
Abstract
Electricity-driven asymmetric catalysis is an emerging powerful tool in organic synthesis. However, asymmetric induction so far has mainly relied on forming strong bonds with a chiral catalyst. Asymmetry induced by weak interactions with a chiral catalyst in an electrochemical medium remains challenging due to compatibility issues related to solvent polarity, electrolyte interference, etc. Enabled by a properly designed phase-transfer strategy, here we have achieved two efficient electricity-driven catalytic asymmetric bromocyclization processes induced by weak ion-pairing interaction. The combined use of a phase-transfer catalyst and a chiral phosphate catalyst, together with NaBr as the bromine source, constitutes the key advantages over the conventional chemical oxidation approach. Synergy over multiple events, including anodic oxidation, ion exchange, phase transfer, asymmetric bromination, and inhibition of Br2 decomposition by NaHCO3, proved critical to the success.
Similar content being viewed by others
Introduction
Organic reactions driven by electricity has emerged as one of the most attractive tools in modern practical organic synthesis owing to their many “green” features, including atom economy, environmental benignity, and mild conditions1,2,3,4,5,6,7,8,9,10,11,12,13,14. The direct oxidation and reduction by the electric current with tunable redox potential constitute the most prominent advantage over conventional processes using stoichiometric chemical oxidants and reductants. Meanwhile, asymmetric synthesis, which provides expedient access to the valuable single-handed chiral molecules, holds an important position in modern organic synthesis15,16. Therefore, merging electrochemistry with asymmetric synthesis represents a highly desirable and enabling synthetic tool.
In the past few decades, substantial efforts have been devoted to asymmetric electrosynthesis. Impressive progress has been achieved by the strategic introduction of different chiral factors, including chiral electrode surface, chiral solvent, chiral electrolyte, chiral auxiliary, chiral mediator, and chiral catalyst17,18,19,20,21,22,23,24,25. Among these strategies, the use of a chiral catalyst for asymmetric amplification represents the most attractive approach19,20,21,22,23,24,25. However, it is not straightforward to implement asymmetric catalysis in an electrochemical context. Various compatibility issues need to be addressed in order to achieve both high efficiency and excellent asymmetric induction. Although a number of efficient electrocatalytic asymmetric transformations have been developed in the past decade, the modes of asymmetric induction are rather limited and there remain unsolved questions19,20,21,22,23,24,25.
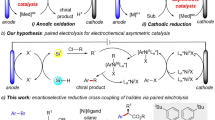
Based on the interaction strength between the chiral catalyst and substrate (or reagent/mediator), modern catalytic asymmetric induction has primarily relied on four types of interactions, namely, covalent bond, dative bond (e.g., metal–ligand coordination), weak bond (e.g., hydrogen bond), and ion-pair interaction, in order of decreasing strength (Fig. 1a)15,16. However, their applicability in electrochemistry varied significantly19,20,21,22,23,24,25. Among them, covalent bonding with a chiral catalyst proved most successful26,27,28,29,30,31,32. For example, chiral amine catalysis via an enamine intermediate has been established in many electrochemical processes, in which the covalently linked substrate-catalyst adduct ensures robust asymmetric induction in the presence of many other interactions in a complex electrochemical system. Likewise, transition metal catalysis relying on the relatively strong coordination between a chiral organometallic catalyst and substrate (or mediator) has also enabled a number of highly enantioselective electrocatalytic processes33,34,35,36,37,38,39,40,41,42. However, weak interactions have met with limited success so far in electrocatalytic asymmetric systems43,44,45,46,47,48. For example, excellent enantioselectivity induced by hydrogen bonding interaction with a small chiral organocatalyst has been achieved only recently46. In fact, to the best of our knowledge, chiral ion-pairing interaction, which is weakest among the above four types of interactions, has found almost no success in inducing high enantioselectivity, in spite of its outstanding performance in non-electrochemical contexts49,50,51. The key challenge lies in the compatibility between these two systems. For example, the weak but essential chiral ion-pair interaction can be easily interrupted by the electrolyte, which is also ionic and must be used in a large amount to ensure conductivity. Thus, the key chiral ion-pair intermediate surrounded by massive achiral ions from the electrolyte would lead to low integrity for asymmetric induction. Moreover, asymmetric ion-pairing catalysis is best operated in nonpolar solvents to guarantee contact ion-pair formation, but electrochemical systems typically favor polar solvents.

a Introduction to different strategies in asymmetric electrochemical catalysis. Covalent bond and dative bond with the chiral catalyst have been established, but weak bond interaction has been less successful. Ion-pairing catalysis remains elusive due to interference by the ionic electrolyte. Cat* chiral catalyst, S/M substrate or mediator. b The design principle of an electricity-driven asymmetric system using phase-transfer ion-pairing strategy. PTC phase-transfer catalyst, A* chiral phosphate anion.
To address the above challenge, we envisioned the use of a phase-transfer catalysis (PTC) strategy (Fig. 1b)52. If the key chiral ion-pair intermediate, once generated, can be spontaneously separated from the electrolyte and function in a different phase, high asymmetric induction could be expected purely from the chiral counter ion. However, the implementation of this concept for electrochemistry entails the choice of a suitable catalytic system. For example, the chiral ion pair prefers to enter the organic phase where asymmetric induction takes place, whereas the electrolyte stays in the aqueous phase to support the redox event. A suitable phase-transfer catalyst may also be needed to impeccably transport the relevant intermediates between phases.
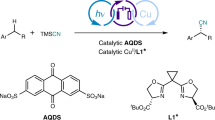
Asymmetric halocyclization is an important family of transformations that have found broad applications in the synthesis natural products and biologically important molecules53. Traditional chemical oxidation approaches typically require the use of stoichiometric amounts of an electrophilic halogenation source, which typically result in a large amount of waste. For example, in 2013, Xie et al. achieved such an elegant process with stoichiometric phase-transfer-catalyst-bound bromination regent54. In contrast, anodic oxidation of aqueous NaBr could serve as a green electrophilic source (Fig. 1b)55. Thus, the PTC-Br2 cation generated in aqueous phase is expected to pair with a large chiral organic anion. Driven by its high lipophilicity, this chiral ion pair enters the organic phase and triggers asymmetric bromination without interference by the electrolyte. After that, the PTC re-enters the aqueous phase to transport Br2 for the next catalytic cycle. The whole process would use only a catalytic amount of PTC and chiral anion, together with a green bromide source.
Here, we describe two efficient asymmetric bromocyclization processes enabled by chiral phosphate anion phase-transfer catalysis and synergistic electrochemical oxidation of an inorganic bromide source.
Results
Reaction development
tert-Butyloxycarbonyl (Boc)-protected tryptamine 1a was employed as the model substrate for the evaluation of reaction parameters in the electrocatalytic asymmetric bromocyclization. Various reaction parameters were systematically investigated, including chiral catalyst, solvent and so on (see Supplementary Information for details). These considerable efforts led to the choice of an operationally simple undivided cell equipped with platinum anode and cathode (Table 1). To simplify this system, NaBr was used as both the bromine source and the supporting electrolyte. The DABCO-derived ammonium salt PTC1 was identified as the best phase-transfer catalyst. Notably, with electricity as the end oxidant, this carrier is only required in a catalytic amount, in sharp contrast to the use of stoichiometric amounts of electrophilic bromine sources in traditional chemical oxidation protocols. A range of chiral phosphoric acids (CPAs) were also compared for asymmetric induction (see Supplementary Tables 2 and 3), which identified a new structure, [H8]BINOL-derivative CPA1 bearing two bulky substituents, as the superior chiral catalyst to provide excellent enantioselectivity (95% e.e.) and quantitative yield in a toluene–water biphasic system under a constant current of 2 mA at room temperature. Notably, almost all the components in this system were crucial for this outcome. Without CPA1, the reaction could proceed, but very slowly to the product in racemic form56,57, implying that the chiral phosphate was not only for asymmetric induction but also playing a phase-transfer role to pull the PTC1-Br2 cation to the organic phase by increasing its solubility, thus facilitating the process. Without PTC1, the reaction proceeded relatively slowly and, more importantly, with only 25% e.e., which was likely a result of the less efficient background reaction directly with Br2 as the bromination agent. Of particular note, NaHCO3 was essential to enhancing the reactivity. Without it or replacing it with other bases, such as Na2CO3 or NaOH, little or no conversion was observed. The origin of sensitivity to base and the critical role of NaHCO3 will be elaborated in mechanistic discussion (vide infra). Moreover, KBr could be used in place of NaBr, but NaCl or NaI did not lead to the corresponding halogenation product. We also evaluated other electrodes, including glassy carbon and nickel form, but they were less efficient. Polar organic solvents, such as EtOAc and DCM, led to dramatic decrease in enantioselectivity (see Supplementary Tables 5). The high solubility of the initially generated PTC1-Br2 cation (with Br−/Cl− as counter anion) in these polar organic solvents allowed the background reaction to take place without involving the chiral phosphate anion. Finally, it is also worth noting that the stirring speed had a minor influence to enantioselectivity. Stirring at 1000 r/min resulted in slightly higher enantioselectivity than that at 660 r/min (from 93% e.e. to 95% e.e.). A higher speed may facilitate ion exchange and subsequent phase transfer of the newly generated PTC1-Br2 salt, which was initially deposited on the electrode surface due to poor solubility in the aqueous phase. To the best of our knowledge, the development of efficient electricity-driven asymmetric processes catalyzed by chiral phosphoric acids remains elusive58,59.
Substrate scope exploration
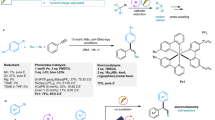
With the optimized conditions, the substrate scope of this electricity-driven asymmetric bromocyclization was examined (Fig. 2a). First of all, different useful N-protective groups were employed for the tryptamine structure, and they were all compatible with the mild conditions, giving rise to excellent yields and enantioselectivities (2a-f) and permitting application in different contexts, such as late-stage modifications. Then, the substitution effect at different positions (C-4 to C-7 positions) of the indole ring was investigated. Regardless of the electron-donating or electron-withdrawing nature of the substituent, these substituted tryptamines all resulted in excellent outcomes (2g-r). Of note, methyl substitution at the C-2 position led to moderate enantioselectivity, albeit with excellent yield. However, this case provided vicinal tetrasubstituted stereogenic centers (2s), a structural motif challenging to assemble in asymmetric synthesis. Moreover, tryptophol derivatives, which bear an internal alcohol nucleophile, could also undergo bromocyclization with excellent efficiency and enantioselectivity (2t-v)60. It is worth noting that all these cases exhibited high Faraday efficiencies (66–80%), illustrating the design rationality of this system. A tryptophan derivative (S)-1w, with an existing chirality, was also examined. In the absence of the CPA catalyst but under otherwise almost identical conditions, the reaction afforded a diastereomeric mixture (2w/2w′ = 5.4:1), suggesting the substrate-directing effect. Interestingly, in the presence of the CPA catalyst, regardless of its absolute configuration, the product diastereoselectivity was significantly enhanced, both favoring the same diastereomer 2w, indicating that the absolute configuration was mainly controlled by the substrate’s existing chirality. The enhanced diastereoselectivity by the CPA catalyst is likely owing to the improved substrate facial sensitivity during C−Br bond formation when a bulky chiral phosphate anion (vs. the small Br−) is bound to the active bromination reagent.

a Reaction scope regarding the tryptamine and tryptophol derivatives. 1 (0.3 mmol), (R)-CPA1 (5 mol %), PTC1 (10 mol %), NaHCO3 (0.1 M in H2O), NaBr (1 M in H2O), toluene (6 mL), H2O (5 mL), Pt anode and cathode (10 mm × 10 mm × 0.2 mm), stirring speed: 1000 r/min, 4 mA (2.5–3.0 F mol−1), rt. Isolated yields were provided, and e.e. values were determined by chiral HPLC. Ts p-toluenesulfonyl, Cbz benzyloxycarbonyl, Alloc allyloxycarbonyl, Fmoc 9-fluorenylmethyl, Bn benzyl + Electric current at 3 mA for 18 h (6.7 F mol−1). b A gram-scale protocol for the synthesis of 2c with reduced catalyst loading and high current and selected natural products that were synthesized from such bromination products.
Finally, this efficient protocol was further applied in a multigram synthesis of 2c to demonstrate its robust practicability (Fig. 2b). With a reduced loading (1 mol %) of the CPA catalyst and a higher current (80 mA), the 10-mmol reaction of 1c proceeded in quantitative yield with excellent enantioselectivity. Such enantioenriched 3-bromoindoline structures have been widely utilized as intermediates toward the syntheses of various cyclotryptamine alkaloids, including (–)-chimonanthine54 and (–)-hodgkinsine61.
Strategy extension
The success of the above electrocatalytic asymmetric protocol enabled by chiral anion phase-transfer strategy prompted us to further study its applicability in other asymmetric halogenation processes. For example, intramolecular asymmetric bromocyclization of 2-amidostyrenes 3 represents another useful reaction providing expedient access to chiral 4H-benzo[d][1,3]oxazines, representing potential chiral ligands and synthetic building blocks (Fig. 3a)62. Minor modifications from the above catalytic system and the conventional chemical oxidation conditions provided excellent reaction efficiency and enantioselectivity for this process. Notably, the use of CPA2 as the chiral catalyst and PTC2 as the phase-transfer catalyst were essential since those previously used catalysts for tryptamine cyclization or chemical oxidation were not able to deliver excellent results in this case (see Supplementary Table 8). Thus, a diverse set of highly enantioenriched chiral 4H-benzo[d][1,3]oxazines bearing different substituents could be synthesized with high efficiency (essentially quantitative yields in all the cases). In addition to terminal olefins, internal and cyclic olefins also reacted with high enantioselectivity and diastereoselectivity (4h and 4i). This robust protocol could also be scaled up for the gram-scale synthesis of 4b with reduced catalyst loading but without obvious erosion in efficiency or stereoselectivity (Fig. 3b). We also carried out two derivatizations of the 4H−3,1-benzoxazine product 4b. Simple reduction by LiAlH4 readily furnished chiral tertiary alcohol 5. Furthermore, thiolation of the bromide unit followed by reduction by NaBH3CN resulted in thioether 6. Combined with the bromocyclization step, these two reactions can be considered as net asymmetric hydration and vicinal thiohydroxylation of olefins, which are still challenging to achieve directly.

a Reaction scale: 3 (0.3 mmol), (R)-CPA2 (5 mol %), PTC2 (20 mol %), NaHCO3 (0.1 M in H2O), NaBr (1.0 M in H2O), toluene (6 mL), H2O (5 mL), Pt anode and cathode (10 mm × 10 mm × 0.2 mm), stirring speed: 1000 r/min, 4 mA (2.5–3.0 F mol−1), rt. Isolated yields were provided, and e.e. values were determined by chiral HPLC. +10 mol % of (R)-CPA2 was used. b Gram-scale protocol with 2 mol % of catalyst and high current for the synthesis of 4b and further transformations.
Mechanistic studies
We next performed a series of mechanistic studies. First of all, mixing Br2 and PTC1 in water and toluene resulted in the immediate formation of the adduct PTC1-Br2 (Fig. 4a, entry 1). However, oxidation of NaBr by electricity in the presence of stoichiometric PTC1 in an undivided cell resulted in trace PTC1-Br2 (Fig. 4a, entry 2), which appeared to contradict the proposed intermediacy of PTC1-Br2 in the standard protocol. However, this experiment may not reflect the whole picture of the standard reaction. We reasoned that the OH– generated by water reduction on the cathode could capture Br2 generated by anodic oxidation by forming NaBrO, which explains the failure to observe PTC1-Br2. In contrast, the bromination event in the organic phase of the standard protocol indeed generates a half equivalent of HBr and contributes to neutralizing the base generated in the cathode. To support this rationale, we performed the same experiment in a divided cell, in which the OH– generated on the cathode is physically isolated from the anode part. As expected, PTC1-Br2 could be successfully obtained in good yield (Fig. 4a, entry 3). Alternatively, the addition of HBr to an undivided cell could also recover the efficient formation of PTC1-Br2, which was in agreement with the above analysis (Fig. 4a, entry 4). These experiments implied that the OH− generated on the cathode is a hidden factor that needs to be considered in such a system.

a Comparison of the PTC1-Br2 intermediate formation conditions. Ar = 3,5-bis(trifluoromethyl)phenyl. b Effect of NaHCO3 and NaOH in the bromination process using preformed PTC1-Br2 as a stoichiometric reagent. When NaOH was used, no conversion of 1a was observed because it decomposed PTC1-Br2. In contrast, NaHCO3 prevented this decomposition and recovered the desired reactivity, which mimicked the standard condition. c, d Cyclic voltammetry measurements in H2O with 0.1 M NaNO3 and 5 mM substrate at room temperature with a scan rate of 100 mVs−1. e Detailed mechanism including the roles of NaHCO3.
Next, we examined the chemical competence of PTC1-Br2 for asymmetric bromination. Mixing PTC1-Br2 with NaHCO3 in the same biphasic media (toluene and aqueous NaBr) followed by the addition of 1a and CPA1 (5 mol%) successfully produced product 2a in >95% yield and 92% e.e., consistent with the standard electricity-driven protocol (Fig. 4b, entry 1). In contrast, if NaOH was used as a base, immediate decomposition of PTC1-Br2 was observed (Fig. 4b, entry 2), and further addition of 1a and CPA1 resulted in no bromination, which proved the detrimental effect of NaOH. Notably, in the standard protocol, NaOH is indeed generated at the cathode anyway and only half of it is neutralized by HBr (generated from bromination). Therefore, the standard protocol must involve others means to consume the excess NaOH, for which NaHCO3 might be responsible. Indeed, when NaHCO3 was used in combination with one equivalent of NaOH (relative to 1a), which mimicked the system of the standard protocol, the decomposition of PTC1-Br2 was inhibited (or retarded) and the bromination reactivity was recovered (Fig. 4b, entry 3). It was believed that NaHCO3 quickly captured NaOH (by forming Na2CO3) like a buffer, which prevented the rapid decomposition of PTC1-Br2. This explained the critical role of NaHCO3 in the standard protocol.
Next, we performed cyclic voltammetry studies to disclose the redox behavior on the electrodes. A comparison of the oxidation potential of PTC1 and NaBr revealed that Br− is easier to oxidize (Ep = 1.12 V vs SCE), which ensured the stability of catalyst PTC1 in the reaction (Fig. 4c). The addition of PTC1 or NaHCO3 into the NaBr solution can stimulate a catalytic current of Br− oxidation (Fig. 4d, red and blue, also see the Supplementary Information for more details). Further comparison indicated that PTC1 can also stimulate the reduction current. This observation revealed that the reaction of PTC1 with Br2 is reversible or the interaction between PTC1 and Br2 is weak. In contrast, NaHCO3 diminishes the reduction current, which might be related to irreversible decomposition of Br2 by the base. However, when PTC1 was added to the mixture of NaBr and NaHCO3, the reduction peak was observed again, suggesting that the formation of PTC1-Br2 and the base-induced decomposition of Br2 are competing with each other (Fig. 4d, green).
Based on the above experiments, a detailed reaction cycle is proposed (Fig. 4e). The reaction starts by anodic oxidation of Br− to form Br2, which is captured by PTC to generate PTC-Br2. Subsequent anion exchange with the chiral phosphate anion (CPA−) forms a more organic-soluble ion pair CPA-PTC-Br2, which initiates asymmetric bromination in the organic phase. In addition to product formation, a molecule of HBr and ammonium salt PTC-CPA are formed. The acid is immediately neutralized by the basic medium, and the latter re-enters the aqueous phase, upon pairing with Br− to regenerate PTC. Alternatively, the bromination step can also directly form PTC and a molecule of chiral phosphoric acid. Importantly, the successful operation of this catalytic cycle relies on the critical use of NaHCO3. Although the exact roles of NaHCO3 are not fully understood, we believe that it not only ensures a slightly basic medium for the phosphate anion to operate, but also efficiently inhibits (or retards) Br2 decomposition by consuming NaOH generated at the cathode.
We have developed a highly efficient electricity-driven catalytic asymmetric protocol employing weak ion-pairing interaction for asymmetric induction. In contrast to the broad success of asymmetric induction by relatively strong bonds asymmetric catalysis induced by weak interactions under electrochemical conditions has been notoriously challenging due to various compatibility issues. For ion-pairing catalysis, particularly problematic is the interference by electrolyte and polar solvents that are typical for electrochemical systems. In the context of this formidable challenge, we have designed an aqueous/organic phase-transfer strategy to permit useful asymmetric bromocyclization to take place with both excellent efficiency and high enantioselectivity. Compared with the conventional chemical oxidation approaches that require a stoichiometric amount of an electrophilic bromination reagent, our electricity-driven system employs only a small amount of a phase-transfer catalyst, together with NaBr as the green bromine source. Two representative asymmetric bromocyclization processes have been developed to showcase this powerful system. The practical utility of this system has also been illustrated by efficient gram-scale electrosynthesis at low catalyst loading. Mechanistic studies revealed that the catalytic cycle impeccably synergizes multiple events, including anodic oxidation, ion exchange, phase transfer, and asymmetric bromination. Of particular note is the critical roles of NaHCO3, which ensures a weakly basic condition and inhibits decomposition of the bromination reagent. This is also a rare demonstration of using chiral phosphoric acids as efficient chiral inducer in electrosynthesis. This system is expected to open a paradigm of electricity-driven asymmetric catalysis via weak ion-pairing interactions.
Methods
General procedure for the synthesis of 2 and 4
To an undivided vial (25 mL) equipped with the platinum anode (10 × 15 × 0.2 mm), platinum cathode (10 × 15 × 0.2 mm), and two magnetic stir bars (oval, 6 × 10 mm) were added substrate 1 or 3 (0.3 mmol), CPA (5 mol %), PTC1 (10 mol %), NaHCO3 (42 mg, 0.5 mmol), NaBr (515 mg, 5.0 mmol), toluene (6 mL), and H2O (5 mL) (note: for substrate 1, catalysts (R)-CPA1 and PTC1 were used; for substrate 3, catalysts (R)-CPA2 and PTC2 were used). This vial was placed on a stir plate with a stirring speed of 1000 r/min. The electrolysis was carried out with a constant current of 4 mA for 4.5–6 h (2.2–3.0 F mol−1). The reaction progress was monitored by thin-layer chromatography. Upon completion, the organic layer of the reaction mixture was separated and the aqueous layer was extracted with EtOAc (5 mL × 2). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated. The residue was purified by silica gel chromatography to yield the desired product 2 or 4.
Data availability
Source data are provided with this paper. The authors declare that all data supporting the findings of this study are available within the article and Supplementary Information files.
References
Schäfer, H. J. Contributions of organic electrosynthesis to green chemistry. C. R. Chim. 14, 745–765 (2011).
Francke, R. & Little, R. D. Redox catalysis in organic electrosynthesis: basic principles and recent developments. Chem. Soc. Rev. 43, 2492–2521 (2014).
Ogawa, K. A. & Boydston, A. J. Recent developments in organocatalyzed electroorganic chemistry. Chem. Lett. 44, 10–16 (2014).
Yan, M., Kawamata, Y. & Baran, P. S. Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev. 117, 13230–13319 (2017).
Kärkäs, M. D. Electrochemical strategies for C–H functionalization and C–N bond formation. Chem. Soc. Rev. 47, 5786–5865 (2018).
Wiebe, A. et al. Electrifying organic synthesis. Angew. Chem. Int. Ed. 57, 5594–5619 (2018).
Yoshida, J.-i, Shimizu, A. & Hayashi, R. Electrogenerated cationic reactive intermediates: the pool method and further advances. Chem. Rev. 118, 4702–4730 (2018).
Wang, H., Gao, X., Lv, Z., Abdelilah, T. & Lei, A. Recent advances in oxidative R1-H/R2-H cross-coupling with hydrogen evolution via photo-/electrochemistry. Chem. Rev. 119, 6769–6787 (2019).
Xiong, P. & Xu, H.-C. Chemistry with electrochemically generated N-centered radicals. Acc. Chem. Res. 52, 3339–3350 (2019).
Jing, Q. & Moeller, K. D. From molecules to molecular surfaces. Exploiting the interplay between organic synthesis and electrochemistry. Acc. Chem. Res. 53, 135–143 (2020).
Yamamoto, K., Kuriyama, M. & Onomura, O. Anodic oxidation for the stereoselective synthesis of heterocycles. Acc. Chem. Res. 53, 105–120 (2020).
Novaes, L. F. T. et al. Electrocatalysis as an enabling technology for organic synthesis. Chem. Soc. Rev. 50, 7941–8002 (2021).
Ackermann, L. Science of Synthesis: Electrochemistry in Organic Synthesis (Thieme, 2021).
Tay, N. E. S., Lehnherr, D. & Rovis, T. Photons or electrons? A critical comparison of electrochemistry and photoredox catalysis for organic synthesis. Chem. Rev. 122, 2487–2649 (2022).
Jacobsen, E. J., Pfaltz, A. & Yamamoto, H. Comprehensive Asymmetric Catalysis I–III (Springer, 1999).
Carreira, E. M. & Yamamoto, H. Comprehensive Chirality, 1st Edition (Elsevier Science, 2012).
Wattanakit, C. Chiral metals as electrodes. Curr. Opin. Electrochem. 7, 54–60 (2018).
Grecchi, S., Arnaboldi, S., Rizzo, S. & Mussini, P. R. Advanced chiral molecular media for enantioselective electrochemistry and electroanalysis. Curr. Opin. Electrochem. 30, 100810 (2021).
Ghosh, M., Shinde, V. S. & Rueping, M. A review of asymmetric synthetic organic electrochemistry and electrocatalysis: concepts, applications, recent developments and future directions. Beilstein. J. Org. Chem. 15, 2710–2746 (2019).
Lin, Q., Li, L. & Luo, S. Asymmetric electrochemical catalysis. Chem. Eur. J. 25, 10033–10044 (2019).
Chang, X., Zhang, Q. & Guo, C. Asymmetric electrochemical transformations. Angew. Chem. Int. Ed. 59, 12612–12622 (2020).
Wang, X. Y., Xu, X. T., Wang, Z. H., Fang, P. & Mei, T. S. Advances in asymmetric organotransition metal-catalyzed electrochemistry. Chin. J. Org. Chem. 40, 3738–3747 (2020).
Chakraborty, P., Mandal, R., Garg, N. & Sundararaju, B. Recent advances in transition metal-catalyzed asymmetric electrocatalysis. Coord. Chem. Rev. 444, 214065 (2021).
Yamamoto, K., Kuriyama, M. & Onomura, O. Asymmetric electrosynthesis: recent advances in catalytic transformations. Curr. Opin. Electrochem. 28, 100714 (2021).
Long, C.-J., He, Y.-H. & Guan, Z. Asymmetric oxidative mannich reactions promoted by photocatalysis and electrochemistry. Org. Biomol. Chem. 20, 2544–2561 (2022).
Bui, N.-N., Ho, X.-H., Mho, S.-I. & Jang, H.-Y. Organocatalyzed α-oxyamination of aldehydes using anodic oxidation. Eur. J. Org. Chem. 2009, 5309–5312 (2009).
Jensen, K. L., Franke, P. T., Nielsen, L. T., Daasbjerg, K. & Jørgensen, K. A. Anodic oxidation and organocatalysis: direct regio- and stereoselective access to meta-substituted anilines by α-arylation of aldehydes. Angew. Chem. Int. Ed. 49, 129–133 (2010).
Fu, N., Li, L., Yang, Q. & Luo, S. Catalytic Asymmetric electrochemical oxidative coupling of tertiary amines with simple ketones. Org. Lett. 19, 2122–2125 (2017).
Li, L., Li, Y., Fu, N., Zhang, L. & Luo, S. Catalytic asymmetric electrochemical α-arylation of cyclic β-ketocarbonyls with anodic benzyne intermediates. Angew. Chem. Int. Ed. 59, 14347–14351 (2020).
Lu, F.-Y. et al. Highly enantioselective electrosynthesis of C2-quaternary indolin-3-ones. Chem. Commun. 56, 623–626 (2020).
Wang, Z.-H. et al. TEMPO-enabled electrochemical enantioselective oxidative coupling of secondary acyclic amines with ketones. J. Am. Chem. Soc. 143, 15599–15605 (2021).
Zhou, P., Li, W., Lan, J. & Zhu, T. Electroredox carbene organocatalysis with iodide as promoter. Nat. Commun. 13, 3827 (2022).
Torii, S., Liu, P., Bhuvaneswari, N., Amatore, C. & Jutand, A. Chemical and electrochemical asymmetric dihydroxylation of olefins in I2−K2CO3−K2OsO2(OH)4 and I2−K3PO4/K2HPO4−K2OsO2(OH)4 systems with sharpless’ ligand. J. Org. Chem. 61, 3055–3060 (1996).
Cai, C.-Y. et al. Photoelectrochemical asymmetric catalysis enables site- and enantioselective cyanation of benzylic C–H bonds. Nat. Catal. 5, 943–951 (2022).
Fu, N. et al. New bisoxazoline ligands enable enantioselective electrocatalytic cyanofunctionalization of vinylarenes. J. Am. Chem. Soc. 141, 14480–14485 (2019).
Huang, X., Zhang, Q., Lin, J., Harms, K. & Meggers, E. Electricity-driven asymmetric lewis acid catalysis. Nat. Catal. 2, 34–40 (2019).
Zhang, Q., Chang, X., Peng, L. & Guo, C. Asymmetric lewis acid catalyzed electrochemical alkylation. Angew. Chem. Int. Ed. 58, 6999–7003 (2019).
Dhawa, U. et al. Enantioselective pallada-electrocatalyzed C−H activation by transient directing groups: expedient access to helicenes. Angew. Chem. Int. Ed. 59, 13451–13457 (2020).
Gao, P.-S. et al. Cu(II)/TEMPO-catalyzed enantioselective C(sp3)–H alkynylation of tertiary cyclic amines via shono-type oxidation. Angew. Chem. Int. Ed. 59, 15254–15259 (2020).
Qiu, H. et al. Enantioselective Ni-catalyzed electrochemical synthesis of biaryl atropisomers. J. Am. Chem. Soc. 142, 9872–9878 (2020).
Song, L. et al. Dual electrocatalysis enables enantioselective hydrocyanation of conjugated alkenes. Nat. Chem. 12, 747–754 (2020).
Xiong, P., Hemming, M., Ivlev, S. I. & Meggers, E. Electrochemical enantioselective nucleophilic α-C(sp3)–H alkenylation of 2-acyl imidazoles. J. Am. Chem. Soc. 144, 6964–6971 (2022).
Gourley, R. N., Grimshaw, J. & Millar, P. G. Electrochemical reduction in the presence of tertiary amines: an asymmetric synthesis of 3,4-dihydro-4-methylcoumarin. Chem. Commun. 24, 1278–1279 (1967).
Nielsen, M. F. et al. Enantioselective cathodic reduction of 4-methylcoumarin: dependence of selectivity on reaction conditions and investigation of the mechanism. Chem. Eur. J. 3, 2011–2024 (1997).
Maekawa, H., Itoh, K., Goda, S. & Nishiguchi, I. Enantioselective electrochemical oxidation of enol acetates using a chiral supporting electrolyte. Chirality 15, 95–100 (2003).
Chang, X., Zhang, J., Zhang, Q. & Guo, C. Merging electrosynthesis and bifunctional squaramide catalysis in the asymmetric detrifluoroacetylative alkylation reactions. Angew. Chem. Int. Ed. 59, 18500–18504 (2020).
Wu, R., Ma, C. & Zhu, Z. Enzymatic electrosynthesis as an emerging electrochemical synthesis platform. Curr. Opin. Electrochem. 19, 1–7 (2020).
Long, C.-J. et al. Merging the von-vatural catalytic activity of lipase and electrosynthesis: asymmetric cxidative cross-coupling of secondary amines with ketones. Angew. Chem. Int. Ed. 61, e202203666 (2022).
Brak, K. & Jacobsen, E. N. Asymmetric ion-pairing catalysis. Angew. Chem. Int. Ed. 52, 534–561 (2013).
Mahlau, M. & List, B. Asymmetric counteranion-directed catalysis: concept, definition, and applications. Angew. Chem. Int. Ed. 52, 518–533 (2013).
Qian, D. & Sun, J. Recent progress in asymmetric ion-pairing catalysis with ammonium Salts. Chem. Eur. J. 25, 3740–3751 (2019).
Rauniyar, V., Lackner, A. D., Hamilton, G. L. & Toste, F. D. Asymmetric electrophilic fluorination using an anionic chiral phase-transfer catalyst. Science 334, 1681–1684 (2011).
Yan, J. et al. The applications of catalytic asymmetric halocyclization in natural product synthesis. Org. Chem. Front. 9, 499–516 (2022).
Xie, W. et al. Highly enantioselective bromocyclization of tryptamines and its application in the synthesis of (−)-chimonanthine. Angew. Chem. Int. Ed. 52, 12924–12927 (2013).
Tang, H.-T., Jia, J.-S. & Pan, Y.-M. Halogen-mediated electrochemical organic synthesis. Org. Biomol. Chem. 18, 5315–5333 (2020).
Liu, K., Deng, Y., Song, W., Song, C. & Lei, A. Electrochemical dearomative halocyclization of tryptamine and tryptophol derivatives. Chin. J. Chem. 38, 1070–1074 (2020).
Wu, J., Abou-Hamdan, H., Guillot, R., Kouklovsky, C. & Vincent, G. Electrochemical synthesis of 3a-bromofuranoindolines and 3a-bromopyrroloindolines mediated by MgBr2. Chem. Commun. 56, 1713–1716 (2020).
Akiyama, T., Itoh, J., Yokota, K. & Fuchibe, K. Enantioselective mannich-type reaction catalyzed by a chiral brønsted acid. Angew. Chem. Int. Ed. 43, 1566–1568 (2004).
Uraguchi, D. & Terada, M. Chiral brønsted acid-catalyzed direct mannich reactions via electrophilic activation. J. Am. Chem. Soc. 126, 5356–5357 (2004).
Liu, H. et al. Highly asymmetric bromocyclization of tryptophol: unexpected accelerating effect of DABCO-derived bromine complex. Org. Lett. 16, 1908–1911 (2014).
Lindovska, P. & Movassaghi, M. Concise synthesis of (−)-hodgkinsine, (−)-calycosidine, (−)-hodgkinsine B, (−)-quadrigemine C, and (−)-psycholeine via convergent and directed modular assembly of cyclotryptamines. J. Am. Chem. Soc. 139, 17590–17596 (2017).
Wang, Y.-M., Wu, J., Hoong, C., Rauniyar, V. & Toste, F. D. Enantioselective halocyclization using reagents tailored for chiral anion phase-transfer catalysis. J. Am. Chem. Soc. 134, 12928–12931 (2012).
Acknowledgements
We are grateful to the National Natural Science Foundation of China (22271242 to J.S.), the Science Technology and Innovation Committee of Shenzhen Municipality (JCYJ20200109141408054 to J.S.), the Hong Kong Research Grants Council (C6012-21G to J.S., 16303420 to J.S., 16309321 to J.S., 16304322 to X.T.) and Innovation and Technology Commission (ITC-CNERC14SC01 to J.S.) for funding support.
Author information
Authors and Affiliations
Contributions
X.T. conceived the project, performed the experiments, and wrote the paper. Q.W. performed the experiments. J.S. conceived and directed the project and wrote the paper. All the authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
X.T. and J.S. are inventors on a Chinese patent application (Application No. 202211411915.2). X.T. and J.S. declare no other competing interests. Q.W. declares no competing interests.
Peer review
Peer review information
Nature Communications thanks Hai-Chao Xu and Xiaoyu Yang for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tan, X., Wang, Q. & Sun, J. Electricity-driven asymmetric bromocyclization enabled by chiral phosphate anion phase-transfer catalysis. Nat Commun 14, 357 (2023). https://doi.org/10.1038/s41467-023-36000-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-36000-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
