
Abstract
The development of transition metal-catalyzed β-C–H bond activation via highly-strained 4-membered metallacycles has been a formidable task. So far, only scarce examples have been reported to undergo β-C–H bond activation via 4-membered metallacycles, and all of them rely on precious metals. In contrast, earth-abundant and inexpensive 3d transition metal-catalyzed β-C–H bond activation via 4-membered metallacycles still remains an elusive challenge. Herein, we report a phosphine oxide-ligated Ni−Al bimetallic catalyst to activate secondary benzylic C(sp3)–H bonds of formamides via 4-membered nickelacycles, providing a series of α,β-unsaturated γ-lactams in up to 97% yield.
Similar content being viewed by others

Introduction
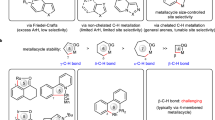
Cyclometallation represents one of the most efficient pathways for transition metal-catalyzed C–H activation, and remarkable progress has been achieved by the combination of a diverse range of directing groups (DG) and transition metals during the past several decades1,2,3,4. However, most examples proceed through stable 5-membered metallacycles, allowing the activation of C−H bonds at the position of γ to a coordinating atom (Fig. 1a). In contrast, the activation of non-γ-C–H bonds via larger or smaller-size metallacycles is faced with great challenges owing to unfavorable entropic effect or ring strain5. Among various non-γ-C–H bonds, proximate β-C–H bonds are in general the most difficult to activate owing to the need of forming highly-strained 4-membered metallacycles. So far, only scarce examples have been achieved for the activation of proximate β-C–H bonds via 4-membered metallacycles. For example, the incorporation of palladium into molecules via alkene carbopalladation or 1,4-palladium shift of aryl halides proves to be an efficient strategy for the activation of β-C(sp3)–H bonds via 4-membered palladacycles (Fig. 1b, I)6,7,8,9,10,11,12,13. Besides, the use of amines or phosphines as monodentate directing groups with proximate sterically-hindered groups is another elegant strategy for the activation of β-C(sp3)–H or β-C(sp2)–H bonds via 4-membered palladacycles, rhodacycles or ruthenacycles (Fig. 1b, II and III)14,15,16,17,18. Most recently, a hydroxyl-containing bidentate directing group is devised to activate β-C(sp3)–H activation via 4-membered palladacycles, allowing various tertiary amines to be used as substrates (Fig. 1b, IV)19. Despite great progress, all these methods have to rely on precious metals such as Pd, Rh, and Ru, while the use of earth-abundant and inexpensive 3d-transition metal as catalysts to activate β-C–H bonds via 4-membered metallacycles still remains an elusive challenge20,21,22,23,24,25,26,27,28,29,30,31,32,33. Although larger-sized metallacycles such as 7- or 10-membered nickelacycles have been devised to enable β-C–H bond activation, these strategies often suffered reduced reactivity and limited scope of C–H bonds, for example, only more reactive β-C(sp2)–H bonds are compatible34,35,36. Thereby, the development of 3d metal-catalyzed unreactive β-C(sp3)−H bond activation via 4-membered metallacycles are highly desirable. Inspired by our previous work on C–C bond activation of cyclopropanes37, in which a phosphine oxide (PO)-ligated Ni–Al bimetallic catalyst can form a bicyclic structure to stabilize a 4-membered nickelacycle intermediate (Fig. 1c, left), we envisioned that the PO–Ni–Al catalytic system may also be used to activate proximate β-C(sp3)–H bond of formamides via a similar 4-membered nickelacycle intermediate (Fig. 1c, right)38,39,40,41,42,43,44,45,46,47,48, thus providing a 3d-metal-catalyzed [3 + 2] cycloaddition of two C–H bonds49,50,51,52,53,54.

a The activation of C–H bonds at different position to the coordinating atom of directing groups is faced with varying levels of difficulty. γ-C–H bonds are the most easily activated via 5-membered metallacycles, while the activation of proximate β-C–H bonds via highly-strained 4-membered metallacycles is extremely challenging. b Only scarce examples on β-C–H bond activation via 4-membered metallacycles have been reported, and all of them rely on precious metals such as Pd, Rh, and Ru. c Proposed secondary β-C(sp3)–H bond activation: PO-ligated Ni–Al bimetal-catalyzed C–H bond activation via 4-membered nickelacycle. d 3d-Transition metal-catalyzed secondary C(sp3)–H bond activation via 4-membered nickelacycle (this work). DG Directing group. M Metal catalyst. FG Functional group. PG Protecting group.
Here, we show a 3d metal-catalyzed unreactive benzylic secondary β-C(sp3)–H bond activation via 4-membered nickelacycles, providing a series of α,β-unsaturated γ-lactams in up to 97% yield through a [3 + 2] cycloaddition of two C–H bonds of formamides with alkynes (Fig. 1d). In the reaction, a phosphine oxide (PO)-ligated Ni–Al bimetallic catalyst plays a crucial role in controlling catalytic reactivity and site-selectivity via the formation of a well-stabilized bicyclic nickelacycle. Preliminary experimental evidence and density functional theory (DFT) calculations reveal that the formyl C–H activation proceeds via ligand-to-ligand H transfer pathway, and the alkyne insertion into 4-membered nickelacycles is a turnover-limiting step of the reaction.
Results
Reaction optimization
N-benzylformamides (1a) and oct-4-yne (2a) were selected as model substrates on the basis of the following considerations: (1) formamides have proved to be good model substrates for acyl group-directed Ni-catalyzed γ-C–H bond activation55,56,57,58,59; (2) to eliminate the interference of more reactive γ- or δ-C(sp3)–H bonds, benzyl group containing only two secondary β-C(sp3)–H bonds was selected as a N-substituent; (3) a removable bulky 2,4,4-trimethylpentan-2-yl (TP) group was selected as another N-substituent to promote β-C(sp3)–H bond activation by steric repulsion and prevent from hydrocarbamoylation side reaction. Through systematic examination on various PO ligands, Al-based Lewis acids and other reaction parameters (Table 1), the optimal condition was eventually achieved: 10 mol% Ni(cod)2, 10 mol% tert-butyl diamine-derived PO (tBu-DAPO), and 40 mol% AlEt3 at 120 °C. Under which, the desired product 3a can be obtained in 81% yield (entry 1), proving that Ni–Al bimetallic catalyst can effectively promote the formation of 4-membered nickelacycles. Control experiments revealed that the combination of Ni(cod)2, tBu-DAPO, and AlEt3 was crucial, and the removal of any of them would inactivate the reaction (entries 2 and 3). Other Ni sources (entries 4 and 5) and traditional ligands such as phosphines (entries 6–8) and N-heterocyclic carbenes (entries 9 and 10) led to poor results. In addition, the reaction was rather sensitive to the structure of PO ligands, for example, other PO ligands gave either no products or low yields (entries 11–16), meaning an extremely narrow range of ligands. The superior reactivity of tBu-DAPO may be ascribed to the presence of two bulky tert-butyl groups that would affect the cone angle of phosphine ligand. Notably, the replacement of AlEt3 by other Lewis acids also resulted in low yields (entries 17–20), suggesting that the reactivity was also highly dependent on the structure and the acidity of Al-based Lewis acids.
Scope of formamides and alkynes
With the optimized reaction conditions established, we first investigated the scope of benzylformamides 1 (Fig. 2). Various electron-donating alkyl groups such as methyl group (3b to 3e), isopropyl group (3f) and tert-butyl group (3g) at different positions of the aromatic ring were in general well compatible with the reaction, providing good to high yield. Different from alkyl substituents, electron-rich methoxy group (3h) gave a significantly decreased yield, which was attributed to the instability of C–O bond when the oxygen atom coordinated to Al-Lewis acid. To inhibit the coordination of oxygen atoms, bulkier benzyloxy group (3i) was examined and the yield was improved to 93%. In addition, due to similar coordination to Al-Lewis acid, electron-rich alkylamino groups also resulted in a little lower yield (3j and 3k). Comparing with electron-donating groups, various electron-withdrawing groups such as CF3O (3l), F (3m and 3n), Cl (3o), CF3 (3p), and carboxylate group (3q) were also well tolerated with the reaction, yet with slightly reduced yields, suggesting that the benzylic β-C(sp3)–H bond activation would proceed via an electrophilic metalation pathway. Except thiophenyl group with a low yield (3t), other (hetero)aryl groups such as biphenyl group (3r), naphthyl group (3s), and carbazolyl group (3u) were well compatible, providing the corresponding products in 80−95% yield. Notably, the presence of carboxylate (3q) and thiophenyl group (3t) resulted in the need of an excess of AlEt3, because these groups had relatively strong coordination with AlEt3. Besides aryl groups, general alkyl groups were also tested, however, these groups would result in undesired γ- or δ-C(sp3)−H activation (vide infra), further confirming that β-C(sp3)–H bonds are much less reactive than γ- or δ-C(sp3)–H bonds. In addition, other protecting groups such as tert-butyl (3v), tert-amyl (3w), and adamantly (3x) were also effective, providing the corresponding products in 58–77% yield, but these less bulky groups would result in partial activation of γ-C–H bonds of protecting groups via 5-membered nickelacycles.

Reaction conditions: 1 (0.2 mmol), 2a (0.6 mmol), toluene (1.0 mL) under N2 for 2 h; yield of isolated products. *AlEt3 (60 mol%). †AlEt3 (80 mol%), 4 h. ‡AlEt3 (100 mol%). nPr = n-propyl. tBu = tert-butyl.
Next, the scope of alkynes was investigated under the optimal conditions (Fig. 3). Linear alkyl alkynes including ethyl (4a), n-butyl (4b), n-hexyl (4c) substituted alkynes and even cyclic alkyl alkynes (4d) were well tolerated, providing the corresponding products in 70−85% yield. In addition, alkynes with branched alkyl group (4e) or functional groups (4f and 4g) also proved to be suitable substrates, affording the desired products in 64–78% yield. However, diphenyl alkynes or aryl alkyl alkynes were not very active in the reaction, delivering the desired product in low yield (4h in 20% yield). In these cases, insoluble precipitation was formed very quickly, resulting in a shutdown of the reaction with most of formamides recovered. We reasoned that insoluble precipitation could be complex of nickel with diaryl alkynes, for example, π-complex, or 5-membered nickelacycle that are generated by nickel with two alkynes60,61. In addition, non-symmetrical alkynes were also compatible, providing the corresponding products in 92% (4i/4i′) and 74% (4j/4j′) yield, respectively, with similar regioisomer ratio of 1.5:1, while further increasing steric bias between two substituents of alkynes would result in no reaction.

Reaction conditions: 1a (0.2 mmol), 2 (0.6 mmol), toluene (1.0 mL) under N2 for 2 h; yield of isolated products. nHex = n-hexyl. TBS = tert-butyldimethylsilyl. TIPS = triisopropylsilyl. nBu = n-butyl.
Reaction utility
To demonstrate the utility of the current method, a range of substrates containing bioactive structural motifs such as menthol (4k), cholesterol (4l), borneol (4m) and diacetonefructose (4n) have been examined, achieving 35–81% yield under the optimal conditions with varying amounts of AlEt3, owing to strong coordination of functional groups with AlEt3 (Fig. 4a). Then, a gram-scale reaction of 1a and 2a was conducted under the standard conditions, and product 3a can be obtained without a significant loss of yield (Fig. 4b).

a Reaction of substrates bearing bioactive structural motifs under the optimal conditions with varying amounts of AlEt3. †AlEt3 (80 mol%). ‡AlEt3 (100 mol%). b Gram-scale reaction and product transformations. TFA = trifluoroacetic acid. DMEDA = N,N’-dimethyl-1,2-ethanediamine. Boc2O = di(tert-butyl)carbonate. DMAP = 4-dimethylamino pyridine. BINOL = (rac)−1,1’-bi(2-naphthol). NaBArF4 = sodium tetrakis(perfluorophenyl)borate.
Upon treatment with trifluoroacetic acid under mild conditions, 2,4,4-trimethylpentan-2-yl (TP) protecting group can be easily removed, affording pyrrolidone 5 in 98% yield. Further treated with aryl iodide and (Boc)2O, compound 5 can be easily transformed into 4-methoxylphenyl-pyrrolidone 6 and Boc-pyrrolidone 7 in 93 and 99% yield, respectively. Under oxidative conditions, the benzylic C−H bond of compound 5 can be facilely oxidized, providing a versatile synthetic precursor, aminal 8, in 58% yield. Aminal 8 underwent in situ dehydration to generate the corresponding imine, which was then attacked by diphenyl phosphonate and indole, providing tetra-substituted carbon-containing compound 9 in 76% yield and 10 in 97% yield, respectively62. In addition, the imine can play a directing group to facilitate Rh or Ir-catalyzed C−H activation, and subsequent cyclization with 1,4-dihydroquinone and alkyne afforded spiro compound 11 and 1263.
Mechanistic investigation
To gain more insights into the mechanism, relevant mechanistic experiments were conducted. Firstly, tBu-DAPO-ligated Ni–Al bimetallic catalyst was prepared and characterized by 1H, 13C, and 31P NMR (Fig. 5a). When the complex was treated with substrates 1a and 2a, 85% yield of product 3a can be obtained, suggesting tBu-DAPO–Ni–Al can catalyze the cycloaddition reaction. Secondly, deuterium-labeling experiment disclosed low isotope effect for formyl C–H bond (kH/kD = 1.2) (Fig. 5b, left). Further intra- and intermolecular competitive experiments on the benzylic C–H bond generated the corresponding kH/kD values, 3.2 and 1.0, respectively (Fig. 5b, middle and right). In addition, parallel reactions also revealed a low isotope effect for benzylic C–H bond (kH/kD = 1.3). These results indicated that neither the formyl C–H bond cleavage nor the benzylic C–H bond cleavage would be involved into the turnover-limiting step, while high kH/kD (3.2) meant that benzylic C−H cleavage could be an irreversible step. Thirdly, we also measured the rate order with regards to alkynes and achieved a first-order reaction for alkynes (see Supplementary Information, note 6), meaning that in turnover-limiting step, only one alkyne was involved, which suggested that the reaction occurred via a 4-membered nickelacycle without bearing an alkyne, otherwise, it would be a second-order reaction for alkynes when the reaction proceeds via a 6‐membered nickelacycle. Fourthly, 3-phenylpropyl group instead of benzyl group of 1a resulted in complete γ-C(sp3)–H bond activation (Fig. 5c), providing the corresponding products 3y′ in 20% yield and 3y″ in 62% yield, without observation of β-C(sp3)–H bond activation. This result further confirmed that the activation of β-C(sp3)–H bond was more difficult than that of common γ-C(sp3)–H bond. In addition, simple methyl group instead of benzyl group of 1a led to γ-C(sp3)–H bond activation product 3z′ in 25% yield and hydrocarbamoylation product 3z″ in 70% yield (Fig. 5d), while tBuCH2 group instead of benzyl group gave no reaction. These results suggested that the presence of phenyl group was critical to β-C(sp3)–H bond activation. On the basis of these mechanistic experiments, possible reaction mechanisms were proposed in Fig. 5e. Substrate 1a coordinates to the bimetallic catalyst, and then formyl C–H activation occurs to produce intermediate B that undergoes benzylic C–H activation to generate 4-membered nickelacycle C (path a). Subsequent alkyne insertion and reductive elimination furnish the desired product 3a and regenerate the bimetallic catalyst. The intermediate B may undergo γ-C(sp3)–H bond activation to form more stable 5-membered nickelacycle (path b), which finally results in side product 3a′. In addition, direct reductive elimination of the intermediate B would provide hydrocarbamoylation product 3a″ (path c). However, the pathway (path a′), wherein the intermediate B undergoes migratory insertion with an alkyne first, followed by benzylic C–H activation, to form a six-membered nickelacycle, would be excluded.

a tBu-DAPO–Ni–Al complex preparation and reactivity. b Kinetic isotope effect of formyl C–H bond (left), and intra- (middle) and intermolecular (right) kinetic isotope effect of benzylic C–H bond. c 3-Phenylpropyl group instead of benzyl group led to complete γ-C(sp3)–H bond activation, suggesting that γ-C(sp3)–H bond were more reactive than β-C(sp3)–H bonds in the reaction. d Methyl group and tBuCH2 group instead of benzyl group led to no β-C(sp3)–H bond activation, suggesting the presence of benzyl group is critical to the reactivity. e Proposed mechanism.
Further DFT calculation shows that ligand-to-ligand H transfer pathway64,65 in the formyl C–H activation step is a more favorable process than oxidative addition pathway (Fig. 6a), which is in accordance with the observed kinetic isotope effect of formyl C–H bond.

The computations were performed at the B3LYP-D3(BJ)/def2- TZVPP-SMD(toluene)//B3LYP-D3(BJ)/def2-SVP level of theory. a Comparison between oxidative addition and ligand-to-ligand H transfer for formyl C–H bond activation, and the result shows that the latter is a preferred pathway. b Product-forming pathway via 4-membered nickelacycle. c Calculation on the activation Gibbs energy of γ-C(sp3)–H bond of TP group, indicating 17.1 kcal/mol (TS-iso), which is a little higher than the activation Gibbs energy of benzylic β-C(sp3)–H bond (15.6 kcal/mol, TS3).
The resulting intermediate IM4 undergoes σ-bond metathesis, a ligand exchange, and a turnover-limiting alkyne insertion via TS4 with an activation Gibbs energy of 23.1 kcal/mol (Fig. 6b), which is also in accordance with the observed kinetic isotope effect of benzylic C–H bond. Notably, DFT calculation shows that intermediate IM4 would not undergo alkyne insertion before the step of benzylic C–H bond activation, otherwise an unstable 8-membered ring intermediate would be formed, resulting in a high energy barrier (33.4 kcal/mol, TS3’). In addition, as shown in Fig. 6c, DFT calculation indicates that the activation Gibbs energy of γ-C(sp3)–H bond of TP group is 17.1 kcal/mol (TS-iso), which is a little higher than the activation Gibbs energy of β-C(sp3)–H bond (15.6 kcal/mol, TS3). This result is also in accordance with the observed selective activation of benzylic β-C(sp3)–H bond over γ-C(sp3)–H bonds of TP group.
In summary, we have developed a Ni-catalyzed benzylic β-C(sp3)–H bond activation of formamides with alkynes via highly-strained 4-membered nickelacycle, providing a series of α,β-unsaturated γ-lactams in up to 97% yield. Bulky protecting group can be easily removed for further elaboration of products. In addition, the formed γ-lactams not only widely exist in bioactive molecules, but also proved to be versatile synthetic precursors. Mechanistic experiments and DFT calculations showed that phosphine oxide-ligated Ni–Al bimetallic catalyst along with removable bulky N-protecting group play critical roles in controlling good efficiency and good selectivity. This method should find wider applications in other 3d metal-involved C–H metalation via 4-membered metallacycles.
Methods
General procedure benzylic β-C(sp3)–H bond
To a 15 mL oven-dried tube were added tBu-DAPO (4.4 mg, 10 mol%), Ni(cod)2 (5.5 mg, 10 mol%), dry degassed toluene (1.0 mL), benzyl formamide (0.2 mmol), alkyne (0.6 mmol) and AlEt3 (1.0 M in toluene, 80 μL, 40 mol%) sequentially in an N2-filled glove-box. The tube was sealed and removed out of the glove-box. After heated at 120 °C in a preheated dry block heater for 2 h, the mixture was cooled to r.t., quenched with 0.1 mL H2O, filtered through a short plug of silica gel (DCM as the eluent) and concentrated in vacuo to afford the crude product. Further purification by flash column chromatography on silica gel (eluting with EtOAc/n-hexane) gave the pure product.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information file. For the experimental procedures, computational details, additional computational results and data of NMR see Supplementary Methods in Supplementary Information file. For computed energies and cartesian coordinates of the stationary points see Supplementary Data 1. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 2089375. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/MyStructures/. Data is available from the corresponding authors upon request.
References
Chen, X., Engle, K, M., Wang, D.-H. & Yu, J.-Q. Palladium(II)-catalyzed C–H activation/C–C cross-coupling reactions: versatility and practicality. Angew. Chem. Int. Ed. 48, 5094–5115 (2009).
Lyons, T. W. & Sanford, M. S. Palladium-catalyzed ligand-directed C–H functionalization reactions. Chem. Rev. 110, 1147–1169 (2010).
Albrecht, M. Cyclometalation using d-block transition metals: fundamental aspects and recent trends. Chem. Rev. 110, 576–623 (2010).
Chen, Z. et al. Transition metal-catalyzed C–H bond functionalizations by the use of diverse directing groups. Org. Chem. Front. 2, 1107–1295 (2015).
Nelson, Y. S. L. et al. Empirical guidelines for the development of remote directing templates through quantitative and experimental analyses. J. Am. Chem. Soc. 144, 2793–2803 (2022).
Liron, F. & Knochel, P. Reactivity of stable neopentyl-Pd intermediates in the absence of nucleophile. Tetrahedron Lett. 48, 4943–4946 (2007).
Kim, H. S., Gowrisankar, S., Kim, S. H. & Kim, J. N. Synthesis of 6-oxacyclopropa[a]indene derivatives starting from Baylis–Hillman adducts via Pd-mediated C(sp3)–H activation. Tetrahedron Lett. 49, 3858–3861 (2008).
Huang, Q. & Larock, R. C. Synthesis of cyclopropanes by Pd-catalyzed activation of alkyl C–H bonds. Tetrahedron Lett. 50, 7235–7238 (2009).
Mao, J., Zhang, S.-Q., Shi, B.-F. & Bao, W. Palladium(0)-catalyzed cyclopropanation of benzyl bromides via C(sp3)–H bond activation. Chem. Commun. 50, 3692–3694 (2014).
Du, W., Gu, Q., Li, Z. & Yang, D. Palladium(II)-catalyzed intramolecular tandem aminoalkylation via divergent C(sp3)–H functionalization. J. Am. Chem. Soc. 137, 1130–1135 (2015).
Gutiérrez-Bonet, A. ́, Juliá-Hernández, F., de Luis, B. & Martin, R. Pd-catalyzed C(sp3)–H functionalization/carbenoid insertion: all-carbon quaternary centers via multiple C–C bond formation. J. Am. Chem. Soc. 138, 6384–6387 (2016).
Chung, D. S. et al. Palladium-catalyzed divergent cyclopropanation by regioselective solvent-driven C(sp3)–H bond activation. Angew. Chem. Int. Ed. 57, 15460–15464 (2018).
Clemenceau, A., Thesmar, P., Gicquel, M., Flohic, A. L. & Baudoin, O. Direct synthesis of cyclopropanes from gem-dialkyl groups through double C–H activation. J. Am. Chem. Soc. 142, 15355–15361 (2020).
McNally, A., Haffemayer, B., Collins, B. S. L. & Gaunt, M. J. Palladium-catalysed C–H activation of aliphatic amines to give strained nitrogen heterocycles. Nature 510, 129–133 (2014).
He, C. & Gaunt, M. J. Ligand-enabled catalytic C–H arylation of aliphatic amines by a four-membered-ring cyclopalladation pathway. Angew. Chem. Int. Ed. 54, 15840–15844 (2015).
Smalley, A. P., Cuthbertson, J. D. & Gaunt, M. J. Palladium-catalyzed enantioselective C–H activation of aliphatic amines using chiral anionic BINOL-phosphoric acid ligands. J. Am. Chem. Soc. 139, 1412–1415 (2017).
Wen, J. et al. Rhodium-catalyzed PIII-directed ortho-C–H borylation of arylphosphines. Angew. Chem. Int. Ed. 58, 2078–2082 (2019).
Wen, J., Dong, B., Zhu, J., Zhao, Y. & Shi, Z. Revealing silylation of C(sp2)/C(sp3)–H bonds in arylphosphines by ruthenium catalysis. Angew. Chem. Int. Ed. 59, 10909–10912 (2020).
Su, B. et al. Palladium-catalyzed oxidation of β‑C(sp3)–H bonds of primary alkylamines through a rare four-membered palladacycle intermediate. J. Am. Chem. Soc. 142, 7912–7919 (2020).
Keim, W. Nickel: an element with wide application in industrial homogeneous catalysis. Angew. Chem. Int. Ed. 29, 235–244 (1990).
Tasker, S. Z., Standley, E. A. & Jamison, T. F. Recent advances in homogeneous nickel catalysis. Nature 509, 299–309 (2014).
Ananikov, V. P. Nickel: the “spirited horse” of transition metal catalysis. ACS Catal. 5, 1964–1971 (2015).
Zweig, J. E., Kim, D. E. & Newhouse, T. R. Methods utilizing first-row transition metals in natural product total synthesis. Chem. Rev. 117, 11680–11752 (2017).
Khake, S. M. & Chatani, N. Nickel-catalyzed C–H functionalization using a non-directed strategy. Chem 6, 1056–1081 (2020).
Gandeepan, P. et al. 3d Transition metals for C–H activation. Chem. Rev. 119, 2192–2452 (2019).
Pototschnig, G., Maulide, N. & Schnerch, M. Direct functionalization of C–H bonds by iron, nickel, and cobalt catalysis. Chem. Eur. J. 23, 9206–9232 (2017).
Yamaguchi, J., Muto, K. & Itami, K. Nickel-catalyzed aromatic C–H functionalization. Top. Curr. Chem. 374, 1–33 (2016).
Nakao, Y. Hydroarylation of alkynes catalyzed by nickel. Chem. Rec. 11, 242–251 (2011).
Liu, Y.-H., Xia, Y.-N. & Shi, B.-F. Ni-Catalyzed chelation-assisted direct functionalization of inert C–H bonds. Chin. J. Chem. 38, 635–662 (2020).
Khake, S. M. & Chatani, N. Chelation-assisted nickel-catalyzed C–H functionalizations. Trends Chem. 1, 524–539 (2019).
Mishra, A. A., Subhedar, D. & Bhanage, B. M. Nickel, cobalt and palladium catalysed C–H functionalization of un-activated C(sp3)–H bond. Chem. Rec. 19, 1829–1857 (2019).
Misal Castro, L. C. & Chatani, N. Nickel catalysts/N,N’-bidentate directing groups: an excellent partnership in directed C–H activation reactions. Chem. Lett. 44, 410–421 (2015).
Su, B., Cao, Z.-C. & Shi, Z.-J. Exploration of Earth-abundant transition metals (Fe, Co, and Ni) as catalysts in unreactive chemical bond activations. Acc. Chem. Res. 48, 886–896 (2015).
Yin, G. et al. Ligand-controlled Ni(0)-Al(III) bimetal-catalyzed C3–H alkenylation of 2-pyridones by reversing conventional selectivity. ACS Catal. 11, 4606–4612 (2021).
Zhang, T., Luan, Y.-X., Zheng, S.-J., Peng, Q. & Ye, M. Chiral aluminum complex controls enantioselective nickel-catalyzed synthesis of indenes: C–CN bond activation. Angew. Chem. Int. Ed. 59, 7439–7443 (2020).
Qi, S.-L., Liu, Y.-P., Li, Y., Luan, Y.-X. & Ye, M. Ni-catalyzed hydroarylation of alkynes with unactivated β-C(sp2)−H bonds. Nat. Commun. 13, 2938–2946 (2022).
Liu, Q.-S. et al. Ni–Al bimetallic catalyzed enantioselective cycloaddition of cyclopropyl carboxamide with alkyne. J. Am. Chem. Soc. 139, 18150–18153 (2017).
Nakao, Y., Kanyiva, K. S. & Hiyama, T. A strategy for C–H activation of pyridines: direct C-2 selective alkenylation of pyridines by nickel/Lewis acid catalysis. J. Am. Chem. Soc. 130, 2448–2449 (2008).
Nakao, Y., Idei, H., Kanyiva, K. S. & Hiyama, T. Direct alkenylation and alkylation of pyridone derivatives by Ni/AlMe3 catalysis. J. Am. Chem. Soc. 131, 15996–15997 (2009).
Tsai, C.-C. et al. Bimetallic nickel aluminum mediated para-selective alkenylation of pyridine: direct observation of η2, η1-pyridine Ni(0)–Al(III) intermediates prior to C–H bond activation. J. Am. Chem. Soc. 132, 11887–11889 (2010).
Nakao, Y., Yamada, Y., Kashihara, N. & Hiyama, T. Selective C-4 alkylation of pyridine by nickel/Lewis acid catalysis. J. Am. Chem. Soc. 132, 13666–13668 (2010).
Diesel, J., Finogenova, A. M. & Cramer, N. Nickel-catalyzed enantioselective pyridone C–H functionalizations enabled by a bulky N-heterocyclic carbene ligand. J. Am. Chem. Soc. 140, 4489–4493 (2018).
Zhang, W. B., Yang, X. T., Ma, J. B., Su, Z. M. & Shi, S. L. Regio- and enantioselective C–H cyclization of pyridines with alkenes enabled by a nickel/N-heterocyclic carbene catalysis. J. Am. Chem. Soc. 141, 5628–5634 (2019).
Li, J.-F. et al. Construction 7-membered ring via Ni–Al bimetal-enabled C–H cyclization for synthesis of tricyclic imidazoles. Nat. Comm. 12, 3070–3078 (2021).
Donets, P. A. & Cramer, N. Diaminophosphine oxide ligand enabled asymmetric nickel-catalyzed hydrocarbamoylations of alkenes. J. Am. Chem. Soc. 135, 11772–11775 (2013).
Wang, Y.-X. et al. Enantioselective Ni−Al bimetallic catalyzed exo-selective C−H cyclization of imidazoles with alkenes. J. Am. Chem. Soc. 140, 5360–5364 (2018).
Wang, Y.-X. & Ye, M. Recent advances in Ni–Al bimetallic catalysis for unreactive bond transformation. Sci. China Chem. 61, 1004–1013 (2018).
Li, J.-F., Luan, Y.-X. & Ye, M. Bimetallic anchoring catalysis for C–H and C–C activation. Sci. China Chem. 64, 1923–1937 (2021).
Park, H. & Yu, J.-Q. Palladium-catalyzed [3+2] cycloaddition via twofold 1,3-C(sp3)–H activation. J. Am. Chem. Soc. 142, 16552–16556 (2020).
Kashanna, J., Kumarb, R. A. & Kishorec, R. Palladium(II)-catalyzed synthesis of indenones through the cyclization of benzenecarbaldehydes with internal alkynes. RSC Adv. 9, 31162–31168 (2019).
Gulías, M. & Mascareñas, J. L. Metal-catalyzed annulations through activation and cleavage of C–H bonds. Angew. Chem. Int. Ed. 55, 11000–11019 (2016).
Tan, P. W., Juwaini, N. A. B. & Seayad, J. Rhodium(III)-amine dual catalysis for the oxidative coupling of aldehydes by directed CH activation: synthesis of phthalides. Org. Lett. 15, 5166–5169 (2013).
Chen, S., Yu, J., Jiang, Y., Chen, F. & Cheng, J. Rhodium-catalyzed direct annulation of aldehydes with alkynes leading to indenones: proceeding through in situ directing group formation and removal. Org. Lett. 15, 4754–4757 (2013).
Kuninobu, Y., Matsuki, T. & Takai, K. Rhenium-catalyzed synthesis of indenones by novel dehydrative trimerization of aryl aldehydes via C-H bond activation. Org. Lett. 12, 2948–2950 (2010).
Nakao, Y., Morita, E., Idei, H. & Hiyama, T. Dehydrogenative [4+2] cycloaddition of formamides with alkynes through double C–H activation. J. Am. Chem. Soc. 133, 3264–3267 (2011).
Anand, M. & Sunoj, R. B. Mechanism of cooperative catalysis in a Lewis acid promoted nickel-catalyzed dual C–H activation reaction. Org. Lett. 14, 4584–4587 (2012).
Chen, H., Wang, Y.-X., Luan, Y.-X. & Ye, M. Enantioselective twofold C–H annulation of formamides and alkynes without built-in chelating groups. Angew. Chem. Int. Ed. 59, 9428–9432 (2020).
Wang, Y.-X., Zhang, F.-P., Luan, Y.-X. & Ye, M. Ligand-enabled Ni–Al bimetallic catalysis for nonchelated dual C–H annulation of arylformamides and alkynes. Org. Lett. 22, 2230–2234 (2020).
Wang, R.-H. et al. Selective C(sp3)–H cleavage of enamides for synthesis of 2‑pyridones via ligand-enabled Ni–Al bimetallic catalysis. ACS Catal. 11, 858–864 (2021).
Barrios-Francisco, R., Benítez-Páez, T., Flores-Alamo, M., Arévalo, A. & García, J. J. Nickel(0) complexes with fluorinated alkyne ligands and their reactivity towards semihydrogenation and hydrodefluorination with water. Chem. Asian J. 6, 842–849 (2011).
Pasynkiewicz, S., Pietrzykowski, A., Kryza-Niemiec, B. & Zachara, J. The first structurally characterized trinickel cluster with an open structure: crystal and molecular structure of CpNi[μ-η4-PhC:C(Ph)-C(Ph):CPh]Ni(μ-η2η2-PhC≡CPh)NiCp. J. Organomet. Chem. 566, 217–224 (1998).
Suneja, A., Unhale, R. A. & Singh, V. K. Enantioselective hydrophosphonylation of in situ generated N-acyl ketimines catalyzed by BINOL-derived phosphoric acid. Org. Lett. 19, 476–479 (2017).
Sharma, S. et al. Rhodium-catalyzed [3+2] annulation of cyclic N-acyl ketimines with activated olefins: anticancer activity of spiroisoindolinones. J. Org. Chem. 82, 3359–3367 (2017).
Guihaumé, J., Halbert, S., Eisenstein, O. & Perutz, R. N. Hydrofluoroarylation of alkynes with Ni catalysts. C–H activation via ligand-to-ligand hydrogen transfer, an alternative to oxidative addition. Organometallics 31, 1300–1314 (2012).
Tang, S., Eisenstein, O., Nakao, Y. & Sakaki, S. Aromatic C–H σ-bond activation by Ni0, Pd0, and Pt0 alkene complexes: concerted oxidative addition to metal vs ligand-to-ligand H transfer mechanism. Organometallics 36, 2761–2771 (2017).
Acknowledgements
We thank the National Natural Science Foundation of China (21871145, M.Y.; 22188101, M.Y.; 22073066, G.H.; 21503143, G.H.; 21975179, G.H.), the Haihe Laboratory of Sustainable Chemical Transformations and “Frontiers Science Center for New Organic Matter”, Nankai University (63181206, M.Y.) for financial support.
Author information
Authors and Affiliations
Contributions
R.-H.W. discovered and developed the reactions. W.-W.X. extended substrate scope and complement mechanistic experiments. Y.L., J.-F.L., and T.Z. performed part of synthetic experiments. H.W. and G.H. performed the DFT calculations and analyzed the computational results; M.Y. conceived, designed the investigations, and wrote the manuscript. R.-H.W. and W.-W.X. wrote the Supplementary Information.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, RH., Xu, WW., Wu, H. et al. Ni-catalyzed benzylic β-C(sp3)–H bond activation of formamides. Nat Commun 13, 7892 (2022). https://doi.org/10.1038/s41467-022-35541-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-35541-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
