓} denote the orbital sites and spins, respectively. The specific forms of 4 × 4 bond matrices Oa(k) and 12 × 12 pairing gap functions \(\hat{\Gamma }({{{{{{{\boldsymbol{k}}}}}}}})\) are tabulated in Table 1, 2. In our mean-field ansatz, the tested CDW order parameters are restricted to the 2 × 2 CDW orders carrying l = 1 angular momentum where the hopping strength modulates with the Q-vectors connecting M1, M2, M3 points in the real space,
where the overall amplitude Φ quantifies the modulation strength and the relative phase of (σAB, σAC, σBC) ∈ { ± 1, ± i} determines the pattern of CDW. Here, we fix the order parameter ϕ ∈ (Φ, Δ) is set as a real value. After constructing the ground state of the mean-field Hamiltonian \(\left|G,\,\phi \right\rangle\) and evaluating the ground state energy E[ϕ] ≡ 〈G; ϕ∣H0 + Hint∣G; ϕ〉, we obtain the phase diagrams presented in Fig. 4 (see Supplementary Notes 8 and 9 for details). 161 grids of the ground energy with ϕi ∈ [ − 0.08, 0.08] are utilized and their numerical integration at given ϕi are carried out with 80 × 80 k-points.
First-principles calculations
We perform density-functional theory (DFT) calculations using the Vienna abinitio simulation package (VASP)57,58 with the projector-augmented wave method59. For the exchange-correlation energy, the generalized-gradient approximation functional of Perdew-Burke-Ernzerhof60 is employed. The van der Waals correction is included within the zero damping DFT-D3 method of Grimme61. The kinetic energy cutoff for the plane wave basis is 300 eV. The force criteria for optimizing the structures is set to 0.01 eV/Å. The monolayer AV3Sb5 is simulated using a periodic supercell with a vacuum spacing of ~ 20 Å. The k-space integration is done with 10 × 17 k-points for the \(\sqrt{3}\times 1\) structure. For the DOS calculation, we use 46 × 69 k-points. The exfoliation energy is calculated using the Jung-Park-Ihm method54. We employ the finite difference method to obtain phonon dispersions. For the pristine monolayer (Fig. 3e), we use 4 × 4 supercell with 9 × 9 k-points with the Phonopy software62. For the ISD-1 phase (Fig. 6b), we use 8 × 8 supercell with 6 × 6 k-points using the nondiagonal supercell method63. We evaluate the (U, V) values in monolayer by using the first-principles constrained random phase approximation (cRPA) method49 with the weighting approach64, as implemented in VASP.
Berry curvature and anomalous Hall conductivity
The berry curvature Ωn(k) associated with the n-th energy band of the TB Hamiltonian H(k) is given by
Here, En(k) and \(\left|{u}_{n{{{{{{{\boldsymbol{k}}}}}}}}}\right\rangle\) are nth eigenvalue and eigenstate of H(k) and the derivative in the momentum space \({\partial }_{i}\equiv \frac{\partial }{\partial {k}_{i}}\) is adopted. The anomalous Hall conductivity is calculated as a function of energy E by integrating the n-band Berry curvatures for En < E over the BZ
The numerical integration is performed by using 500 × 500 k points.
Data availability
The data that support the findings of this study are available within the paper and Supplementary Information. Additional relevant data are available from the corresponding authors upon request.
Code availability
The code that supports the findings of this study is available from the corresponding authors upon request.
References
Ortiz, B. R. et al. New kagome prototype materials: discovery of KV3Sb5, RbV3Sb5, and CsV3Sb5. Phys. Rev. Mater. 3, 094407 (2019).
Ortiz, B. R. et al. CsV3Sb5: a Z2 topological kagome metal with a superconducting ground state. Phys. Rev. Lett. 125, 247002 (2020).
Jiang, Y.-X. et al. Unconventional chiral charge order in kagome superconductor KV3Sb5. Nat. Mater. 20, 1353–1357 (2021).
Zhao, H. et al. Cascade of correlated electron states in the kagome superconductor CsV3Sb5. Nature 599, 216–221 (2021).
Li, H. et al. Observation of unconventional charge density wave without acoustic phonon anomaly in kagome superconductors AV3Sb5 (A = K, Rb, Cs). Phys. Rev. X 11, 031050 (2021).
Denner, M. M., Thomale, R. & Neupert, T. Analysis of charge order in the kagome metal AV3Sb5 (A = K, Rb, Cs). Phys. Rev. Lett. 127, 217601 (2021).
Feng, X., Jiang, K., Wang, Z. & Hu, J. Chiral flux phase in the Kagome superconductor AV3Sb5. Sci. Bull. 66, 1384–1388 (2021).
Park, T., Ye, M. & Balents, L. Electronic instabilities of kagome metals: saddle points and Landau theory. Phys. Rev. B 104, 035142 (2021).
Lin, Y.-P. & Nandkishore, R. M. Complex charge density waves at van Hove singularity on hexagonal lattices: Haldane-model phase diagram and potential realization in the kagome metals AV3Sb5 (A = K, Rb, Cs). Phys. Rev. B 104, 045122 (2021).
Christensen, M. H., Birol, T., Andersen, B. M. & Fernandes, R. M. Theory of the charge density wave in AV3Sb5 kagome metals. Phys. Rev. B 104, 214513 (2021).
Ortiz, B. R. et al. Fermi surface mapping and the nature of charge-density-wave order in the kagome superconductor CsV3Sb5. Phys. Rev. X 11, 041030 (2021).
Kang, M. et al. Twofold van Hove singularity and origin of charge order in topological kagome superconductor CsV3Sb5. Nat. Phys. 18, 301–308 (2022).
Luo, H. et al. Electronic nature of charge density wave and electron-phonon coupling in kagome superconductor KV3Sb5. Nat. Commun. 13, 273 (2022).
Liang, Z. et al. Three-dimensional charge density wave and surface-dependent vortex-core states in a kagome superconductor CsV3Sb5. Phys. Rev. X 11, 031026 (2021).
Chen, H. et al. Roton pair density wave in a strong-coupling kagome superconductor. Nature 599, 222–228 (2021).
Xiang, Y. et al. Twofold symmetry of c-axis resistivity in topological kagome superconductor CsV3Sb5 with in-plane rotating magnetic field. Nat. Commun. 12, 6727 (2021).
Tan, H., Liu, Y., Wang, Z. & Yan, B. Charge density waves and electronic properties of superconducting kagome metals. Phys. Rev. Lett. 127, 046401 (2021).
Wang, Y. et al. Proximity-induced spin-triplet superconductivity and edge supercurrent in the topological kagome metal, K1−xV3Sb5. arXiv:2012.05898 (2020).
Zhao, C. C. et al. Nodal superconductivity and superconducting domes in the topological kagome metal CsV3Sb5.arXiv:2102.08356 (2021).
Wu, X. et al. Nature of unconventional pairing in the kagome superconductors AV3Sb5 (A = K, Rb, Cs). Phys. Rev. Lett. 127, 177001 (2021).
Xu, H.-S. et al. Multiband superconductivity with sign-preserving order parameter in kagome superconductor CsV3Sb5. Phys. Rev. Lett. 127, 187004 (2021).
Mu, C. et al. S-Wave superconductivity in kagome metal CsV3Sb5 revealed by 121/123Sb NQR and 51V NMR measurements. Chin. Phys. Lett. 38, 077402 (2021).
Duan, W. et al. Nodeless superconductivity in the kagome metal CsV3Sb5. Sci. China Phys. Mech. Astron. 64, 107462 (2021).
Yang, S.-Y. et al. Giant, unconventional anomalous Hall effect in the metallic frustrated magnet candidate, KV3Sb5. Sci. Adv. 6, eabb6003 (2020).
Yu, F. H. et al. Concurrence of anomalous Hall effect and charge density wave in a superconducting topological kagome metal. Phys. Rev. B 104, L041103 (2021).
Kenney, E. M., Ortiz, B. R., Wang, C., Wilson, S. D. & Graf, M. J. Absence of local moments in the kagome metal KV3Sb5 as determined by muon spin spectroscopy. J. Phys. Condens. Matter 33, 235801 (2021).
Neupert, T., Denner, M. M., Yin, J.-X., Thomale, R. & Hasan, M. Z. Charge order and superconductivity in kagome materials. Nat. Phys. 18, 137–143 (2022).
Jiang, K. et al. Kagome superconductors AV3Sb5 (A = K, Rb, Cs). arXiv:2109.10809 (2021).
Ye, L. et al. Massive dirac fermions in a ferromagnetic kagome metal. Nature 555, 638–642 (2018).
Kang, M. et al. Dirac fermions and flat bands in the ideal kagome metal FeSn. Nat. Mater. 19, 163–169 (2020).
Yin, J.-X. et al. Quantum-limit chern topological magnetism in TbMn6Sn6. Nature 583, 533–536 (2020).
Gong, C. et al. Discovery of intrinsic ferromagnetism in two-dimensional van der waals crystals. Nature 546, 265–269 (2017).
Fatemi, V. et al. Electrically tunable low-density superconductivity in a monolayer topological insulator. Science 362, 926–929 (2018).
Song, Y. et al. Competition of superconductivity and charge density wave in selective oxidized CsV3Sb5 thin flakes. Phys. Rev. Lett. 127, 237001 (2021).
Song, B. Q. et al. Competing superconductivity and charge-density wave in kagome metal CsV3Sb5: evidence from their evolutions with sample thickness. arXiv:2105.09248 (2021).
Wang, T. et al. Enhancement of the superconductivity and quantum metallic state in the thin film of superconducting kagome metal KV3Sb5. arXiv:2105.07732 (2021).
Mermin, N. D. & Wagner, H. Absence of ferromagnetism or antiferromagnetism in one- or two-dimensional isotropic heisenberg models. Phys. Rev. Lett. 17, 1133–1136 (1966).
Hohenberg, P. C. Existence of long-range order in one and two dimensions. Phys. Rev. 158, 383–386 (1967).
Coleman, S. There are no goldstone bosons in two dimensions. Commun. Math. Phys. 31, 259–264 (1973).
Yao, H. & Yang, F. Topological odd-parity superconductivity at type-II two-dimensional van hove singularities. Phys. Rev. B 92, 035132 (2015).
Yuan, N. F., Isobe, H. & Fu, L. Magic of high-order van hove singularity. Nat. Commun. 10, 1–7 (2019).
Qin, W., Li, L. & Zhang, Z. Chiral topological superconductivity arising from the interplay of geometric phase and electron correlation. Nat. Phys. 15, 796–802 (2019).
Kiesel, M. L. & Thomale, R. Sublattice interference in the kagome hubbard model. Phys. Rev. B 86, 121105 (2012).
Wang, W.-S., Li, Z.-Z., Xiang, Y.-Y. & Wang, Q.-H. Competing electronic orders on kagome lattices at van hove filling. Phys. Rev. B 87, 115135 (2013).
Ye, Z., Luo, A., Yin, J.-X., Hasan, M. Z. & Xu, G. Structural instability and charge modulations in the kagome superconductor AV3Sb5. Phys. Rev. B 105, 245121 (2022).
Wu, S. et al. Charge density wave order in the kagome metal AV3Sb5 (A = Cs, Rb, K). Phys. Rev. B 105, 155106 (2022).
Si, J.-G., Lu, W.-J., Sun, Y.-P., Liu, P.-F. & Wang, B.-T. Charge density wave and pressure-dependent superconductivity in the kagome metal CsV3Sb5: A first-principles study. Phys. Rev. B 105, 024517 (2022).
Zhou, S. & Wang, Z. Chern Fermi pocket, topological pair density wave, and charge-4e and charge-6e superconductivity in kagomé superconductors. Nat. Commun. 13, 7288 (2022).
Aryasetiawan, F. et al. Frequency-dependent local interactions and low-energy effective models from electronic structure calculations. Phys. Rev. B 70, 195104 (2004).
Oey, Y. M. et al. Fermi level tuning and double-dome superconductivity in the kagome metal CsV3Sb5−xSnx. Phys. Rev. Mater. 6, L041801 (2022).
Isobe, H., Yuan, N. F. Q. & Fu, L. Unconventional superconductivity and density waves in twisted bilayer graphene. Phys. Rev. X 8, 041041 (2018).
Li, H. et al. Observation of unconventional charge density wave without acoustic phonon anomaly in kagome superconductors AV3Sb5 (A = Rb, Cs). Phys. Rev. X 11, 031050 (2021).
Liu, G. et al. Observation of anomalous amplitude modes in the kagome metal CsV3Sb5. Nat. Commun. 13, 1–8 (2022).
Jung, J. H., Park, C.-H. & Ihm, J. A rigorous method of calculating exfoliation energies from first principles. Nano Lett. 18, 2759–2765 (2018).
Zhao, S., Li, Z. & Yang, J. Obtaining two-dimensional electron gas in free space without resorting to electron doping: an electride based design. J. Am. Chem. Soc. 136, 13313–13318 (2014).
Song, J. et al. Creation of two-dimensional layered zintl phase by dimensional manipulation of crystal structure. Sci. Adv. 5, eaax0390 (2019).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Computer. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Lloyd-Williams, J. H. & Monserrat, B. Lattice dynamics and electron-phonon coupling calculations using nondiagonal supercells. Phys. Rev. B 92, 184301 (2015).
Şaşıoğlu, E., Friedrich, C. & Blügel, S. Effective coulomb interaction in transition metals from constrained random-phase approximation. Phys. Rev. B 83, 121101 (2011).
Acknowledgements
This work was supported by the Korean National Research Foundation (NRF) Basic Research Laboratory (NRF-2020R1A4A3079707). Y.K. acknowledges the support from the NRF Grant numbers (NRF-2021R1A2C1013871, NRF-2021M3H3A1038085). E.-G.M. acknowledges the support from the NRF funded by the Ministry of Science and ICT (No. 2019M3E4A1080411, No. 2021R1A2C4001847, No. 2022M3H4A1A04074153), National Measurement Standard Services and Technical Services for SME funded by Korea Research Institute of Standards and Science (KRISS -2022 - GP2022-0014). The computational resource was provided by the Korea Institute of Science and Technology Information (KISTI) (KSC-2020-CRE-0108) and the Cambridge Tier-2 system operated by the University of Cambridge Research Computing Service and funded by EPSRC [EP/P020259/1].
Author information
Authors and Affiliations
Contributions
E.G.M. and Y.K. designed and organized the research. S.W.K. and H.O. performed all calculations. E.G.M. and Y.K. supervised the research. All authors discussed the results and contributed to writing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, SW., Oh, H., Moon, EG. et al. Monolayer Kagome metals AV3Sb5. Nat Commun 14, 591 (2023). https://doi.org/10.1038/s41467-023-36341-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-36341-2
This article is cited by
-
Two-fold symmetric superconductivity in the Kagome superconductor RbV3Sb5
Communications Physics (2024)
-
Artificial kagome lattices of Shockley surface states patterned by halogen hydrogen-bonded organic frameworks
Nature Communications (2024)
-
Anomalous enhancement of charge density wave in kagome superconductor CsV3Sb5 approaching the 2D limit
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
