
Abstract
CMOS-like circuits in bioelectronics translate biological to electronic signals using organic electrochemical transistors (OECTs) based on organic mixed ionic-electronic conductors (OMIECs). Ambipolar OECTs can reduce the complexity of circuit fabrication, and in bioelectronics have the major advantage of detecting both cations and anions in one device, which further expands the prospects for diagnosis and sensing. Ambipolar OMIECs however, are scarce, limited by intricate materials design and complex synthesis. Here we demonstrate that judicious selection of p- and n-type materials for blend-based OMIECs offers a simple and tunable approach for the fabrication of ambipolar OECTs and corresponding circuits. These OECTs show high transconductance and excellent stability over multiple alternating polarity cycles, with ON/OFF ratios exceeding 103 and high gains in corresponding inverters. This work presents a simple and versatile new paradigm for the fabrication of ambipolar OMIECs and circuits with little constraints on materials design and synthesis and numerous possibilities for tunability and optimization towards higher performing bioelectronic applications.
Similar content being viewed by others

Introduction
The detection and amplification of biological signals has been an increasingly important subject of research in the past few decades. For an artificial electronic device to record brain activity1, detect ionic biological markers in body fluids2, identify organic molecules in tumor cells3, etc., it must sense and transmit in real time the changes in an electrochemical potential during a biological process. Coupling the ion-based biological signals to electron-based devices requires materials that support the transport of both ions and electrons, combined with circuit designs that can provide on-site signal amplification. Organic mixed ionic-electronic conductors (OMIECs) are promising in this field because they can couple electronic and ionic currents, are easy to process into circuit architectures and offer mechanical compatibility with soft tissues4,5. The most common and versatile device that utilizes OMIECs for the amplification of biological signals is the Organic Electrochemical Transistor (OECT)6,7,8 that relies on the injection/extraction of ions from an electrolyte, regulated by a gate electrode, to modulate the bulk conductivity in an OMIEC channel. The technology for amplification and transfer of the biological signals harnesses the CMOS-like circuit architectures. This technology is based on the layout and wiring of p-type and n-type transistors to perform analog and digital processes. Therefore, both p-type and n-type OMIECs are required to fabricate and wire n-type and p-type OECTs into complex circuits. Most OMIECs, as a subset of organic semiconductors, are however p-type in nature with only a handful of n-types emerging in recent years9,10,11,12,13,14. The design of these n-type OMIECs is usually inspired by semiconducting materials that perform well in n-type Organic Field-Effect Transistors (OFETs), but with sidechains that support ionic transport, yet polymers that bear no side-chains are also a rising class in this group15.
OFET technology offers not only p-type and n-type performances for conventional CMOS circuit design, but also ambipolar character, i.e. devices that can act as both p-type and n-type. Ambipolar OFETs, therefore, offer the fabrication of programmable logic circuits with polymorphic functionality16,17,18, and were also shown to significantly reduce the complexity, processing steps and manufacturing costs of complementary-like inverters and logic circuits fabrication, while maintaining reasonably high gains19,20,21. It was very recently shown that ambipolarity in OECTs can also reduce the device footprint and increase device density in circuits, for example through cofacial architecture22. In the case of bioelectronics, ambipolarity has another major advantage, the detection of both cations and anions in one device, which further expands the prospects of ambipolar OMIECs as biosensors23,24,25,26,27,28. It was recently found that modification of the glycolated side chains can drive the initially n-type material to perform in a p-type OECTs structure11. The p-type character comes on expense of the n-type performance so that the OECT was ambipolar, i.e., the channel can shuttle both holes and electrons between source and drain, but performed quite poorly. A very recent study demonstrated that controlling the dihedral angle and charge delocalization by co-monomer backbone replacement can also transform the OMIEC polarity from p- to n-type29. Therefore, the common approach to expand the ambipolar OMIECs material library is the design and synthesis of new materials that involves deep theoretical studies considering the complex interplay between energy level position, chemical structure and resulting film morphology that can support both ionic and electronic transport. This is a challenging route that so far did not lead to the demonstration of well performed and stable ambipolar OECTs, as reviewed in Table 1, and evident from the scarcity of ambipolar OMIECs in previous studies10,22,30,31.
Inspired by other organic electronic devices, mainly Organic Photovoltaics (OPVs), but also OFETs, we suggest a new design strategy for ambipolar OECTs: blending p- and n-type materials in a single layer, each responsible for the transport of a different charge carrier32,33 with a bulk-heterojunction structure. This offers great versatility in material selection as well as excellent control over morphology through processing, without the need to synthesize new materials. In this report we demonstrate, for the first time, blend-based ambipolar OECT devices and corresponding inverters. Building on the vast experience and knowledge gained in OPV research, specifically the insightful understanding of processing-morphology-performance relationships in fullerene:polymer blends, we implemented an analogous blend of judiciously-selected polymer and fullerene-derivative mixed conductors in OECTs. We controlled blend composition, processing conditions and thermal treatments to direct the crystallinity and morphology of the materials in the active layer to support ambipolar transport across the device. These OECTs, as well as the inverters made of two identical ambipolar OECTs, show high current and voltage amplification factors and excellent stability over multiple alternating polarity cycles.
Results
Organic electrochemical transistors
Our selected p-type component is the poly(2-(3,3′-bis(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)-[2,2’-bithiophen]−5-yl)thieno[3,2-b]thiophene) (p(g2T-TT)) polymer (Fig. 1a, bottom) which has been thoroughly studied for OECTs and its characteristics are well established34,35. The n-type fullerene derivative, C60,N,N,N-trimethyl-1-(2,3,4-tris(2-(2-methoxyethoxy)ethoxy)phenyl)methanaminium monoadduct (PrC60MA, Fig. 1a, top), was “borrowed” from the field of perovskite solar cells and high-k dielectrics36,37,38, and although it was not used in OECTs, a very similar fullerene derivative, C60-TEG, already demonstrated n-type OECT performance12. The performances of p(g2T-TT) and C60-TEG are listed in Table 1 for comparison with the current study. In contrast to OPVs where the energy band alignment should encourage charge transfer at the fullerene:polymer interface, here we aimed for a substantial misalignment between the HOMO and LUMO levels to avoid charge transfer at the n:p interface39. We speculate that under such conditions the figure of merit for the n-type and p-type performances of the OECT are almost independent, and hence ambipolarity, i.e. balanced performances, can be achieved using a simple rule of mixtures.

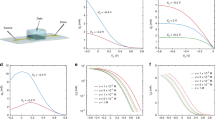
a Chemical structures of the materials used in this study: n-type fullerene derivative PrC60MA (top, red) and the p-type polymer p(g2T-TT) (bottom, blue). b Schematic of the OECT device with 3-terminals (Au source and drain, Ag/AgCl pellet gate), using a PrC60MA:p(g2T-TT) blend as the active channel in a 0.1 M KCl aqueous electrolyte. c Output characteristics (ID-VD) of the ambipolar device (length (L) = 30 \(\mu m\), width (W) = 1000 \(\mu m\), thickness (d) = 60 nm) for VG varying from +0.2 V to −0.3 V at the p-type (black to blue curves in bottom left quadrant) and from +0.4 V to +0.9 V at the n-type (black to red curves in top right quadrant) with a step of 0.05 V. d Transfer curves for VD = −0.4 V (p-type, blue) and VD = +0.4 V (n-type, red) in log scale (solid lines) and \(\sqrt{{I}_{D}}\) scale (dashed lines). Presented characteristics are averaged over four devices. e Typical stability over 100 cycles operation in alternating polarities, each pulsed to ON state for ~3 s with a ~3 s interval time between pulses. f Stability measurement of 7 cycles out of total 100 presented in (e) (pink rectangle). In the n-type VG is pulsed from +0.55 V to +0.9 V and back to +0.55 V while VD = +0.4 V, while in the p-type VG is pulsed from +0.05 V to −0.3 V and back to +0.05 V while VD = −0.4 V. All of device data presented here was acquired with a blend ratio of 95:5 (w:w) PrC60MA:p(g2T-TT).
Considering the measured ~15-fold higher OECT figure of merit of p(g2T-TT) polymer relative to that of the PrC60MA fullerene (Supplementary Table 1, Supplementary Fig. 1) as well as reported values (Table 1)12,34, and the high fullerene content required for efficient electron percolation, evident from blend-based ambipolar OFET studies40,41,42, we fabricated OECTs based on PrC60MA -rich blends. By comparing the p- and n-type performances for OECTs based on several blend compositions (Supplementary Table 1), we found that similar n and p figures of merit are obtained for a blend ratio of 95:5 w:w. Namely, for this composition the \({{{{{\rm{\mu }}}}}}{{{{{{\rm{C}}}}}}}^{*}\), where μ is the carrier mobility and \({C}^{*}\) the volumetric capacitance, are similar for both positive and negative charge carriers. Maintaining the channel dimensions (where W, L and d are channel width, length and thickness, respectively) and the difference between maximum transconductance gate voltage \({{{{{{\rm{V}}}}}}}_{{{{{{\rm{G}}}}}}}\) and threshold voltage \({{{{{{\rm{V}}}}}}}_{{{{{{\rm{th}}}}}}}\) similar for all fabricated OECTs, we can use Eq. 1 to calculate the transconductance (\({{{{{{\rm{g}}}}}}}_{{{{{{\rm{m}}}}}}}\)) which is directly proportional to \({{{{{\rm{\mu }}}}}}{{{{{{\rm{C}}}}}}}^{*}\).
The obtained ambipolar device characteristics display a well-balanced and low-hysteresis behavior, both in output and transfer curves (Fig. 1c, d), with good reproducibility between devices (see Supplementary Fig. 2 and Table 2). It can be noted from Table 2 and Supplementary Table 1 that the threshold voltages of the blend are shifted further away from the OFF voltage, with respect to OECTs based on the pristine materials, indicating that for both polarities doping becomes more difficult upon blending. Interestingly, both the \({{{{{{\rm{g}}}}}}}_{{{{{{\rm{m}}}}}}}\) and \({{{{{\rm{\mu }}}}}}{{{{{{\rm{C}}}}}}}^{*}\) of the p-type performance decrease linearly with p(g2T-TT) content, suggesting that the performance of the polymer is proportional to the number of ion-accessible monomer units in the channel. It has already been demonstrated that semiconducting polymers can be incorporated in blends with extremely low content and still retain mobility in OFETs, i.e. the percolation threshold for charge transport is extremely low43,44,45. Therefore, if the hole mobility in our system is also retained when decreasing the polymer content in the blend, it might suggest that the capacitance decreases with the number of ion-accessible monomer units. On the contrary, the n-type performance is significantly impaired even with the slight decrease in PrC60MA concentration in the blend, suggesting that the presence of the long p(g2T-TT) polymer strands obstructs the electron percolation pathways and reduces electron mobility. Comparing the figure-of-merit, \({{{{{\rm{\mu }}}}}}{{{{{{\rm{C}}}}}}}^{*}\), of the blend presented in Table 2 to the best performing materials reveals that although impressive, it still has a long way to reach state-of-the-art p-type (~500 \([{{{{{\rm{F}}}}}}\,{{{{{\rm{c}}}}}}{{{{{{\rm{m}}}}}}}^{-1}{{{{{{\rm{V}}}}}}}^{-1}{{{{{{\rm{s}}}}}}}^{-1}]\))46,47 and a small leap for state-of-the-art n-type (~25 \([{{{{{\rm{F}}}}}}\,{{{{{\rm{c}}}}}}{{{{{{\rm{m}}}}}}}^{-1}{{{{{{\rm{V}}}}}}}^{-1}{{{{{{\rm{s}}}}}}}^{-1}]\))15.
The high fullerene content in our blend has been a subject for concern in terms of device stability, as the reduced fullerene species with an adjacent metal cation may form a soluble product48. This was demonstrated in a recent study on C60-TEG that found that the a C60-TEG film quickly degrades over repeated cycling12. However, in our case, after 100 cycles in alternating polarities we noticed no significant current degradation and the current ON/OFF ratios that were extracted from the stability measurements remained above 103 (Fig. 1e, f). A previous study on small molecule p-type OECTs blended with high molecular weight PEO showed a similar trend of improved film stability over cycles upon blending, yet an explanation was not provided49. Examining the current for both polarities reveals that the p-type transient response time, \({{{{{{\rm{\tau }}}}}}}_{{{{{{\rm{ON}}}}}}}=466\pm 56{{{{{\rm{ms}}}}}}\), is slower than that of the n-type, \({{{{{{\rm{\tau }}}}}}}_{{{{{{\rm{ON}}}}}}}=20\pm 3{{{{{\rm{ms}}}}}}\), by a factor of ~25 (Supplementary Table 2, Supplementary Fig. 3). These differences are attributed to the low content of the polymer in the film, making the pathways for anion injection and doping scarcer. This correlates well with the slightly higher hysteresis of the device characteristic in p-type operation compared to n-type operation (blue and red curves in Fig. 1d, respectively). Notably, the p-type response time of the ambipolar OECT is slower than that of unipolar OECTs based on the same material, probably due to the very small amount of the p-type polymer in the blend. In contrast, the n-type response time of the ambipolar OECT is the fastest reported for small molecule n-type OECTs although it is blended with the polymer.
Electrochemical and optical properties
The ambipolar nature of the selected blend in also reflected in steady-state spectroelectrochemistry (SEC) measurements recorded in the same voltage range as the OECTs. Taken at similar thicknesses, spectral changes are calculated via a simple rule-of-mixtures according to the weight percent of each material, and compared to the measured blend (Fig. 2a, b). The changes in the absorption spectrum of the blend as a function of applied voltage corroborate the doping of both p- and n-type components and show that the measured changes comply well with the calculated, namely the spectroelectrochemical changes are additive. Compared to the calculated spectrum, both \(\pi -{\pi }^{*}\) and polaronic peaks of p(g2T-TT) exhibit a slight red shift in the measured spectra vs the calculated, and a higher A0-0/A0-1 vibronic peaks ratio (Supplementary Fig. 5). Together, these observations suggest a higher degree of polymer J-aggregation in the blend than in the pristine film50,51, namely, the polymer adopts a more fibrous structure in the blend. A possible explanation is that the p(g2T-TT) strands in the blend are surrounded by the less polar fullerene matrix that induces more J-aggregation. A similar observation was reported for polythiophene co-polymers with polar glycolated sidechains where J-aggregation increases when polarity is reduced52.

Steady-state spectroelectrochemical absorption change of the ambipolar films (a) measured in the 95:5 blend, and b calculated from a rule of mixtures of the steady-state spectroelectrochemical absorption change of the pristine materials (Supplementary Fig. 4). For all measurements the voltage is applied vs an Ag/AgCl pellet electrode at a range of + 0.3 V < VWE < −0.9 V with steps of 0.05 V. Absorption changes are displayed with respect to the recorded spectrum at VWE = −0.4 V. Blue curves denote changes that occur as a result of spectral changes in p(g2T-TT), whereas red curves originate from spectral changes in PrC60MA. Formation of the positive polymer polaron is visible in the blue curves peak at ~830 nm, rising on the expense of the \(\pi -{\pi }^{*}\) interactions peaks at 600–650 nm34. In the PrC60MA red curves, the broad absorption change is due to the formation of a radical anion that stretches into the NIR region61,62. As the HOMO of p(g2T-TT) and LUMO of PrC60MA are ~0.7 eV apart (Supplementary Fig. 6), the changes in absorption for each of the components are independent and spectrally resolved. This is also manifested in c Cyclic voltammetry of the ambipolar 95:5 blend in an EQCM cell at 0.1 V/sec, showing the p- and n-doped current as well as a “neutral” zone where there is no doping. d Mass changes in the 95:5 blend while performing CV in an EQCM cell at 0.01 V/sec, red areas belong to PrC60MA response with injection/expulsion of K+ cations, and blue to p(g2T-TT) response with injection/expulsion of Cl- anions.
Cyclic Voltammetry (CV) confirmed the electrochemical activity of both species in the blend, with three distinguishable regions for n-doping, p-doping and film neutrality (Fig. 2c). Electrochemical Quartz Crystal Microbalance (EQCM) measurements demonstrated the expected mass gain on both polarities, within the same voltage range as the OECTs, SEC and CV measurements (Fig. 2d). Notably, the mass gain on the p-type is notably lower than on the n-type (~100 vs ~700 \([{{{{{\rm{ng}}}}}}/{{{{{{\rm{cm}}}}}}}^{2}]\)), as a result of the significantly lower content of the polymer in the blend. To corroborate the assignment of the low mass gain on the p-type to chloride injection/extraction, we compared it to the EQCM responses of pristine material and a blend with higher polymer content (Supplementary Figs. 7, 8). Thus, the injection and expulsion of both anions and cations as doping agents in the same film is demonstrated here via EQCM for the first time, to the best of our knowledge. The overall spectroelectrochemical and electrochemical results imply that not only there is no chemical interaction between the blended materials, but also that the steady-state doping of each material is mostly unaffected by the presence of the other, as hypothesized by us at the materials selection stage. It is worth noting that the reduction current of PrC60MA consists of a large Oxygen Reduction Reaction (ORR) contribution53 as a result of the relatively shallow LUMO level. The simple decoupling between p-type and n-type signals is a result of the significant energy gap between PrC60MA LUMO and p(g2T-TT) HOMO and opens possibilities for independent tunability.
Microstructural characterization
To gain understanding on the correlation between blend morphology and device performance we performed structural characterization. Grazing Incidence Wide Angle X-ray Scattering (GIWAXS) measurements indicate that the blend film is highly crystalline with a strong preferential orientation relative to the substrate, similarly to neat PrC60MA films (Supplementary Fig. 9) and other fullerene derivatives12,54, though it is significantly less crystalline than the neat PrC60MA film (Fig. 3a). While the lamellar stacking d-space is practically unchanged (Table 3), it is found that the out-of-plane coherence length of PrC60MA is reduced by a staggering 20% (Fig. 3b, Supplementary Fig. 10). This decrease in coherence length and crystallinity lowers the mobility and overall performance of the semiconductor in the blend55 leading to the obtained two-fold reduction of the PrC60MA figure of merit upon blending with 5 wt% polymer only.

a Out-of-plane XRD of pristine materials and the 95:5 w:w PrC60MA:p(g2T-TT) blend and the Grazing Incidence Wide-Angle X-ray Scattering (GIWAXS) measurement of the blend (inset). Dotted lines at 2θ = 3.1° and 5.7° are associated with the (100) peaks of PrC60MA and p(g2T-TT), respectively. All other peaks are higher (h00) reflections of PrC60MA. b Scherrer analysis of the coherence length (Lc) of PrC60MA crystals in pristine and 95:5 blend films using a pseudo-voigt function fitting. In this analysis the FWHM of each (h00) reflection, \({\triangle }_{q}\), is plotted vs the square reflection order, m2, and the extrapolation to the 0th order provides the Lc55,63 (Supplementary Fig. 10). c Back-Scattered Electrons (BSE) detector cross-section High Resolution Scanning Electron Microscopy (HRSEM) images of the p(g2T-TT) (left), 95:5 blend (middle), and PrC60MA (right) films following a Vapor Phase Infiltration (VPI) ‘staining’ process. The bright contrast is due to nucleation and growth of ZnO particles. The distribution of ZnO particles throughout the blend film, in contrast to the pristine PrC60MA film, corroborates the bulk-heterojunction morphology. The scale bar is 200 nm.
As a complementary tool to GIWAXS, pristine and blend films were exposed to a Vapor-Phase Infiltrated (VPI) process of diethyl zinc (DEZ) and water which to reveal the nanoscale morphology via High Resolution Scanning Electron Microscopy (HRSEM). Visualization of different phases in organic films is challenging, however in VPI the ZnO precursors selectively infiltrate different phases, resulting in a Z-contrast ‘staining’ detectable by cross section HRSEM. Figure 3c shows that ZnO precursors easily infiltrate p(g2T-TT) films, evident from the strong bright contrast, but generally accumulated on the surface of PrC60MA films with limited infiltration. The cross section HRSEM image of the blend film shows presence of ZnO particles evenly distributed throughout the film. This measurement confirms that the nanoscale morphology of the ambipolar layer is indeed a uniform bulk-heterojunction-like, and not, for example, a phase-separated bilayer56. Moreover, the visible interpenetrating network agrees with the reduction of coherence length of PrC60MA alongside retainment of connectivity between p(g2T-TT) chains.
Inverter logic circuit
Finally, to demonstrate the feasibility of the symmetrical performance for circuitry we fabricated inverters (NOT logic gates) using two identical ambipolar OECTs (Fig. 4a). Applying both positive and negative \({{{{{{\rm{V}}}}}}}_{{{{{{\rm{DD}}}}}}}\), we were able to measure the voltage transfer characteristics (VTC) of the inverter in both quadrants (Fig. 4b), showing small hysteresis (\(\Delta {{{{{{\rm{V}}}}}}}_{{{{{{\rm{in}}}}}}}=0.05{{{{{\rm{V}}}}}}\)) that suggests a small overpotential is required to turn off the OECTs. This might be due to the different energetic barriers for injection and expulsion of the two ions, or possibly due to different ion kinetics. Since the operating voltages of this blend system are symmetrical around \({{{{{{\rm{V}}}}}}}_{{{{{{\rm{G}}}}}}}=0.3{{{{{\rm{V}}}}}}\) (Fig. 1d) rather than 0 V, the indication for a well-balanced ambipolar inverter is that the voltage step occurs at \(\left(\frac{{{{{{{\rm{V}}}}}}}_{{{{{{\rm{DD}}}}}}}}{2}+0.3{{{{{\rm{V}}}}}}\right)\). Very high gains are calculated from the VTC (Fig. 4c), slightly lower than those reported for the state-of-the-art OECT inverters15,57 but still outperforming most13,22,31,58,59. Stability of the inverter is also recorded over 20 cycles and found to be excellent (Fig. 4d, e). It is important to note that while processing becomes simpler in ambipolar inverters, it is not free of problems compared to traditional CMOS inverters. The fact that ambipolar transistors cannot be totally switched off causes a Z-shape in the VTC which, in turn, is translated to higher static power consumption and lower static noise margins. Still, the improved performance and stability of the system reported here, and its unique tunability possibilities open new directions for exploiting ambipolar blend-based OECT circuitry.

a Wiring diagram of an inverter consisting of two identical ambipolar OECTs connected in series, one grounded and the other connected to VDD, with a common gate electrode applying Vin and an output voltage Vout. The range for Vin was selected to be the same as VG of the blend OECT presented in Fig. 1. b Voltage Transfer Characteristics (VTC), showing activity in both quadrants centering around Vin = +0.3 V at a voltage scan rate of 0.1 V/sec; small hysteresis of ~0.05 V is observed in all scans. c Gains calculated from the slope of the VTC, with forward and back peaks of 62, 35 (VDD = 0.7 V); 74, 48 (VDD = 0.8 V); 82, 56 (VDD = 0.9 V); 33, 49 (VDD = −0.7 V); 62, 49 (VDD = −0.8 V); 56, 51 (VDD = −0.9 V). d, e Shows a 20-cycle response of the inverter to input voltage square pulses for VDD = −0.8 V and + 0.8 V, respectively. All of device data presented here was acquired with a blend ratio of 95:5 (w:w) PrC60MA:p(g2T-TT).
Discussion
We have demonstrated the bulk-heterojunction methodology as a viable and promising tool in the ambipolar OECTs toolbox that opens exciting new venues with a broad matrix of tunable parameters and relationships to be unraveled. We show, for the first time, an ambipolar OECT based on a blend of two materials with opposite polarities, a fullerene derivative and a polythiophene, with improved performances compared to current ambipolar OMIECs. Our study shows that not only both components are optically, electrochemically and electrically active, but also do not interfere with one another. We demonstrate the utilization of this blend in a single OECT as well as an inverter made of two identical OECTs, easily fabricated using a single solution-processing step. Importantly, film integrity and electrical stability are very good due to the bulk-heterojunction morphology with the interpenetrating network. A main advantage of the blend approach over the synthesis of new ambipolar materials is that each component can be independently optimized, overcoming the built-in p-n trade-off that is inherent to a single ambipolar material. Moreover, the continuously growing library of OECT materials offers multiple combinations and possibilities to tune and improve the OECT performances. We suggest that when selecting the blend components, one should consider a few aspects. First, from the morphology point of view, material phase separation and the interplay between amorphous and crystalline domains should be controlled and optimized to support the transport of both ionic and electronic charge carriers. Second, both components should attain similar performances so that blend composition could be relatively balanced. Third, the threshold voltages (determined by the energy levels) should be symmetrical around 0 V so that the device can serve as a building block in advanced CMOS-like circuitry. Finally, we expect the ambipolar OECT to pave the way toward the design and fabrication of a new generation of simply-processed, compact and dynamically reconfigurable advanced bioelectronic devices as well as biologic sensors with dual detection of cations and anions for bodily fluids and soft tissues.
Methods
Materials
Poly(2-(3,3′-bis(2-(2-(2- methoxyethoxy)ethoxy)ethoxy)-[2,2’-bithiophen]−5-yl)thieno [3,2-b]thiophene) (p(g2T-TT), Mn = 8.1 kDa, Mw = 18.3 kDa from GPC in DMF at 40 oC) was synthesized by the McCulloch group according to previous reports and used as received34. C60,N,N,N -trimethyl-1-(2,3,4-tris(2-(2-methoxyethoxy)ethoxy)phenyl)methanaminium monoadduct (PrC60MA, Solenne BV), KCl (Sigma-Aldrich Israel Ltd.), Diethylzinc (packaged for use in deposition systems, Sigma-Aldrich Israel Ltd.) were used as received.
Film Preparation
All substrates were cleaned prior to film deposition in an ultrasonic bath in acetone, methanol and isopropanol (15 min each) and blow-dried in N2 (99.995%). p(g2T-TT) and PrC60MA were dissolved in chloroform (anhydrous, contains amylene as stabilizer, ≥99%, Sigma-Aldrich Israel Ltd.) at concentrations of 5 mg/ml and either 10 mg/ml or 30 mg/ml at room temperature, respectively. For blend preparation, pristine material solutions were mixed in the appropriate amounts to achieve 90:10, 95:5 and 99:1 (w:w) blend ratios. All solutions were spin-coated at room temperature onto substrates at 1000 rpm for 60 s followed by 3000 rpm for 10 s, to obtain ~60–70 nm film thicknesses, when using 5 mg/ml p(g2T-TT), 10 mg/ml PrC60MA and 9.5 mg/ml of the blend. For VPI measurements we prepared thicker films, ~175 nm, using 5 mg/ml p(g2T-TT) with 500 rpm for 60 s, 30 mg/ml PrC60MA with 1000 rpm for 60 s, and 24 mg/ml blend with 1000 rpm for 60 s. All films were subsequently annealed on a hot plate at 120 oC for 20 min. Film preparation was executed entirely under inert atmosphere inside a glovebox. Film thicknesses were measured using a stylus profilometer (Bruker DektakXT) with a \(12.5\mu m\) radius of the needle tip.
Spectroelectrochemistry (SEC) and Cyclic Voltammetry (CV)
Fluorine-doped tin oxide (FTO) coated glass (surface resistivity ~7 Ω/sq, Sigma-Aldrich Israel Ltd.) was used as a conductive transparent WE and coated with materials as described above. The FTO-coated substrates were dipped in a 0.1 M KCl solution in a glass cuvette (3/G/10, Starna Scientific Ltd.) with a Pt wire as CE and a Ag/AgCl pellet (E206 Warner Instruments, LLC) RE. The setup was put inside a UV-Vis spectrophotometer (Cary 100 Scan, Agilent Technologies, Inc.) and connected to an external potentiostat (Palmsens4, PalmSens BV) as a controller. To calculate the expected spectrum of the blend a rule of mixtures was applied according to the equation:
Electrochemical quartz-crystal microbalance (EQCM)
Gold-coated quartz crystal sensors (5 MHz for Q-sense E1/E4, RenLux Crystal Ltd.) was used as WE and coated with materials as described above. The sensor was placed in a dedicated cell (QWEM 401, Q-sense Explorer, Biolin Scientific) with a Pt wire CE and Ag/AgCl (3 M KCl) RE. The sensor was put in the module and aqueous 0.1 M KCl solution was continuously pumped into the chamber at a flow rate of 100 μl/min. CV in the range + 0.3 V to −0.9 V was performed for several cycles at a scan rate of 10 mV/sec and the frequencies recorded over 7 overtones. As the films are thin they were treated as rigid, therefore we could use the Sauerbrey equation to convert Δf/n to the change in areal mass Δm:
This equation uses \(C=17.7 \big[\frac{{ng}}{c{m}^{2}\cdot {Hz}}\big]\) as a constant for a 5 MHz crystal, a constant that is dependent on the wave velocity in quartz plate, the density of quartz and the fundamental resonance frequency. Only the 3rd overtone was used in calculations; therefore, it is put into the equation as well as \(n=3\).
Vapor Phase Infiltration (VPI)
Vapor Phase Infiltration (VPI) process of Zinc Oxide into the organic films was performed in an Atomic Layer Depositions system (Savannah® S200, Veeco Instruments Inc.). Silicon substrates were cleaned followed by film deposition via spin-coating as described above. The samples were then put in the ALD system and exposed to Diethylzinc vapors and DI water in a pulse-exposure-purge sequence. The cycle was repeated 80 times, with N2 (99.99%) as the carrier and purging gas. The reaction chamber was heated to 60 oC and the base flow rate of the reactor was 20 sccm.
High Resolution Scanning Electron Microscopy (HRSEM)
High-Resolution Scanning Electron Microscopy (HRSEM) cross-section images of films on Silicon substrates were recorded using a Zeiss Ultra-Plus FEG-SEM at 1.5 keV, with a Back-scattered Electrons (BSE) detector for Z-contrast observation between ZnO grains and unstained organic regions in the film.
X-ray Diffraction (XRD)
Out-of-plane \(\theta /2\theta\) XRD measurements of films on silicon substrates were recorded using a Rigaku SmartLab 9 kW x-ray diffractometer with D/tex Ultra250 0D detector and Cu Kα radiation (λ = 1.5418Å). The conversion of \(2\theta\) to d-space was performed using regular bragg’s equation:
To calculate the coherence length, we applied a pseudo-voigt function fitting on each peak of PrC60MA (in both the pristine and blend films). The fitting function is of the form of a linear combination of a Gaussian and a Lorentzian with a similar FWHM:
where \(I\) is the overall intensity, \({I}_{0}\) offset intensity, A the area, \({m}_{u}\) profile shape factor, \(q\) the scattering vector, \({q}_{c}\) scattering vector peak center, and w the FWHM of the peak. Since finite grain size and cumulative disorder is common in organic films, diffraction peaks that belong to the same set of crystal planes, namely (h00), displayed a successive broadening at higher orders. The extracted FHWM was plotted against the square peak order m2 and the extrapolated intersection at m2 = 0 was taken as \(\frac{2\pi }{{L}_{c}}\), where \({L}_{c}\) is the coherence length, following a method of analysis described elsewhere55.
For 2D-GIWAXS measurements we used Rigaku SmartLab 9 kW x-ray diffractometer with Cu Kα radiation (λ = 1.54186Å), equipped with a HyPix-3000 2D detector and an aperture slit installed. Incident angle was \(\omega=0.2^\circ\).
Organic Electrochemical Transistors (OECTs) device fabrication and measurement
Ultra-flat quartz-coated glass 15 mm x 20 mm substrates were cleaned as described above and blow-dried in N2 (99.995%). Then, substrates were placed in an OFET shadow mask (E291, Ossila Ltd) with \(W=1000\mu m,L=30\mu m\). The assembly was put inside a metal thermal evaporator (Edwards Auto 500), where 5 nm chromium and 50 nm gold were evaporated at a pressure of ~6E-6 Torr. Substrates were additionally cleaned for 15 min in isopropanol in an ultrasonic bath, blow dried with N2 gun and coated with the organic film. The film was dry-wiped from most of the metal contacts except for the channel area to minimize device crosstalk. Finally, Kapton® tape was used to manually mask the areas where the metal contacts were exposed to avoid contact between electrolyte and metal.
For OECT device characterization, a dual-channel source measure unit (SMU) (B2902A, Keysight Technologies, Inc.) was used with a dedicated software (EasyExpert group+). The SMU was connected to source & drain gold contacts as well as Ag/AgCl pellet (E206 Warner Instruments, LLC) that was used as the gate electrode. The electrolyte was 0.1 M KCl and dropped onto the devices prior to measurement. Output and Transfer curves were acquired with a double-sweep measurement. Calculations were based on an average of 4 devices per composition, each device averaged over 3 operation cycles, using a custom-made MATLAB script. The threshold voltage \({V}_{{th}}\) was calculated by using a linear fit of \(\sqrt{{I}_{D}}\) vs VG from the transfer curves and extraction of the intercept point between the fit and \(\sqrt{{I}_{D}}=0\). Transconductance and \(\mu {C}^{*}\) were calculated from transfer curves at maximum VG for each polarity (\(-0.3V\) for p-type, \(+0.9V\) for n-type) according to the equation:
For stability measurements the same setup as OECT characterization was used, yet square wave pulses were applied instead of regular sweep. The pulses were applied in another dedicated software (Quick I/V Measurement).
For inverter measurements 2 transistors were connected in series at the probing pads with silver paste, channel 1 was connected to Vin and ground, channel 2 connected to Vout, and another SMU (2612B, Keithley Instruments, Inc.) connected to VDD. The voltage transfer characteristics were recorded at a rate of 0.1 V/s, 5 cycles for each curve, using Quick I/V Measurement software. The gain was calculated as \(\frac{\delta {V}_{{out}}}{\delta {V}_{{in}}}\) from voltage transfer characteristics.
Response time of the ambipolar 95:5 blend was extracted from the stability measurement using an exponential fit:
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its supplementary information files.
References
Khodagholy, D. et al. In vivo recordings of brain activity using organic transistors. Nat. Commun. 4, 1575 (2013).
Kergoat, L. et al. Detection of glutamate and acetylcholine with organic electrochemical transistors based on conducting Polymer/Platinum nanoparticle composites. Adv. Mater. 26, 5658–5664 (2014).
Braendlein, M. et al. Lactate detection in tumor cell cultures using organic transistor circuits. Adv. Mater. 29, 1605744 (2017).
Rivnay, J., Owens, R. M. & Malliaras, G. G. The rise of organic bioelectronics. Chem. Mater. 26, 679–685 (2014).
Owens, R. M. & Malliaras, G. G. Organic electronics at the interface with biology. MRS Bull. 35, 449–456 (2010).
Rashid, R. B., Ji, X. & Rivnay, J. Organic electrochemical transistors in bioelectronic circuits. Biosens. Bioelectron. 190, 113461 (2021).
Strakosas, X., Bongo, M. & Owens, R. M. The organic electrochemical transistor for biological applications. J. Appl. Polym. Sci. 132, 41735 (2015).
Nawaz, A., Liu, Q., Leong, W. L., Fairfull-Smith, K. E. & Sonar, P. Organic electrochemical transistors for in vivo bioelectronics. Adv. Mater. 33, 2101874 (2021).
Sun, H., Gerasimov, J., Berggren, M. & Fabiano, S. N-Type organic electrochemical transistors: Materials and challenges. J. Mater. Chem. C. 6, 11778–11784 (2018).
Giovannitti, A. et al. N-type organic electrochemical transistors with stability in water. Nat. Commun. 7, 13066 (2016).
Ohayon, D. et al. Influence of side chains on the n-type organic electrochemical transistor performance. ACS Appl. Mater. Interfaces 13, 4253–4266 (2021).
Bischak, C. G., Flagg, L. Q., Yan, K., Li, C.-Z. & Ginger, D. S. Fullerene active layers for n-type organic electrochemical transistors. ACS Appl. Mater. Interfaces 11, 28138–28144 (2019).
Sun, H. et al. Complementary logic circuits based on high-performance n-type organic electrochemical transistors. Adv. Mater. 30, 1704916 (2018).
Rivnay, J. et al. Organic electrochemical transistors. Nat. Rev. Mater. 3, 17086 (2018).
Wu, H. Y. et al. Influence of molecular weight on the organic electrochemical transistor performance of ladder-type conjugated polymers. Adv. Mater. 34, 2106235 (2022).
Wu, P., Reis, D., Sharon Hu, X. & Appenzeller, J. Two-dimensional transistors with reconfigurable polarities for secure circuits. Nat. Electron. 4, 45–53 (2021).
Sou, A. et al. Programmable logic circuits for functional integrated smart plastic systems. Org. Electron. 15, 3111–3119 (2014).
Ishida, K. et al. User Customizable Logic Paper (UCLP) With Sea-Of Transmission-Gates (SOTG) of 2-V Organic CMOS and Ink-Jet Printed Interconnects. IEEE J. Solid-State Circuits 46, 285 (2011).
Ren, Y. et al. Recent advances in ambipolar transistors for functional applications. Adv. Funct. Mater. 29, 1902105 (2019).
Zulkarnaen Bisri, S. et al. Outlook and emerging semiconducting materials for ambipolar transistors. Adv. Mater. 26, 1176–1199 (2014).
Gentili, D. et al. Logic-gate devices based on printed polymer semiconducting nanostripes. Nano Lett. 13, 3643–3647 (2013).
Rashid, R. B. et al. Ambipolar inverters based on cofacial vertical organic electrochemical transistor pairs for biosignal amplification. Sci. Adv. 7, eabh1055 (2021).
Wustoni, S. et al. Membrane-free detection of metal cations with an organic electrochemical transistor. Adv. Funct. Mater. 29, 1904403 (2019).
Flagg, L. Q., Giridharagopal, R., Guo, J. & Ginger, D. S. Anion-dependent doping and charge transport in organic electrochemical transistors. Chem. Mater. 30, 5380–5389 (2018).
Lin, P., Yan, F. & Chan, H. L. W. W. Ion-sensitive properties of organic electrochemical transistors. ACS Appl. Mater. Interfaces 2, 1637–1641 (2010).
Flagg, L. Q. et al. P-type electrochemical doping can occur by cation expulsion in a high-performing polymer for organic electrochemical transistors. ACS Mater. Lett. 2, 254–260 (2020).
Cendra, C. et al. Role of the anion on the transport and structure of organic mixed conductors. Adv. Funct. Mater. 29, 1807034 (2018).
Nair, R. R. Organic electrochemical transistor on paper for the detection of halide anions in biological analytes. Flex. Print. Electron. 5, 045004 (2020).
Parr, Z. S. et al. From p- to n-Type Mixed Conduction in Isoindigo-Based Polymers through Molecular Design. Adv. Mater. 34, 2107829 (2022).
Zhang, Y. et al. High‐performance organic electrochemical transistors and neuromorphic devices comprising naphthalenediimide‐dialkoxybithiazole copolymers bearing glycol ether pendant groups. Adv. Funct. Mater. 32, 2201593 (2022).
Samuel, J. J. et al. Single-Component CMOS-Like Logic using Diketopyrrolopyrrole-Based Ambipolar Organic Electrochemical Transistors. Adv. Funct. Mater. 31, 2102903 (2021).
Li, G., Zhu, R. & Yang, Y. Polymer solar cells. Nat. Photonics 6, 153–161 (2012).
Meijer, E. J. et al. Solution-processed ambipolar organic field-effect transistors and inverters. Nat. Mater. 2, 678–682 (2003).
Giovannitti, A. et al. Controlling the mode of operation of organic transistors through side-chain engineering. Proc. Natl. Acad. Sci. 113, 12017–12022 (2016).
Savva, A. et al. Balancing ionic and electronic conduction for high‐performance organic electrochemical transistors. Adv. Funct. Mater. 30, 1907657 (2020).
Shao, S. et al. Elimination of the light soaking effect and performance enhancement in perovskite solar cells using a fullerene derivative. Energy Environ. Sci. 9, 2444–2452 (2016).
Jahani, F. et al. Fullerene derivatives with increased dielectric constants. Chem. Commun. 50, 10645 (2014).
Li, C.-Z. et al. Effective interfacial layer to enhance efficiency of polymer solar cells via solution-processed fullerene-surfactants. J. Mater. Chem. 22, 8574–8578 (2012).
Cho, S., Yuen, J., Kim, J. Y., Lee, K. & Heeger, A. J. Ambipolar organic field-effect transistors fabricated using a composite of semiconducting polymer and soluble fullerene. Appl. Phys. Lett. 89, 153505 (2006).
Geens, W., Shaheen, S. E., Brabec, C. J., Poortmans, J. & Sariciftci, N. S. Field-effect mobility measurements of conjugated polymer/fullerene photovoltaic blends. in AIP Conference Proceedings vol. 544 516–520 (AIP Publishing, 2000).
von Hauff, E., Parisi, J. & Dyakonov, V. Field effect measurements on charge carrier mobilities in various polymer-fullerene blend compositions. Thin Solid Films 511–512, 506–511 (2006).
Janasz, L. et al. Balanced ambipolar organic field-effect transistors by polymer preaggregation. ACS Appl. Mater. Interfaces 9, 20696–20703 (2017).
Goffri, S. et al. Multicomponent semiconducting polymer systems with low crystallization-induced percolation threshold. Nat. Mater. 5, 950–956 (2006).
Qiu, L. et al. Organic Thin-Film Transistors Based on Blends of Poly(3-hexylthiophene) and Polystyrene with a Solubility-Induced Low Percolation Threshold. Chem. Mater. 21, 4380–4386 (2009).
Ford, M. J. et al. High Mobility Organic Field-Effect Transistors from Majority Insulator Blends. Chem. Mater. 28, 52 (2016).
Moser, M. et al. Side Chain Redistribution as a Strategy to Boost Organic Electrochemical Transistor Performance and Stability. Adv. Mater. 2002748 https://doi.org/10.1002/adma.202002748 (2020).
Wu, X. et al. Enhancing the Electrochemical Doping Efficiency in Diketopyrrolopyrrole-Based Polymer for Organic Electrochemical Transistors. Adv. Electron. Mater. 7, 2000701 (2021).
Chlistunoff, J., Cliffel, D. & Bard, A. J. Electrochemistry of fullerene films. Thin Solid Films 257, 166–184 (1995).
Parr, Z. S. et al. Semiconducting small molecules as active materials for p‐type accumulation mode organic electrochemical transistors. Adv. Electron. Mater. 6, 2000215 (2020).
Spano, F. C. The spectral signatures of Frenkel Polarons in H-and J-aggregates. Acc. Chem. Res. 43, 429–439 (2010).
Spano, F. C. & Silva, C. H-and J-Aggregate Behavior in Polymeric Semiconductors. Annu. Rev. Phys. Chem. 65, 477–500 (2014).
Finn, P. A. et al. Effect of polar side chains on neutral and p-doped polythiophene. J. Mater. Chem. C. 8, 16216 (2020).
Surgailis, J. et al. Mixed Conduction in an N‐Type Organic Semiconductor in the Absence of Hydrophilic Side‐Chains. Adv. Funct. Mater. 31, 2010165 (2021).
Verploegen, E., Miller, C. E., Schmidt, K., Bao, Z. & Toney, M. F. Manipulating the Morphology of P3HT−PCBM Bulk Heterojunction Blends with Solvent Vapor Annealing. Chem. Mater. 24, 3923–3931 (2012).
Rivnay, J., Mannsfeld, S. C. B., Miller, C. E., Salleo, A. & Toney, M. F. Quantitative determination of organic semiconductor microstructure from the molecular to device scale. Chem. Rev. 112, 5488–5519 (2012).
Björström, C. M. et al. Vertical phase separation in spin-coated films of a low bandgap polyfluorene/PCBM blend-Effects of specific substrate interaction. Appl. Surf. Sci. 253, 3906–3912 (2007).
Romele, P., Ghittorelli, M., Miklós Kovács-Vajna, Z. & Torricelli, F. Ion buffering and interface charge enable high performance electronics with organic electrochemical transistors. Nat. Commun. 10, 3044 (2019).
Travaglini, L. et al. Single‐Material OECT‐based flexible complementary circuits featuring polyaniline in both conducting channels. Adv. Funct. Mater. 31, 2007205 (2021).
Hutter, P. C., Rothlander, T., Scheipl, G. & Stadlober, B. All screen-printed logic gates based on organic electrochemical transistors. IEEE Trans. Electron Devices 62, 4231–4236 (2015).
Liao, H. et al. Efficient n-type small-molecule mixed ion-electron conductors and application in hydrogen peroxide sensors. ACS Appl. Mater. Interfaces 14, 16477–16486 (2022).
Reed, C. A. & Bolskar, R. D. Discrete Fulleride Anions and Fullerenium Cations. Chem. Rev. 3, 1075–1120 (2000).
Khaled, M. M., Carlin, R. T., Trulove, P. C., Eaton, G. R. & Eaton, S. S. Electrochemical Generation and Electron Paramagnetic Resonance Studies of C60-, C602-, and C603-. J. Am. Chem. Soc. 116, 3465–3474 (1994).
Rivnay, J., Noriega, R., Kline, R. J., Salleo, A. & Toney, M. F. Quantitative analysis of lattice disorder and crystallite size in organic semiconductor thin films. Phys. Rev. B 84, 045203 (2011).
Acknowledgements
This project was partially funded by the European Union’s Horizon 2020 research and innovation program under grant agreement No 964677 (MITICS). The authors thank Professor Jonathan Rivnay for fruitful discussions.
Author information
Authors and Affiliations
Contributions
E.S. conceived, designed, and performed OECT and Inverters fabrication and characterization, XRD, Electrochemistry and Spectroelectrochemistry measurement and analysis, as well as HRSEM and EQCM analysis, and wrote the manuscript under the supervision of G.L.F. O.N. performed the HRSEM imaging. M.S., under the guidance of V.F., performed the EQCM measurement. I.M.P., under the supervision of I.M., synthesized the polymer used in this study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stein, E., Nahor, O., Stolov, M. et al. Ambipolar blend-based organic electrochemical transistors and inverters. Nat Commun 13, 5548 (2022). https://doi.org/10.1038/s41467-022-33264-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-33264-2
This article is cited by
-
Designing organic mixed conductors for electrochemical transistor applications
Nature Reviews Materials (2024)
-
Organic mixed conductors for bioinspired electronics
Nature Reviews Materials (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
