
Abstract
Hydrogen energy is critical for achieving carbon neutrality. Heterostructured materials with single metal-atom dispersion are desirable for hydrogen production. However, it remains a great challenge to achieve large-scale fabrication of single atom-anchored heterostructured catalysts with high stability, low cost, and convenience. Here, we report single iron (Fe) atom-dispersed heterostructured Mo-based nanosheets developed from a mineral hydrogel. These rationally designed nanosheets exhibit excellent hydrogen evolution reaction (HER) activity and reliability in alkaline condition, manifesting an overpotential of 38.5 mV at 10 mA cm−2, and superior stability without performance deterioration over 600 h at current density up to 200 mA cm−2, superior to most previously reported non-noble-metal electrocatalysts. The experimental and density functional theory results reveal that the O-coordinated single Fe atom-dispersed heterostructures greatly facilitated H2O adsorption and enabled effective adsorbed hydrogen (H*) adsorption/desorption. The green, scalable production of single-atom-dispersed heterostructured HER electrocatalysts reported here is of great significance in promoting their large-scale implementation.
Similar content being viewed by others

Introduction
Hydrogen (H2) is an attractive source of energy due to its high energy density and emission of few pollutants when combusted in air1,2,3. Among the wide range of H2 generation methods, the electrochemical HER is particularly facile and economical4,5. The high cost of commercial noble metal-based HER electrocatalysts motivates researchers to develop low-cost high-activity electrocatalysts with high electrical conductivity and high stability on a large scale, which remains a challenging task6,7. Single-atom catalysts (SACs) have been extensively explored for use in catalytic HER application due to their high activity, maximized atomic efficiency and minimized catalyst usage of a given process8,9. Many efforts had been devoted to constructing various kinds of single-atom catalysts, such as single-atom/carbon, single-atom/metal oxide, single-atom/metal sulphide, single-atom alloy10.
Single-atom catalysts were usually fabricated by anchoring targeted single-atom metal on different substrates by introducing the targeted single-atom metal to the substrate precursor and following a post-heating treatment11,12. In order to achieve favorable loading of the single atom and strong interaction between single-atoms and the substrates, large number of defects, heterogenous atom dopants, favorable facets or abundant interfaces in the substrates are required,which greatly increase their fabrication difficulty11. Thus, simplifying the preparing process for their substrates and increasing the universality in dispersing targeted single atoms are vital to expand the diversity and prevailingness of single-atom materials. Nevertheless, most of reported methods suffer from complex fabrication process, low dispersion of single-atom metal, high post-treatment temperature (usually higher than 700 °C), and environmentally unfriendly issues involved, impeding the practical use of SACs. In addition, to maximize the exposure of the single atomic sites, the substrates prefer to low dimensional, porous, and abundant grain boundaries, which brings even higher requirements for SACs preparation. For example, using the porous metal-organic frameworks (MOFs) or covalent organic frameworks (COFs) as a precursor to construct SACs is recently a promising method13,14. However, the synthesis and purification of MOFs/COFs, as well as the follow-up metal species absorption process are extremely a time-consuming process. In other practices, the targeted metal species were added in advance during the MOFs/COFs preparation process, which may disturb the frameworks formation of MOFs/COFs. In addition, the utilization of organic, complex synthetic parameters, high and precisely-control of pyrolysis temperature, release of organic exhaust gas during MOFs/COFs preparation and their carbonization process, are regarded as unsustainable, unfriendly, and tedious methods, seriously restricting the flexibility and universality of this strategy15. These issues substantially impede the feasibility of this method. Oppositely, the synthesis of heterostructure constructed by metal oxides/phosphides for SACs preparation could be easier and more convenient16. Heterostructure featured with the low dimensional and porous characters can vastly increase the loading of single-atoms and the expose of active sites, making it an ideal single-atom carrier. Therefore, it is urgent to develop robust single-atom-dispersed heterostructured nanosheets featuring simplicity, environmentally benignity, and high effiency17,18.
Mineral hydrogel is made by inorganic matter, inorganic salt and water, and can self-assemble into gel networks with various morphologies via a rapid and straightforward process of mixing and gelation under mild conditions19,20. The coordinated metal ions, which are singly dispersed in the gel networks, can convert into a heterostructure of metal oxides/phosphides/sulphides following a post-heating treatment. A single Fe atoms N-doped carbon was successfully prepared with a Fe-Polypyrrole supramolecular hydrogel by Yu’s group according to the highly dispersed metal character21. However, the conversion of supramolecular hydrogel to a carbon usually need a high temperature and the yield of carbonization is usually low. Encouragingly, some metals can keep single dispersion state in the heterostructured material derived from mineral hydrogel due to the strong interaction with the abundant oxygen/phosphorus atoms and plentiful interfaces, and can convert to heterostructured material at a low temperature. Therefore, mineral hydrogel could act as an excellent candidate for single-atom catalyst synthesis. In addition, the strong ionic character of the mineral hydrogel can easily accommodate highly dispersed metal ionic additives and transition metal salts which means that the targeted single-atom can be easily manipulated. These additives can be added before the gelation process, avoiding the long-time metal absorption procedure of the porous precursor. More importantly, we can expand the metal selection range to cheaper mineral metals to construct mineral hydrogel, alleviating the high cost of precious metals and unfriendly environmental problems during the synthetic process. In summary, compared with other common single atom substrate precursors (porous frameworks and carbon), mineral hydrogels show great advantages in terms of synthetic route, environment-friendly, production efficiency, tunability, raw materials, sustainability, university, and cost-effectiveness (Fig. 1a and Supplementary Table 1).

a General characteristics comparison of the common substrate precursors for SACs (more details presented in Table S1); b schematic of synthesis of the Fe/SAs@Mo-based-HNSs eletrocatalyst; c SEM image, d TEM image, with inset of the SAED pattern, e high-resolution TEM image, and f STEM image of an FePMoG with an Fe3+: PMo molar ratio of 25:1, and the corresponding EDS elemental mapping of Fe (green), Mo (violet), phosphorus (P; orange), and oxygen (O; cyan).
In this work, we propose using Mo-based mineral hydrogel as a precursor for designing a novel carbon (C)-free single Fe-atom-dispersed heterostructured nanosheets. Firstly, a novel 2D wrinkle-like iron–phosphomolybdic acid mineral hydrogel nanosheets (FePMoG) was simply synthesized in one step as a precursor, which was directly converted to carbon (C)-free single Fe-atom-dispersed heterostructured Mo-based nanosheets (Fe/SAs@Mo-based-HNSs) via a one-step low-temperature phosphorisation process. The 2D wrinkle-like nanosheet morphology of FePMoG is entirely different and new in mineral hydrogel, and the preparation of FePMoG is simple and environmentally benign through a self-assembly inorganic–inorganic coordination process: that is, in water at room temperature without the addition of any surfactants or additives. The porosity and heterostructure of the phosphorised nanosheets derived from the FePMoG nanosheets were easily regulated during the phosphorisation process by adjusting the reaction temperature and time. It is important to mention that this single-atom-dispersed heterostructured nanosheets was first developed from a mineral hydrogel. We found that Fe/SAs@Mo-based-HNSs exhibited excellent electrocatalytic activity and long-term durability in the HER: an overpotential of 38.5 mV at 10 mA cm−2 with a Tafel slope of 35.6 mV dec−1 and a negligible increase in polarisation potential and overlapping polarisation curves upon 600 h continuous operation at 200 mA cm−2. This is one of the highest performances in the HER demonstrated by any nanostructured Mo-based electrocatalyst, and is attributable to the optimized single atom-dispersed heterostructure of Fe/SAs@Mo-based-HNSs, with a large active surface area and high porosity. This heterostructure contains numerous interfaces and a facile O-coordinated monoatomic dispersed Fe atoms, and thus has high H2O adsorption ability ad appropriate H* adsorption/desorption ability.
Results
Synthesis and characterization of as-prepared FePMoG and Fe/SAs@Mo-based-HNSs eletrocatalyst
Figure 1b illustrates our novel coordination-induced self-assembly process for preparing the FePMoGs nanosheets. This process involved simply mixing a solution of polyoxometallate acid (PMo) and ferric ions (Fe3+) at room temperature, which is much more convenient than previously reported processes that typically require a template or solvothermal treatment22,23. A suspension of homogeneously distributed FePMoG nanosheets was formed by this process of coordination-induced self-assembly, and was subsequently centrifuged (Centrifugation speeds: 447 g) to remove excess water to enable the next step (Supplementary Fig. 1). During the self-assemble process, the empty orbital in Fe3+ species coordinated with the molybdate groups of PMo, resulting in the self-assembly of FePMoG compounds. A 50-fold amplification of the synthesis experiment for this mineral hydrogel was successfully conducted (Supplementary Fig. 2), together with the simple experimental condition and procedure, indicating our method is amenable for large-scale production. Concentration-dependent experiment with various molar ratios of Fe3+: PMo shows that the mixture comprising a 25:1 molar ratio of Fe3+: PMo can form wrinkled nanosheet (Fig. 1c, Supplementary Fig. 3–5) with a width of micrometres and a thickness of ~50 nm, whose surface was otherwise smooth. Time-dependent experiment of the self-assembly FePMoG nanosheet (25:1) revealed that the wrinkled nanosheets gradually evolved from apophyses (Fig. 1b, Supplementary Fig. 6, 7). To demonstrate the applicability of the mineral hydrogel for producing other monatomic dispersed catalysts, different ions (e.g., Co2+, Ni2+, Cu2+, Ag+, or Mn3+, ~ 3 at.% of the sum of Fe and Mo atoms) were added to the Fe3+ solution and mixed with the phosphomolybdic acid to prepare different mineral hydrogels. As shown in Supplementary Fig. 8, the addition of the other ions did not affect the formation of the mineral hydrogel.
The X-ray diffraction (XRD) pattern of the FePMoG (25:1) has a prominent broad peak at 27.5° and a weak broad peak at 55° (Supplementary Fig. 9a), which are completely distinct from the sharp peaks of the crystalline structures of pure PMo and iron (III) nitrate nonahydrate24,25, indicating the amorphous state and lamellar structure of this FePMoG and the strong interactions between the phosphomolybdate and metallic ions26. Fourier-transform infrared (FTIR) spectroscopy of FePMoG (25:1) was performed to determine the interactions of its chemical bonds (Supplementary Fig. 9b), which revealed a distinct set of bands characteristic of a Keggin structure derived from PMo27,28,29, and strong interactions between Mo and various forms of O as well as between Fe3+ and PMo–heteropolyanions27,28. The slight shift of the Mo (VI) oxidation state to a higher binding energy (from 232.3 to 232.4 eV for Mo6+ 3d3/2 and from 235.4 to 235.6 eV for Mo6+ 3d5/2 in the Mo 3d X-ray photoelectron spectroscopy (XPS) curve) in the FePMoG is also attributable to the coordination between phosphomolybdate and Fe3+ (Supplementary Fig. 10)30. These FTIR and XPS data indicate that this FePMoG is formed by strong interactions between Fe3+ and PMo.
Transmission electron microscopy (TEM) was used to further analyse the structure and elemental distribution of FePMoG. The TEM image of the FePMoG in Fig. 1d demonstrates the 2D structure of FePMoG composites, and the selected-area electron diffraction (SAED) patterns (inset in Fig. 1d) contain a broad and diffuse ring, confirming this compound’s amorphous nature. High-resolution TEM images (Fig. 1e) show the disordered arrangement of atoms in the amorphous FePMoG. The scanning transmission electron microscopy (STEM) image and corresponding energy-dispersive spectroscopy (EDS) elemental mapping (Fig. 1f) of FePMoG reveal the uniform distribution of Fe, Mo, P, and O atoms within its structure, indicating that it consists of uniform interactions between Fe3+ and PMo.
The structural evolution of FePMoG nanosheets after phosphorization is shown in Fig. 2 and Supplementary Fig. 11. FeMoP-T represents samples generated at various pyrolysis temperatures (T); for example, FeMoP-500 represents Fe/SAs@Mo-based-HNSs generated by pyrolysis at 500 °C. XRD patterns of Fe/SAs@Mo-based-HNSs reveal the characteristic diffraction peaks of molybdenum dioxide (MoO2, JCPDS No. 78-1069), molybdenum phosphide (MoP, JCPDS No. 24-0771), and molybdenum diphosphide (MoP2, JCPDS No. 89-2678) (Fig. 2a). We also determined that the heterostructure of the interconnected nanoparticle subunits could be tailored by varying the pyrolysis temperature (Supplementary Fig. 11 and Supplementary Table 2). Iron phosphide (FeP; JCPDS No. 89-2597) and MoO2 phases were formed when the pyrolysis temperature was <450 °C and <550 °C, respectively, whereas MoP and MoP2 phases were formed when the pyrolysis temperature was ≥400 °C and ≥450 °C, respectively. The 2D porous nanosheet structure of the Fe/SAs@Mo-based-HNSs maintained its initial nanosheet morphology during the process of self-templated phosphorisation (Fig. 2b), whereas a simple mixture of an Fe salt and PMo (denoted as bulk FeMoP-500) subjected to phosphorisation produced micrometre-scale aggregates of heterostructured nanoparticles (Supplementary Fig. 12). This indicates that the formation of the strongly coordinated FePMoG plays a decisive role in the phosphorisation-mediated formation of the 2D porous nanosheet structure of Fe/SAs@Mo-based-HNSs. The corresponding SAED pattern contains rings ascribable to MoO2, MoP, and MoP2 (Fig. 2c). A high-magnification TEM image shows that Fe/SAs@Mo-based-HNSs (Fig. 2d) comprise an interconnected structure of pores with an average diameter of <10 nm. The lattice fringes in Fig. 2e were found to be 0.281, 0.243, 0.278, 0.209, 0.258, and 0.217 nm wide, corresponding to the interplanar distance of MoO2 (−102), MoO2 (020), MoP (100), MoP (101), MoP2 (111), and MoP2 (131), respectively, indicating the heterostructure of Fe/SAs@Mo-based-HNSs. The HAADF-STEM image of the Fe/SAs@Mo-based-HNSs and the corresponding EDS elemental mapping (Fig. 2f) reveal the uniform distribution of Fe, Mo, P, and O atoms and the Fe atoms were independently dispersed in the lattice, validating that an in-situ transformation occurs during the phosphorisation process and the single atoms dispersion of Fe. The samples formed at other pyrolysis temperatures have similar heterostructures, e.g., FeMoP-450 has four phases (FeP, MoO2, MoP, and MoP2; Supplementary Fig. 13), and FeMoP-550 has three phases (MoO2, MoP, and MoP2; Supplementary Fig. 14), respectively. This demonstrates that the heterostructure in the phosphorised materials were easily obtained and modulated by controlling the annealing temperatures, which is accordance with the above XRD results.

a XRD patterns; b low-magnification TEM image, c SAED pattern, d high-magnification TEM image, e HRTEM image, and f HAADF-STEM image and the corresponding EDS elemental mapping of Fe (green), Mo (violet), P (orange) and O (cyan).
The Brunauer–Emmett–Teller (BET) and pore-size distribution of the Fe/SAs@Mo-based-HNSs show a surface area of 89.6 m2 g−1 with many mesopores (pore diameters 1.3–12 nm) (Supplementary Fig. 15). This mesoporous and interconnected 2D structure is likely responsible for the efficient electrocatalysis of the HER by Fe/SAs@Mo-based-HNSs, as this structure would greatly enhance the accessibility of the rich active sites of Fe/SAs@Mo-based-HNSs and increase the infiltration of electrolytes, thereby facilitating charge transfer.
XPS and synchrotron-radiation-based hard X-ray absorption near-edge structure (XANES) were performed to characterise the chemical composition, valence and coordination states, and electronic structure of Fe/SAs@Mo-based-HNSs (Fig. 3). The full-scan XPS spectrum confirms that Fe/SAs@Mo-based-HNSs comprise Fe, Mo, P, and O atoms (Supplementary Fig. 16). As depicted in Fig. 3a, the high-resolution XPS spectrum of Mo 3d consists of six peaks representing three ionic species: 231.66 and 228.34 eV (Mo3+), 232.58 and 229.68 eV (Mo4+), and 235.63 and 233.49 eV (Mo6+), corresponding to MoP, MoO2, and MoP2, respectively31,32,33. The Mo K-edge XANES spectrum shows that the near-edge absorption energy of Fe/SAs@Mo-based-HNSs is intermediate between that of Mo foil and MoO2, demonstrating that the average oxidation state of Mo is between Mo0 and Mo4+ (Fig. 3d). The corresponding extended X-ray absorption fine structure (EXAFS) reveals that dispersed Mo atoms in Fe/SAs@Mo-based-HNSs are coordinated with P and O atoms, with no Mo–Mo bonding apparent (Fig. 3e, f, Supplementary Fig. 17, and Supplementary Table 3).

a Mo 3d XPS spectra; b Fe 2p XPS spectra; c P 2p XPS spectra; d Mo K-edge XANES spectra; e corresponding k2-weighted FT of EXAFS spectra; f wavelet transforms for k2-weighted EXAFS signals at Mo K-edge; g Fe K-edge XANES spectra; h corresponding k3-weighted FT of EXAFS spectra; and i wavelet transforms for k3-weighted EXAFS signals at Fe K-edge.
In terms of the oxidation state of Fe, the high-resolution XPS spectrum of Fe (Fig. 3b) has peaks at 710.7 eV (Fe 2p3/2) and 724.1 eV (Fe 2p1/2), which are characteristic of Fe3+, and an Fe3+ 2p satellite peak at 715.6 eV. Similarly, the Fe K-edge XANES spectrum also shows that the valency of Fe is close to that of Fe3+ (Fig. 3g). The corresponding EXAFS results delineate the coordination environment of Fe atoms: the strong peak at ~1.5 Å is attributable to Fe–O bonding, and no Fe–Fe peak is present at 2.47 or 2.85 Å in the Fourier-transform-EXAFS (Fig. 3h), suggesting that the Fe is largely isolated in Fe/SAs@Mo-based-HNSs, and is mostly coordinated with O (Fig. 3i and Supplementary Fig. 18)34,35. Specifically, one Fe atom is coordinated with 5.9 O atoms, and the mean Fe–O bond distance is 1.98 Å (Supplementary Fig. 18 and Supplementary Table 4). The XPS peak centred at 130.39 and 129.54 eV in the P 2p region (Fig. 3c) represents P bonded to Mo in MoP and MoP2, while the P 2p3/2 peak at 134.18 eV represents the typical oxidation peak of P in residual phosphate36. The XPS peak at 454.7 eV in the O 1 s spectrum corresponds to O2− (Supplementary Fig. 19), confirming the presence of MoO2. The Fe content in the FeMoP-T catalysts substantially decreased as the annealing temperature was increased (Supplementary Table 5–7), which suggests slight evaporation of Fe species at high pyrolysis temperatures. Although the greater loss of Fe species in a higher pyrolysis temperature (e.g., in 550 °C), the residual Fe atoms were also in the form of monoatomic dispersion (Supplementary Fig. 20, Supplementary Table 8).
Electrocatalytic alkaline HER performances
To highlight the electrocatalytic advantages of the synthetic heterostructured nanosheets, the performance of Fe/SAs@Mo-based-HNSs in the HER was investigated using a typical three-electrode system in 1 M KOH solution. The linear sweep voltammograms shown in Fig. 4a and Supplementary Fig. 21a illustrate that the Fe/SAs@Mo-based-HNSs exhibited excellent performance in the HER: an overpotential of 38.5 mV vs reversible hydrogen electrode (RHE) at 10 mA cm−2, which is much lower than those of other FeMoP-T samples and commercial 20 wt% Pt/C. The overpotential of Fe/SAs@Mo-based-HNSs slightly increase to 109.9 mV when the current density up to 200 mA cm−2, significantly lower than that of commercial 20 wt% Pt/C (299.3 mV). The polarization curves of three independent Fe/SAs@Mo-based-HNSs prepared from different batches almost overlapped (Supplementary Fig. 22), indicating good repeatability of Fe/SAs@Mo-based-HNSs. The electrocatalytic HER mechanisms of the Fe/SAs@Mo-based-HNSs were examined by constructing Tafel plots (Fig. 4b, Supplementary Fig. 21b, and Supplementary Table 9), wherein it can be seen that they have a smaller slope (35.6 mV dec−1) than those of bulk FeMoP-500 (89.3 mV dec−1) and other FeMoP-T samples, close to that of 20% Pt/C (36.1 mV dec−1). This indicates that a fast Heyrovsky step dominates the Volmer–Heyrovsky process during Fe/SAs@Mo-based-HNSs-electrocatalysed H2 evolution, which is attributable to the strong electronic coupling within the single dispersed atoms and heterostructure.

a Polarisation curves at a scan rate of 5 mV s−1 with potential error (iR) correction; b corresponding Tafel plots; c Electrochemical impedance spectroscopy (EIS) of the FeMoP-T samples and 20% Pt/C at an overpotential of 200 mV; d calculated electrochemical double-layer capacitance for selected as-prepared materials; e Faradaic efficiency of Fe/SAs@Mo-based-HNSs at different applied potentials. The error bar reflects the three device results; f Hydrogen production at a specific constant current density of 10 mA/cm2. The error bar reflects the three device results; g long-duration curves at a constant current density of 200 mA cm−2.
Low charge-transfer resistance is a characteristic of an effective electrocatalyst, especially a C-free electrocatalyst37,38. Thus, electrochemical impendence spectroscopy was conducted at an overpotential of 200 mV to obtain charge-transfer data. As shown in Fig. 4c, the Fe/SAs@Mo-based-HNSs have a much smaller semicircular diameter than the other phosphide samples, which is very similar to that of 20 wt% Pt/C; this suggests that the unique heterostructure with single-atom dispersed nature of Fe SAs@Mo-based HNSs, in addition to their 2D porous nanosheet morphology, contribute substantially to their optimised electronic structure. As variation in the surface area of electrocatalysts may affect their HER performance, the electrochemical surface areas of these electrocatalysts were estimated by measuring their electrochemical double-layer capacitances (Cdl) by cyclic voltammetry, which is typically considered representative of electrochemically active surface area (Supplementary Fig. 23). The Cdl of Fe/SAs@Mo-based-HNSs was found to be 78.4 mF cm−2 (Fig. 4d), which is much higher than that of the directly phosphatised bulk FeMoP-500 (10.9 mF cm−2) and those of the FeMoP-450 and FeMoP-550 samples (36.1 and 30.8 mF cm−2, respectively). Moreover, although the Fe/SAs@Mo-based-HNSs have a similar morphology, structure, and specific surface area to FeMoP-450 and FeMoP-550, the catalytic current density of Fe/SAs@Mo-based-HNSs at an overpotential of 100 mV is 14.2 times that of FeMoP-450, which is in turn only 3.6 times that of the bulk FeMoP-500. This suggests that the high HER electrocatalytic activity of Fe/SAs@Mo-based-HNSs is largely attributable to the optimised electronic coupling in their abundant heterostructured active sites, and is supported by the very high electrolyte-accessible surface area of their 2D porous networks. Subsequently, gas chromatography was employed to detect the H2 production and to determine the Faradaic efficiency. As can be seen in Fig. 4e, the Faradaic efficiency of Fe/SAs@Mo-based-HNSs is nearly 100% under a wide range of potentials. Controlled-current electrolyses at −10 mA cm−2 were further conducted to compare the Faradaic efficiency with commercial 20 wt% Pt/C and bulk FeMoP-500, wherein it can be seen that the yield of H2 of Fe/SAs@Mo-based-HNSs increases linearly over time and is significantly higher than that of commercial 20 wt% Pt/C and bulk FeMoP-500 (Fig. 4f and Supplementary Table 10). The Faradaic efficiency of Fe/SAs@Mo-based-HNSs after a 2-h period is 99.26%, which is higher than those of commercial 20 wt% Pt/C (89.46%) and bulk FeMoP-500 (52.74%).
The stability of the Fe/SAs@Mo-based-HNSs was also found to be excellent: they maintained a current density of 200 mA cm−2 in the HER over a 600 h durability test, which represents ultrareliable performance. Specifically, as shown in Fig. 4g, the polarisation potential of Fe/SAs@Mo-based-HNSs exhibits little amplification of overpotential, that is only ~30 mV decreases at 200 mA cm−2. The Supplementary Fig. 24 depicts the initial and post-500 h test polarisation curves of Fe/SAs@Mo-based-HNSs at 20 mA cm−2, showing that they are extremely similar except in the area of the boosted onset potential. Moreover, the TEM (Supplementary Fig. 25), XRD (Supplementary Fig. 26), and XPS measurements (Supplementary Fig. 27) show that the Fe/SAs@Mo-based-HNSs retained porous multinary heterostructures with disordered edges after this durability test at 20 mA cm−2, demonstrating that their heterostructure and valence states were well preserved and attesting to their excellent stability during the HER. The concentrations of elemental Fe, Mo, and P dissolved in solution after the durability test were measured by inductively coupled plasma–optical emission spectrometry (ICP-OES) (Supplementary Table 11). The concentration of all elements was close to the limit of reporting for the ICP-OES instrument, which is in accordance with the only slight change in the elemental composition of Fe/SAs@Mo-based-HNSs seen in the XPS analysis (Supplementary Table 5), and further illustrates the high stability of Fe/SAs@Mo-based-HNSs.
To further highlight the excellent electrocatalytic performance of our Fe/SAs@Mo-based-HNSs, we compared their overpotentials at a current density of 10 mA cm−2 and the corresponding Tafel slope with those of various other electrocatalysts that have been reported for use in the HER in an alkaline medium (more details presented in Table S12). We found that the overpotential and the Tafel slope of the Fe/SAs@Mo-based-HNSs were superior to those of many non-precious-metal HER electrocatalysts and comparable to that of most of noble metal-based electrocatalysts. Thus, the ultrastability of our novel C-free porous 2D heterostructured Mo-based electrocatalysts may indicate they electrocatalyse the HER in alkaline media via a new and as-yet underdeveloped mechanism. If so, this could possibly be exploited for the design of high-performance HER electrocatalysts.
Theoretical investigations
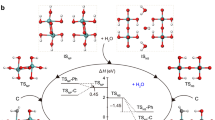
Previously reported heterostructured and single atom-dispersed HER electrocatalysts have been based on either active N-coordinated or C-coordinated metal sites, and no experimental or theoretical studies have been published describing O-coordinated single metal-atom dispersion heterostuctured HER electrocatalysts. We thus performed systematic DFT calculations to determine the molecular basis of the outstanding electrocatalytic activity of Fe/SAs@Mo-based-HNSs in the HER. It is well known that a key factor in the HER performance of electrocatalysts is the magnitude of the energy required for the adsorption of water molecules onto catalytic sites (ΔEH2O), especially in alkaline media39. Thus, ΔEH2O values of various sites extracted from our experimental observations were calculated. This involved the construction of three single-phase models (MoP, MoP2, MoO2), three heterostructured interfacial models (MoP/MoP2, MoP/MoO2, MoP2/MoO2), and two monoatomic-dispersed models (Fe@MoO2−1, Fe@MoO2−2), based on the microstructures we identified (Figs. 2 and 3). The results presented in Fig. 5a clearly show that all of the interfacial models have higher ΔEH2O values than the single-phase models proving that the heterostructured interfaces effectively promote H2O adsorption. Moreover, the substantially different H2O adsorption abilities of monoatomic dispersed Fe atoms bonded with one (Fe@MoO2−1) or two (Fe@MoO2−2) O atoms in MoO2 indicate that the local bonding environment of monoatomic dispersed Fe atoms greatly affects the H2O adsorption behaviour of electrocatalytic sites40,41,42. Furthermore, both the single-phase and heterostructured interfacial models show stronger H2O adsorption ability than Pt (111), demonstrating that MoP, MoP2, and MoO2 nanocrystals and the heterostructured interfaces are critical for efficient H2O adsorption. To further explain the principle of enhancing H2O molecule adsorption, the DFT simulations of atomic configurations and the corresponding electron density difference after H2O adsorption at Mo or Fe sites are shown in Fig. 5b. The H2O adsorption energy, adsorption site and the bonding distance between active site and O atom in H2O was summarized in Supplementary Table 13. These results show that the heterostructured interfaces and single Fe atom sites presents a significant depletion of electrons when adsorbing the H2O molecules, demonstrating the effective promotion of electrons transfer from active sites to H2O molecules43. In addition, the partial density of states (PDOS) of Mo/Fe active sites and O atoms in H2O molecules was also calculated (Supplementary Fig. 28). The PDOS at s-, p-, d-orbitals of Mo/Fe and the s-, p-orbitals of O at heterostructured interface models are rather different compared to the single-phase models. This result explains the variation in the electron-transfer ability of different structures, and thus highlights the microstructures that are critical for efficient H2O adsorption.

a DFT-calculated ΔEH2O on various exposed surfaces of MoP, MoP2, MoO2, MoP/MoP2, MoP/MoO2, MoP2/MoO2, Fe@MoO2−1, and Fe@MoO2−2, respectively. As a reference, the ΔEH2O of the Pt(111) surface is marked by the grey dashed line. b DFT-optimised atomic configurations and corresponding electron density differences for MoP (a1 and a2), MoP2 (b1 and b2), MoO2 (c1 and c2), MoP/MoP2 (d1 and d2), MoP/MoO2 (e1 and e2), MoP2/MoO2 (f1 and f2), Fe@MoO2−1 (g1 and g2), and Fe@MoO2−2 (h1 and h2), after H2O adsorption at their surface sites. Yellow isosurfaces and blue isosurfaces represent the depletion and segregation of electrons, respectively. c ΔGH* profiles of various catalytic sites at the surfaces of MoP, MoP2, MoO2, MoP/MoP2, MoP/MoO2, MoP2/MoO2, Fe@MoO2−1, and Fe@MoO2−2. d Representative atomic configurations after H* adsorption at the surface sites of MoP/MoP2, MoP/MoO2, MoP2/MoO2, Fe@MoO2−1, and Fe@MoO2−2, with corresponding ΔGH*. e DFT-calculated 2D electron density differences after adsorption of H* on active sites in heterostructured interface models and monoatomic dispersed models. Red backgrounds and blue backgrounds represent the depletion and accumulation of electrons (e/Å3), respectively.
As an important rate-determining factor for HER44,45,46, the H2O dissociation performance on the surfaces of the above constructed models are further investigated and compared with Pt(111) (Supplementary Fig. 29)46. The H2O dissociation energies, either thermodynamically upward with low energy barrier or thermodynamically downward, indicate that H2O dissociation is easy to occur on the surface of catalysts. The results show that the H2O dissociation on the surface sites of MoP2, MoP/MoO2 and MoP2/MoO2 are thermodynamically upward, but much lower than that on the surface of Pt(111), while the others, especially MoP/MoP2 and Fe@MoO2−1, are thermodynamically downward, indicating the H2O dissociation is easy to occur on the surface of our catalyst in which MoP/MoP2 and Fe@MoO2−1 shows the greatest H2O dissociation capability. Combined with the excellent H2O adsorption energy of MoP/MoP2 and Fe@MoO2−1 (Fig. 5a), it can be concluded that the origin of efficient H2O adsorption and dissociation capability of our catalyst owes in part, to MoP/MoP2 and Fe@MoO2−1 promoting proton transfer to accelerate the Volmer step of HER.
To examine the second H* adsorption/H2 desorption step (Heyrovsky step), the Gibbs free energies (ΔGH*) for all possible active sites were calculated (Fig. 5c), and the corresponding atomic configurations are shown in Fig. 5d and Supplementary Fig. 30. ΔGH* is a key descriptor of HER performance under alkaline conditions, and HER activity increases as ΔGH* approaches zero47,48. Figure 5c shows that the ΔGH* values of H* adsorption onto all of the active sites in single-phase models are out of the range of −0.3–0.3 eV, indicating poor HER performance. In contrast, the ΔGH* values for most of the active sites in heterostructured interfacial and monoatomic dispersed models are in the range of −0.19–0.22 eV, except for MoP/MoP2. In particular, both the <Mo top> and <Mo-P bridge> sites in MoP2/MoO2 have ΔGH* values very close to zero (0.04 eV and −0.05 eV) (Fig. 5d). Thus, the heterostructured interfaces and monoatomic dispersed sites underpin the excellent HER performance of the Fe SAs@Mo-based HNSs.
To clarify the fundamental physical mechanism of H* adsorption, the magnitude of electron transfer between H* and active sites was analysed by investigating the 2D electron density differences resulting from H* adsorption onto the surface sites of single-phase (Supplementary Fig. 31), heterostructured interface, and monoatomic dispersed models (Fig. 5e). In single-phase models, the electron transfer between H* and active sites that is either too strong or too weak, which results in these models having poor H* adsorption/desorption abilities. While in the heterostructured interface models, the electron transfer from the Mo top at the MoP2/MoO2 interface to H* is between that of MoP/MoP2 (ΔGH* = −0.37 eV) and MoP/MoO2 (ΔGH* = 0.22 eV), and results in an ideal ΔGH* value (0.04 eV). A comparison of <MoP2 (Mo-P bridge)> (ΔGH* = 0.30 eV) and <MoP2/MoO2 (MoP bridge)> (ΔGH* = −0.05 eV) shows that the electron transfer from Mo to H* is greatly promoted at the MoP2/MoO2 interface. In the Fe monoatomic dispersed models, we found that the electron transfer from Fe to H* is greater at the Fe site in Fe@MoO2−1 than that in Fe@MoO2−2. This is consistent with the difference in their ΔGH* values (−0.19 eV for Fe@MoO2−1 and 0.16 eV for Fe@MoO2−2). These favourable ΔGH* values indicate that monoatomic dispersed Fe atoms bonded with either one or two O atoms in MoO2 possess appropriate H* adsorption/desorption ability for effective HER electrocatalysis.
To further explain the excellent HER performance of these sites, the d-orbital PDOS was also calculated (Supplementary Fig. 32). The results clearly show that the centres of d-orbital PDOSs of Mo sites in MoP(−1.23 eV) and MoP2 (−1.40 eV) are either too far away from, or in MoO2 (−0.75 eV) are too close to the Fermi level, resulting in too weak or too strong interactions between catalytic sites and H*. In contrast, the centres of d-orbital PDOSs of Mo sites in MoP/MoO2 and MoP2/MoO2 interfaces have appropriate values (−1.13 eV and −0.99 eV, respectively), which account for their better ΔGH* values. Moreover, the change in the centres of d-bands is consistent with the variation in ΔGH* values (Supplementary Table 14) indicate that the appropriate d-band centre for Mo top sites is ~1.0 eV. These results revealed that the Heyrovsky reaction occurs most efficiently at MoP2/MoO2 interfaces and monoatomic dispersed Fe locations. This is perfectly consistent with the experimental results that the best HER performance of Fe/SAs@Mo-based-HNSs was achieved at an appropriately ‘medium’ temperature (500 °C), owing to its highest ratio of MoP2/MoO2 interfaces and monoatomic dispersed Fe locations.
Based on the DFT and experimental results and analysis, the superior HER performance of the Fe/SAs@Mo-based-HNSs is attributable to the following reasons. (1) The heterostructured interfaces of Fe/SAs@Mo-based-HNSs lead to optimised electronic structures and H* adsorption energies, increasing their intrinsic electrocatalytic activity. (2) The monoatomic dispersed Fe locations contribute to efficient H2O adsorption and dissociation capability, promoting proton transfer to accelerate the HER performance. (3) The unique 2D and hierarchical porous architecture of Fe/SAs@Mo-based HNSs leads to effective mass transport within their structures and high exposure of active sites to electrolyte, thereby enhancing their electrocatalytic activity. Fe/SAs@Mo-based-HNSs is the best-performing single-atom dispersed heterostructured HER electrocatalyst.
Discussion
In summary, we have devised a highly efficient HER electrocatalyst composed of porous Fe/SAs@Mo-based-HNSs, which is formed via a novel low-temperature phosphorisation of environmentally benign and simple self-assembled inorganic–inorganic coordinated FePMoG nanosheets. The porosity and the heterogeneous interfaces of the heterostructured nanosheets derived from the FePMoG nanosheets were easily regulated during the phosphorisation process by adjusting the temperature and time. Consequently, our Fe/SAs@Mo-based-HNSs exhibited an overpotential of 38.5 mV at a current density of 10 mA cm−2, a Tafel slope of 35.6 mV dec−1, and long-term durability with negligible increase in polarisation potential over 600 h of continuous operation at 200 mA cm−2, which is one of the best HER performances reported to date for nanostructured Mo-based electrocatalysts. This outstanding performance is attributable to the Fe/SAs@Mo-based-HNSs’ optimised electronic structure, enriched interface and boundary phases, large active surface areas and porosities, and the synergetic effect of their single dispersed atoms and heterostructures. Experiments and DFT calculations reveal that the heterostructured interface and the single dispersed Fe atoms of Fe/SAs@Mo-based-HNSs facilitate H2O adsorption and appropriate H* adsorption/desorption during the HER, and their porous 2D network means that their active sites are highly accessible.
Thus, this work describes a simple, new approach for fabricating 2D nanosheet precursors of nanostructured C-free porous heterostructured nanosheets, and for optimising their electronic structures via a multi-phase strategy to increase their electrocatalytic performance in the HER. This high-performance C-free electrocatalyst represents an effective alternative to corrosion-prone C-containing catalysts for use in proton-exchange membrane fuel cells and other state-of-the-art energy technologies.
Methods
Preparation of wrinkle-like FePMoG nanosheets
In a typical procedure, 12 ml of aqueous Fe(NO3)3·9H2O (0.5 M) solution was added to 30 ml phosphomolybdic acid (PMo, 15 mg ml−1), and the solution was kept still for a designated time. As the reaction proceeded, the transparent yellow solution gradually became turbid due to the formation of nanoparticles or nanosheet self-assembled by Fe3+ and PMo. After reacting for 20 h, the suspension was centrifuged and washed with distilled water three times. The centrifugation procedure (Centrifugation speeds: 447 g) is used to remove the excessive water toward the formation of FePMoG mineral hydrogel. The morphology of Fe3+-phosphomolybdic acid assemblies can be easily controlled by changing the reactant concentration.
Preparation of Fe/SAs@Mo-based-HNSs
The as-obtained wrinkle-like FePMoG mineral hydrogel was re-dispersed in 30 ml ethanol and 30 g NaCl was added into the dispersion under magnetic stirring to avoid the aggregation of the nanosheets. The ethanol was evaporated under 60 °C with continuous magnetic stirring and obtained the FePMoG and NaCl mixture (denoted as FePMoG-NaCl). The dried FePMoG-NaCl was annealed at 500 °C in Ar gas flow for 2.5 h with a heating rate of 3.5 °C min−1 with the sodium hypophosphite in the upstream as phosphorus source and to produce the Fe/SAs@Mo-based-HNSs. For comparison, the control samples were prepared at different annealing temperature.
Characterizations
The samples were characterized with scanning electron microscopy (SEM, Philips XL-30 FESEM) and high-resolution transmission electron microscopy (TEM, JEOL TEM 2100F FEG operated with an accelerating voltage of 200 kV). Atomic-resolution scanning transmission electron microscopy (STEM) images and energy-dispersive spectrometry (EDS) maps were acquired on a Titan FEI Themis G60-300 S/TEM (fitted with a high-brightness field emission gun (X-FEG), probe Cs corrector and super-X EDS with four windowless silicon drift detectors). The X-ray diffraction (XRD) patterns were collected using an X-ray diffractometer (Rigaku SmartLab) with Cu Kα (λ = 1.5418 Å). Fourier transform infrared spectroscopy (FT-IR) spectra were documented with KBr pellets from a Bruker Model R 200-L spectrophotometer. Brunauer-Emmett-Teller (BET) experiments were tested on a Quantachrome NOVA 1200e Gas Adsorption Analyzer at 77 K. X-ray photoelectron microscopy (XPS) was performed on a ESCALAB 250 photoelectron spectrometer (ThermoFisher Scientific) with Al Ka (1486.6 eV) as the X-ray source set at 150 W and a pass energy of 30 eV for high-resolution scan, The base pressure was 3 × 10−9 mbar, and the binding energies were referenced to C1s line at 284.8 eV from adventitious carbon. The Fe/Mo K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) experiments for the Fe/SAs@Mo-based-HNSs were carried out at BL07A1 beamline of National Synchrotron Radiation Reaearch Center (NSRRC). The data were collected in fluorescence mode using a Lytle detector while the corresponding reference sample were collected in transmission mode. The incident beam was monochromatized by using a Si (111) fixed-exit, double-crystal monochromator, and a harmonic rejection mirror was applied to cut off the high-order harmonics. The obtained XAFS data was processed in Athena (version 0.9.26) for background, pre-edge line and post-edge line calibrations. Then Fourier transformed fitting was carried out in Artemis (version 0.9.26). The k3 weighting, k-range of 3−14 Å−1 and R range of 1–~3 Å were used for the fitting of Mo/Fe foil; k-range of 3−12 Å−1 and R range of 1–~3.5 Å were used for the fitting of Mo samples, k-range of 3−10 Å−1 and R range of 1- ~2 Å were used for the fitting of Fe samples. The four parameters, coordination number, bond length, Debye-Waller factor, and E0 shift (CN, R, ΔE0) were fitted without anyone was fixed, the σ2 was set. For Wavelet Transform analysis, the χ(k) exported from Athena was imported into the Hama Fortran code. The parameters were listed as follow: R range, 1−4 Å; k range, 0−13 Å−1 for Mo and 0−15 Å−1 for Fe; k weight is 2 and 3 for Mo and Fe, respectively; and Morlet function with κ = 3, σ = 1 was used as the mother wavelet to provide the overall distribution.
Electrochemical measurements
Prior to use, the carbon paper was cut into cut into small pieces, then the carbon paper chips were washed ultrasonically in ethanol and dried in oven. First, 4 mg catalyst was ultrasonically dispersed in 400 µL of 0.5 wt% Nafion solution (ethanol: water = 3: 1) to form homogeneous slurry. The slurry was then coated onto a carbon paper with a catalyst loading of 670 μg cm−2. The catalyst-coated carbon paper electrode, saturated calomel electrode (SCE), and graphite rod were used as the working, reference, and counter electrodes, respectively. Electrochemical measurements were conducted at room temperature on a CHI760e electrochemical station with a three-electrode cell system. Linear sweep voltammetry was recorded in H2-saturated 1 M KOH at a scan rate of 5 mV s−1 to obtain the polarization curves. The long-term stability tests were performed by continuously applying a current density of 200 mA cm−2 to the working electrode and used a Hg/HgO to alter the SCE as reference electrode (The H2 wasn’t supplied in the stability test as the H2 constantly being produced in the working electrode therefore the electrolyte in the working electrode side can be considered as H2 saturated). All the data presented were corrected for iR losses and background current, and the potentials were later converted to the reversible hydrogen electrode (RHE) scale (for SCE reference in 1 M KOH, VRHE = VSCE + VSCEθ + 0.059 pH). Cyclic voltammograms (CV) were obtained with different scan rates from 4 to 40 mV s−1 in the potential range of 0-0.1 V (vs. RHE).
The Faradaic efficiency (FE) of catalysts is defined as the ratio of the amount of experimentally determined H2 to that of the theoretically expected H2 from the reaction, which was measured at different potentials (−0.04, −0.08, −0.12, −0.16 V, and −0.20 vs. RHE) and at a specific constant current density of 10 mA/cm2 by gas chromatography (GC-2014, Shimadzu). The experiments were conducted in a custom-made two compartment cell (single cell: 50 mL) separated by a Nafion 117 membrane, each compartment of the cell was filled with 35 mL 1.0 m KOH. The H2 gas was purged out from the cell by using 100 μL syringe and injected into gas chromatography. For the specific applied potentials, the gas was sampled after one hour potential supply; for the specific constant current density of 10 mA/cm2, the gas was took a sample every 15 min. FE was calculated according to following relationship
Where, nH2 is the amount of hydrogen (mol), F is the Faraday’s constant (96485 C mol−1), and Q is the total amount of charge passed through the cell (C).
DFT calculations
Density functional theory (DFT) calculations, as one of the most versatile tools to study electrocatalytic mechanism, are conducted to investigate hydrogen evolution reaction (HER) behaviors of Fe/SAs@Mo-based-HNSs using the Cambridge Sequential Total Energy Package (CASTEP) module in Materials Studio software49. The generalized gradient approximation (GGA) method and Perdew-Burke-Ernzerh (PBE) function are employed to describe the exchange and corrections of interaction between atoms50. The ultrasoft pseudo-potential method is utilized to describe the interactions between valence electrons and ionic cores51. A plane-wave basis set is assigned with a cutoff energy of 400 eV. The Brillouin zone is sampled by a Monkhorst-Pack grid52. The tolerances of energy, force and displacement for structure optimization are 10−6 eV/atom, 0.02 eV/Å and 0.001 Å respectively. The self-consistence field (SCF) is set as 5×10−6 eV/atom. The effect of van der Waals interaction is taken into account by the semi-empirical DFT-D force-field approach53.
The Gibbs free energies for hydrogen adsorption (ΔGH*) are calculated from the Eq. 1:
Where the \(\triangle {E}_{H*}\), \(\triangle {ZPE}\), \(T\) and \(\triangle S\) represent the binding energy, zero-point energy change, temperature and entropy change of H adsorption system, respectively.
The vibration entropy is H at the adsorbed states is negligible. Thus, \(\triangle S\) can be obtained from the following Eq. 2:
Where \({S}_{{H}_{2}}\) is the entropy of H2 in the gas phase at the standard conditions.
Besides, \(\triangle {ZPE}\) can be calculated from the Eq. 3:
Thus, the free energy of the adsorbed state can be calculated using the simplified Eq. 448:
The water adsorption energies (ΔEH2O) at the surface of catalysts are calculated by the Eq. 5:
Where the \({E}_{{surf}}\) and \({E}_{{{surf}+H}_{2}0}\) are the total energies of the surface before and after water adsorption, and \({E}_{{H}_{2}O}\) is the energy of a free water molecule.
Therefore, the HER performance of catalysts can be evaluated by DFT calculations of ΔEH2O and ΔGH*, which are the key descriptors for the Volmer-Heyrovsky reaction efficiency.
According to the experimental characterization information, a series of representative single-phase, heterostructured interface, and monoatomic dispersed models are built for DFT study of their HER performance with corresponding atomic mechanism investigation. Specifically, (1) MoP, MoP2 and MoO2 single-phase models; (2) MoP/MoP2, MoP/MoO2 and MoP2/MoO2 heterostructured interface models and (3) two kinds of monoatomic dispersed Fe onto MoO2 surface models are constructed, bonding with one or two O atoms in MoO2. The initial atomic configurations of single-phase MoP, MoP2 and MoO2 are from crystallography open database. In order to build the MoP/MoP2, MoP/MoO2 and MoP2/MoO2 interfaces, the heterogeneous plane structure consisting of four layers of MoP2 or MoO2 with (2×2) supercells in the bottom, with the addition of MoP or MoP2 clusters onto the top of them54. Since the DFT calculation results are very sensitive to the local coordination structure of the metallic centers40,41,42, a series of initial configurations with Fe atoms coordinating different number of O atoms are constructed to demonstrate the representative stable models with monoatomic dispersed Fe onto MoO2.Two different stable monoatomic dispersed Fe onto MoO2 surface models (bonding with one or two O atoms, named Fe@MoO2−1 and Fe@MoO2−2 respectively) are obtained via a series of structure optimization test. The constructed initial configurations with Fe atoms coordinating more O atoms are unstable. It should be noted that the monoatomic Fe sites only coordinate with 1 or 2 O atoms in our DFT models which is much smaller than the result obtained from EXAFS. However, it should be noted that the data obtained from EXAFS is the information of average coordination number, which mainly be determined by the coordination number of Fe atoms in MoO2 interior. Since the Fe active sites contributing to HER are the monoatomic Fe atoms at the surface of the material, their coordination number should be smaller than that in the material interior, existing in the form of unsaturated coordination. Therefore, the DFT models only containing 1 or 2 O coordination atoms are taken as the representative monoatomic Fe models. A vaccum gap of 15 Å is employed to create free surface for the simulation of hydrogen evolution reaction (HER) behaviors for all the models. The k points are set as 2 × 4 × 1 for MoP, MoP2, MoP/MoP2, and 5 × 4 × 1 for MoO2, MoP/MoO2, MoP2/MoO2, Fe@MoO2−1, Fe@MoO2−2 to sample the Brillouin zone.
Data availability
Source data are provided with this paper. The data used to generate the Figs. 4a, g shown in this study, and other two independent LSV test data for Fe/SAs@Mo-based-HNSs have been deposited. Further data supporting the findings of this study are available in the Supplementary Information. All other relevant source data are available from the corresponding author upon request. Source data are provided with this paper.
References
Liao, G. et al. Semiconductor polymeric graphitic carbon nitride photocatalysts: the “holy grail” for the photocatalytic hydrogen evolution reaction under visible light. Energy Environ. Sci. 12, 2080–2147 (2019).
Jacobson, M. Z., Colella, W. & Golden, D. Cleaning the air and improving health with hydrogen fuel-cell vehicles. Science 308, 1901–1905 (2005).
Shi, Y. & Zhang, B. Recent advances in transition metal phosphide nanomaterials: synthesis and applications in hydrogen evolution reaction. Chem. Soc. Rev. 45, 1529–1541 (2016).
Chen, Z., Qing, H., Zhou, K., Sun, D. & Wu, R. Metal-organic framework-derived nanocomposites for electrocatalytic hydrogen evolution reaction. Prog. Mater. Sci. 108, 100618 (2020).
Jia, Z. et al. A Novel Multinary Intermetallic As An Active Electrocatalyst For Hydrogen Evolution. Adv. Mater. 32, 2000385 (2020).
Rao, C. N. R. & Chhetri, M. Borocarbonitrides as metal‐free catalysts for the hydrogen evolution reaction. Adv. Mater. 31, 1803668 (2019).
Liang, S. X. et al. Compelling rejuvenated catalytic performance in metallic glasses. Adv. Mater. 30, 1802764 (2018).
Wang, Y. et al. Advanced electrocatalysts with single-metal-atom active sites. Chem. Rev. 120, 12217–12314 (2020).
Zhao, D. et al. Atomic site electrocatalysts for water splitting, oxygen reduction and selective oxidation. Chem. Soc. Rev. 49, 2215–2264 (2020).
Qi, K., Chhowalla, M. & Voiry, D. Single atom is not alone: Metal–support interactions in single-atom catalysis. Mater. Today 40, 173–192 (2020).
Zheng, X. et al. Non-carbon-supported single-atom site catalysts for electrocatalysis. Energy Environ. Sci. 14, 2809–2858 (2021).
Zhou, K. L. et al. Platinum single-atom catalyst coupled with transition metal/metal oxide heterostructure for accelerating alkaline hydrogen evolution reaction. Nat. Commun. 12, 1–10 (2021).
Zhang, Y., Jiao, L., Yang, W., Xie, C. & Jiang, H. L. Rational fabrication of low‐coordinate single‐atom Ni electrocatalysts by MOFs for highly selective CO2 reduction. Angew. Chem. Int. Ed. 60, 7607–7611 (2021).
Dong, P. et al. Platinum single atoms anchored on a covalent organic framework: boosting active sites for photocatalytic hydrogen evolution. ACS Catal. 11, 13266–13279 (2021).
Sun, Z. et al. Multistimuli‐responsive, moldable supramolecular hydrogels cross‐linked by ultrafast complexation of metal ions and biopolymers. Angew. Chem. Int. Ed. 127, 8055–8059 (2015).
Chen, G. et al. Layered metal hydroxides and their derivatives: Controllable synthesis, chemical exfoliation, and electrocatalytic applications. Adv. Energy Mater. 10, 1902535 (2020).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
Duan, J., Chen, S. & Zhao, C. Ultrathin metal-organic framework array for efficient electrocatalytic water splitting. Nat. Commun. 8, 15341 (2017).
Lyu, F. & Lu, J. Rational Construction of Low-Dimensional Transition Metal Based Material for Energy Storage and Conversion. Ph.D. Dissertation, City University of Hong Kong, Hong Kong (2020).
Li, B. et al. Mineral hydrogel from inorganic salts: biocompatible synthesis, all‐in‐one charge storage, and possible implications in the origin of life. Adv. Funct. Mater. 32, 2109302 (2021).
Jin, Z. et al. Understanding the inter-site distance effect in single-atom catalysts for oxygen electroreduction. Nat. Catal. 4, 615–622 (2021).
Zeng, X. et al. Ice‐templated assembly strategy to construct 3D boron nitride nanosheet networks in polymer composites for thermal conductivity improvement. Small 11, 6205–6213 (2015).
Hong, X. et al. AuAg nanosheets assembled from ultrathin AuAg nanowires. J. Am. Chem. Soc. 137, 1444–1447 (2015).
El-Wahab, M. A. & Said, A. Phosphomolybdic acid supported on silica gel and promoted with alkali metal ions as catalysts for the esterification of acetic acid by ethanol. J. Mol. Catal. A: Chem. 240, 109–118 (2005).
Qiu, J. et al. Oxidative desulfurization of diesel fuel using amphiphilic quaternary ammonium phosphomolybdate catalysts. Fuel Process. Technol. 90, 1538–1542 (2009).
Guo, H. et al. Self‐templated conversion of metallogel into heterostructured TMP@ Carbon Quasiaerogels boosting bifunctional electrocatalysis. Adv. Funct. Mater. 1903660 (2019).
Liu, R. et al. Enhanced proton and electron reservoir abilities of polyoxometalate grafted on graphene for high-performance hydrogen evolution. Energy Environ. Sci. 9, 1012–1023 (2016).
Tan, C. Self-assembly, aggregates morphology and ionic liquid crystal of polyoxometalate-based hybrid molecule: From vesicles to layered structure. J. Mol. Struct. 1148, 34–39 (2017).
Zhai, Q. et al. A novel iron-containing polyoxometalate heterogeneous photocatalyst for efficient 4-chlorophennol degradation by H2O2 at neutral pH. Appl. Surf. Sci. 377, 17–22 (2016).
Dubal, D. P., Ballesteros, B., Mohite, A. A. & Gómez‐Romero, P. Functionalization of polypyrrole nanopipes with redox‐active polyoxometalates for high energy density supercapacitors. ChemSusChem 10, 731–737 (2017).
Liu, B. et al. Few layered N, P dual‐doped carbon‐encapsulated ultrafine MoP nanocrystal/MoP cluster hybrids on carbon cloth: an ultrahigh active and durable 3D self‐supported integrated electrode for hydrogen evolution reaction in a wide pH range. Adv. Funct. Mater. 28, 1801527 (2018).
Yue, Q. et al. MoP is a novel, noble-metal-free cocatalyst for enhanced photocatalytic hydrogen production from water under visible light. J. Mater. Chem. A 3, 16941–16947 (2015).
Li, S. et al. Metal‐organic precursor–derived mesoporous carbon spheres with homogeneously distributed molybdenum carbide/nitride nanoparticles for efficient hydrogen evolution in alkaline media. Adv. Funct. Mater. 29, 1807419 (2019).
Zhang, S. et al. Electrocatalytically active Fe‐(O‐C2)4 single‐atom sites for efficient reduction of nitrogen to ammonia. Angew. Chem. Int. Ed. 132, 13525–13531 (2020).
Li, X. et al. Identification of the electronic and structural dynamics of catalytic centers in single-Fe-atom material. Chem 6, 3440–3454 (2020).
Lyu, F. et al. Lamellarly stacking porous N, P Co‐Doped Mo2C/C nanosheets as high performance anode for lithium‐ion batteries. Small 15, 1805022 (2019).
Jia, Z. et al. A self‐supported high‐entropy metallic glass with a nanosponge architecture for efficient hydrogen evolution under alkaline and acidic conditions. Adv. Funct. Mater. 31, 2101586 (2021).
Zhang, L.-C., Jia, Z., Lyu, F., Liang, S.-X. & Lu, J. A review of catalytic performance of metallic glasses in wastewater treatment: Recent progress and prospects. Prog. Mater. Sci. 105, 100576 (2019).
Jiao, Y., Zheng, Y., Jaroniec, M. & Qiao, S. Z. Design of electrocatalysts for oxygen-and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 44, 2060–2086 (2015).
Wu, Z. P. et al. Manipulating the local coordination and electronic structures for efficient electrocatalytic oxygen evolution. Adv. Mater. 33, 2103004 (2021).
Cheng, X. et al. Engineering local coordination environment of atomically dispersed platinum catalyst via lattice distortion of support for efficient hydrogen evolution reaction. Mater. Today Energy 20, 100653 (2021).
Tomboc, G. M., Kim, T., Jung, S., Yoon, H. J. & Lee, K. Modulating the local coordination environment of single‐atom catalysts for enhanced catalytic performance in hydrogen/oxygen evolution reaction. Small 18, 2105680 (2022).
Cheng, Y., Ma, E. & Sheng, H. Atomic level structure in multicomponent bulk metallic glass. Phys. Rev. Lett. 102, 245501 (2009).
Sun, H. et al. Topotactically transformed polygonal mesopores on ternary layered double hydroxides exposing under-coordinated metal centers for accelerated water dissociation. Adv. Mater. 32, 2006784 (2020).
Zhang, B. et al. Interface engineering: The Ni(OH)2/MoS2 heterostructure for highly efficient alkaline hydrogen evolution. Nano Energy 37, 74–80 (2017).
Li, F. et al. Construction of porous Mo3P/Mo nanobelts as catalysts for efficient water splitting. Angew. Chem. Int. Ed. 57, 14139–14143 (2018).
Greeley, J., Jaramillo, T. F., Bonde, J., Chorkendorff, I. & Nørskov, J. K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. mater. 5, 909–913 (2006).
Nørskov, J. K. et al. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 152, J23 (2005).
Clark, S. J. et al. First principles methods using CASTEP. Z. f.ür. Kristallographie-Crystalline Mater. 220, 567–570 (2005).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892 (1990).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Grimme, S. Semiempirical GGA‐type density functional constructed with a long‐range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Zhao, H. et al. Heterostructured CoP/MoO 2 on Mo foil as high-efficiency electrocatalysts for the hydrogen evolution reaction in both acidic and alkaline media. J. Mater. Chem. A 8, 6732–6739 (2020).
Acknowledgements
This work is supported by Shenzhen-Hong Kong Science and Technology Innovation Cooperation Zone Shenzhen Park Project (HZQB-KCZYB-2020030) to J.L.; the National Key R&D Program of China (Project No. 2017YFA0204403) to J.L.; Hong Kong Innovation and Technology Commission via the Hong Kong Branch of National Precious Metals Material Engineering Research Center; the National Natural Science Foundation of China (Project No. 12002108) to L.S.; the Guangdong Basic and Applied Basic Research Foundation (Project No. 2020A1515110236, 2022A1515011402) to L.S.; and the Science, Technology, and Innovation Commission of Shenzhen Municipality (Project No. GXWD20201230155427003-20200824105236001, ZDSYS20210616110000001) to L.S.
Author information
Authors and Affiliations
Contributions
F.L., Y.Y.L., and J.L. designed the research. S.Z., Z.J., F.M., and Z.M. prepared and characterised the samples. L.C. and J.P. conducted SEM and XPS. F.L., Y.B., and Y.B. conducted TEM. L.S. performed the theoretical calculations. F.L., S.Z., L.S., Z.J., Y.Y.L., and J.L. wrote the manuscript. All authors discussed the results and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhi-Peng Wu and the other anonymous reviewer(s) for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lyu, F., Zeng, S., Jia, Z. et al. Two-dimensional mineral hydrogel-derived single atoms-anchored heterostructures for ultrastable hydrogen evolution. Nat Commun 13, 6249 (2022). https://doi.org/10.1038/s41467-022-33725-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-33725-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
