
Abstract
The Subantarctic Southern Ocean has long been thought to be an important contributor to increases in atmospheric carbon dioxide partial pressure (pCO2) during glacial-interglacial transitions. Extensive studies suggest that a weakened biological pump, a process associated with nutrient utilization efficiency, drove up surface-water pCO2 in this region during deglaciations. By contrast, regional influences of the solubility pump, a process mainly linked to temperature variations, have been largely overlooked. Here, we evaluate relative roles of the biological and solubility pumps in determining surface-water pCO2 variabilities in the Subantarctic Southern Ocean during the last deglaciation, based on paired reconstructions of surface-water pCO2, temperature, and nutrient utilization efficiency. We show that compared to the biological pump, the solubility pump imposed a strong impact on deglacial Subantarctic surface-water pCO2 variabilities. Our findings therefore reveal a previously underappreciated role of the solubility pump in modulating deglacial Subantarctic CO2 release and possibly past atmospheric pCO2 fluctuations.
Similar content being viewed by others


Introduction
The Southern Ocean is widely regarded as a crucial source of carbon dioxide (CO2) to the atmosphere during glacial terminations because this region serves as a window for CO2 exchanges between the atmosphere and the ocean interior1,2,3,4. In the Southern Ocean, prevailing southern hemisphere westerly winds drive upwelling of carbon-rich deep waters surrounding Antarctica, some of which are transported northward to the Subantarctic Zone (SAZ)5,6. The surface SAZ exposes the newly upwelled carbon-rich deep waters to the atmosphere enabling CO2 outgassing, before these waters are entrained to form intermediate and mode waters5,6,7.
Changes in the SAZ have been thought to be critical to deglacial atmospheric pCO2 rises, with a contribution estimated to be around 40 ppm8,9,10,11. In the SAZ, CO2 tends to escape to the atmosphere due to elevated surface-water CO2 partial pressures (pCO2) driven by high surface-water dissolved inorganic carbon (DIC) concentrations12,13 associated with the newly upwelled deep waters surrounding Antarctica. CO2 outgassing in the SAZ is somewhat alleviated by biologically driven carbon sequestration that exports carbon to depths in the form of organic matter, a process called the biological pump1,14,15. In addition to this biological process, it is important to note that surface-water pCO2 is further affected by CO2 solubility determined by seawater temperature and salinity, a process known as the solubility pump14,16. Changes in the solubility pump have been shown by modeling studies to contribute substantially to atmospheric pCO2 variability2,17,18,19. Everything else being equal, warming lowers CO2 solubility in seawater and thus increases surface-water pCO2 with an effect to cause CO2 outgassing from the ocean to the atmosphere16. Surface-water pCO2 increases with increasing salinity, but the salinity effect on the CO2 solubility is generally smaller than the temperature effect in most regions20 (Supplementary Fig. 2).
During glacial terminations, it has been proposed that the biological pump efficiency was lowered, driving up CO2 outgassing in the SAZ1,11,21,22,23. The deglacial SAZ biological pump efficiency decline has been linked to reduced supplies of micronutrients such as iron via eolian lithogenic fluxes10,11,24. In this case, weakened biological pump would leave more carbon unused in the SAZ surface, raising surface-water pCO2 which would stimulate CO2 outgassing1. On the other hand, as manifested by Southern Ocean temperature reconstructions25,26,27, deglacial SAZ warming, in theory, should weaken the solubility pump, raise surface-water pCO2, and promote CO2 outgassing16 (Supplementary Fig. 1). Existing reconstructions in the SAZ indeed show elevated surface-water pCO2 during the last deglaciation (~18-11 ka BP)22,23. Yet, it remains unknown regarding the respective roles of biological and solubility pump changes in affecting these past surface-water pCO2 rises in the SAZ and by extension atmospheric pCO2 changes.
Here, we systematically investigate the contributions of the biological and solubility pumps to the SAZ surface-water pCO2 changes in the modern ocean and during the last deglaciation. For the deglacial investigation, we have generated a surface-water pCO2 record using a sediment core from the Southwest Pacific, paired with nutrient utilization efficiency and sea surface temperature (SST) reconstructions. We evaluate the relative roles of biological and solubility pumps in regulating deglacial surface-water pCO2 changes at our site location. The same approach is further applied to published records at three additional SAZ locations. Moreover, we examine simulated early deglacial carbon cycle changes in an earth system model28 to distinguish effects of the two pumps during the early deglaciation. All our investigations suggest a strong solubility pump effect on the SAZ surface-water pCO2 changes, urging a rethinking of mechanisms underlying deglacial CO2 outgassing from the SAZ and, by extension, past atmospheric pCO2 variabilities.
Results
Solubility pump in the modern SAZ
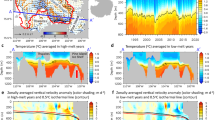
In the modern SAZ, both biological and solubility pumps strongly control spatial distribution and seasonal variability of surface-water pCO212,13,29. Regarding the spatial distribution, Fig. 1a shows small northward declines in annual mean surface-water pCO2 within the SAZ, where nutrient (nitrate) is progressively utilized equatorward (Fig. 1b). Enhanced nutrient consumption would lower surface-water pCO2, but this biological effect is compensated by the opposing influence of equatorward surface-water warming in the SAZ (Fig. 1c). Consequently, surface-water pCO2 changes caused by nutrient consumption is largely balanced by decreasing CO2 solubility, and the total northward surface-water pCO2 decline is marginal. Regarding the seasonal variability, Fig. 1d shows minimal monthly surface-water pCO2 deviation from the annual mean levels in the SAZ. By contrast, seasonal changes in the biological and solubility pumps each cause surface-water pCO2 to fluctuate ~40 ppm around the annual mean level (Fig. 1e, f)12,29. Because the strong (weak) biological pump occurs during warm (cold) seasons with low (high) CO2 solubility, effects from the biological and solubility pumps generally cancel each other, leading to little seasonal surface-water pCO2 variability. Combined, these spatial and temporal patterns suggest that the solubility pump plays a critical role, comparable to that of the biological pump, in determining the SAZ surface-water pCO2 fluctuations in the modern ocean. Next, we move on to explore the impact of the solubility pump on past surface-water pCO2 changes, a topic that has rarely been studied by proxy reconstructions.

a Annual mean surface-water pCO2 during years 1985–2018. The map is made from data presented in Gregor and Gruber29. https://creativecommons.org/licenses/by/4.0/. b Annual mean surface-water nitrate concentration. The map is made from data presented in Garcia et al.48, accessible from https://www.ncei.noaa.gov/access/world-ocean-atlas-2018/. c Annual mean sea surface temperature. Two black curves indicate the modern positions of the Subtropical Front (STF; the northern curve) and the Subantarctic Front (SAF; the southern curve), respectively, and the region between them is the Subantarctic Zone (SAZ). The red star represents the location of our study site MD97-2106, and squares represent locations with published δ11B-based surface-water pCO2 reconstructions in the SAZ. The map is made from data presented in Locarnini et al.49, accessible from https://www.ncei.noaa.gov/access/world-ocean-atlas-2018/. d Monthly surface-water pCO2 variability within the SAZ (year 1985–2018) calculated from the OceanSODA-ETHZ dataset29. e Monthly surface-water pCO2 variability attributed to biological pump changes within the SAZ calculated from the OceanSODA-ETHZ dataset29. f Monthly surface-water pCO2 variability attributed to solubility pump changes within the SAZ calculated from the OceanSODA-ETHZ dataset29. In d–f, shadings show ±1σ standard deviation ranges of observations at discrete locations (represented by dots). As can be seen from d–f, the solubility pump plays a critical role in stabilizing SAZ surface-water pCO2 by countering effects due to biological pump changes.
New deglacial SAZ surface-water pCO2 and δ15N records
We present records of surface-water pCO2 and nutrient utilization, respectively, based on boron isotopes (δ11B)22,30,31 and foraminifera-bound nitrogen isotopes (δ15NFB) of mixed-layer-dwelling planktic foraminifera species Globigerina bulloides for site MD97-2106 from the Southwest Pacific Ocean (Fig. 1). Site MD97-2106 (45.15°S, 146.28°E) is located in the northern part of the SAZ. Compared to today, the Subtropical Front, the northern boundary of the SAZ, possibly shifted northward to Tasmania during the Last Glacial Maximum (LGM; ~22-18 ka)32,33. During the last deglaciation, the Subtropical Front might migrate southwards32,33, but unlikely to the south of our site. This is because the Subtropical Front marks a ~4 °C SST gradient34, while reconstructions at our site only show ~3 °C SST change during the entire deglaciation35,36. Consequently, our site was likely located within the SAZ during the entire last deglaciation, ideal for investigating deglacial SAZ surface conditions. The age model of site MD97-2106 during the last deglaciation is based on new radiocarbon dates and tuning of SST at this site to Antarctic temperatures26 (Fig. 2a, b; Supplementary Table 1, Supplementary Fig. 4). See Methods for analytical details.

a Sea surface temperature (SST) reconstructed from planktic foraminiferal Mg/Ca36 (blue circles, left axis). b Antarctic temperature changes represented by δD26 (gray curve, right axis). Crosses and triangles at the bottom are age control points based on planktic radiocarbon dating and SST-δD matching, respectively. c Planktic foraminiferal δ11B and seawater borate δ11B with error bars showing ±2σ uncertainties. d Reconstructed surface-water pCO2 at site MD97-2106 (red circles) compared with atmospheric pCO2 recorded in the Antarctic ice cores (black curve)43, 44 (left axis). e Total surface-water pCO2 change relative to 18 ka (δpCO2TOTAL) at site MD97-2106 (right axis). f Deglacial surface-water pCO2 attributed to the solubility and biological pumps (δpCO2SOL and δpCO2BIO, blue and orange dots, respectively) at site MD97-2106. g Foraminifera-bound δ15N, a proxy reflecting surface nutrient utilization. Curves in d–g are derived from a LOESS smoother. In d, light and dark envelopes, respectively, represent 2.3–97.7% (roughly ±2σ) and 15.9–84.1% (roughly ±1σ) uncertainty ranges of timeseries incorporating uncertainties from measurements, all individual parameters used for calculations, and age models. In f and g only 15.9–84.1% uncertainty ranges are shown for clarify. The vertical pale orange bars represent Heinrich Stadial 1 (HS1; ~18.0–14.6 ka BP) and the Younger Dryas (YD; ~12.8–11.7 ka BP). The vertical pale blue bar represents the Antarctic Cold Reversal (ACR; ~14.6–12.8 ka BP). δpCO2SOL and δpCO2BIO (f) show similar structures to SST (a) and foraminifera-bound δ15N (g), respectively.
Our reconstructed surface-water pCO2 at site MD97-2106 fluctuated between ~210 and ~270 ppm during the last deglaciation, reached ~270 ppm at the onset of the Holocene, and increased by ~20 ppm from the early to middle Holocene (Fig. 2d). The range of deglacial surface-water pCO2 at site MD97-2106 is comparable to previous reconstructions22,23. At our site, surface-water pCO2 briefly dropped below the atmospheric pCO2 values during the Antarctic Cold Reversal (ACR; 14.6–12.8 ka BP) and the Younger Dryas (YD; 12.8–11.7 ka BP) (Fig. 2d).
In addition to our surface-water pCO2 reconstructions, we employ δ15NFB to infer nitrate utilization efficiency at site MD97-2106. δ15NFB reflects δ15N of surface-water nitrate taken up by foraminifera which increases as surface nitrate is progressively consumed by photosynthetic algae11,37,38. During Heinrich Stadial 1 (HS1; 18.0-14.6 ka BP) when the age model of our core is well constrained by a warming phase, δ15NFB at our site remained roughly unchanged (Fig. 2b, g). This observation is consistent with a precisely dated coral-bound δ15N record from the same region showing minimal δ15N variability over the same period21 (Supplementary Fig. 9). Maxima of δ15NFB occurred during the ACR and the YD, coinciding with surface-water pCO2 minima. During the Holocene, δ15NFB declined by about 2.0‰, consistent with other fossil-bound δ15N records from the Southern Ocean11,21,39,40,41.
Evaluating past biological and solubility effects at site MD97-2106
We partition surface-water pCO2 changes at site MD97-2106 into two components caused by changes in biological and solubility pumps, using a similar method previously applied to investigate modern surface-water pCO2 variability12,16,29. Firstly, we calculate total in-situ surface-water pCO2 changes (noted as δpCO2TOTAL) relative to the reference age of 18 ka; choosing a different reference age has little effect on our conclusions (Supplementary Fig. 6). Secondly, surface-water pCO2 is recalculated using carbonate chemistry (i.e., DIC and alkalinity) fixed at the reference age, but using varying SST and sea surface salinity (SSS) based on our reconstructions (Methods). Thirdly, this recalculated surface-water pCO2 is used to compute changes relative to 18 ka (the reference age). The relative surface-water pCO2 changes calculated in this way are only driven by SST and SSS, and thus are attributed to the solubility pump effect (noted as δpCO2SOL). Fourthly, we calculate the difference between δpCO2TOTAL and δpCO2SOL, which is defined as the biology-driven surface-water pCO2 change (noted as δpCO2BIO). We also provide an alternative approach to directly calculate δpCO2BIO, which yields consistent results with those presented in the main text (Methods; Supplementary Fig. 8). Any influence of external processes on surface-water pCO2, such as changes associated with frontal shift, is embedded in our method, because these external processes affect surface-water pCO2 via the carbonate chemistry, SST-SSS, or both. See “Methods” for calculation details.
As can be seen from Fig. 2f, δpCO2BIO at site MD97-2106 fluctuated between ~−20 ppm and ~+40 ppm from the LGM to the early Holocene, followed by a ~20-ppm increase during the Holocene. From 18 to 15 ka, δpCO2BIO showed little net change, suggesting marginal influence of biological processes on surface-water pCO2 variations at our site. During the ACR and the YD, δpCO2BIO exhibits mostly negative values, indicating a strengthened biological pump that would lower surface-water pCO2 during these times. The evolution of δpCO2BIO at our site is well corroborated by our δ15NFB record from the same core. Little net δpCO2BIO change concurred with stable δ15NFB during HS1, while negative δpCO2BIO during the ACR and the YD coincided with higher δ15NFB values which indicate more complete nutrient utilization (Fig. 2f–g). We note that, in addition to nitrate utilization in the SAZ, our δ15NFB might have also been affected by δ15N of nitrate supplied to our site, which depends on the nitrate utilization of the nitrate source11,21,41,42. Despite these complications, similar deglacial structures in δ15NFB and independently derived δpCO2BIO suggest that δ15NFB at site MD97-2106 reflects local nitrate utilization efficiency. Moreover, given their independent methods, consistent patterns of our δpCO2BIO and δ15NFB lend strong support to our inference about past biological pump changes at this site. In stark contrast to previous findings in the SAZ22,23,24, our δpCO2BIO and δ15NFB suggest that the biological pump played a minor role in contributing to surface-water pCO2 and thus air-sea CO2 exchange at site MD97-2106 during HS1 and the YD when atmospheric pCO2 rose substantially43,44.
Compared with δpCO2BIO, our calculated δpCO2SOL, which reflects solubility pump effects, shows a different history (Fig. 2f). As expected, deglacial δpCO2SOL increased in response to warming, although this warming effect was slightly counteracted by a declining SSS (Supplementary Fig. 5). From 18 to 10 ka, δpCO2SOL showed a net increase of ~20 ppm, contributing to about one-third of the δpCO2TOTAL at site MD97-2106 over the same period. More specifically, the δpCO2SOL increase dominated the δpCO2TOTAL rise from 18 to 15 ka, in contrast to little net change in the concurrent δpCO2BIO. This highlights the critical role of the weakened solubility pump in maintaining positive sea-air pCO2 gradients, which would contribute to the atmospheric pCO2 rise during HS1. During the YD, rising δpCO2SOL helped reverse biological effects (shown by δpCO2BIO and δ15NFB) to limit the development of negative sea-air pCO2 gradient (Fig. 2f-g), contributing to the contemporary atmospheric pCO2 rise.
By separating influences of the biological and solubility pumps downcore, we demonstrate that a substantial portion of deglacial surface-water pCO2 rise at our site originated from variations in the solubility pump. This suggests that the solubility pump, which has been neglected in previous investigations in the region, played an important role in regulating deglacial surface-water pCO2 changes in the Pacific SAZ.
Proxy and model data evaluation for broader SAZ
To quantify past influences of biological and solubility pumps in broader SAZ regions, we reanalyze deglacial surface-water pCO2 changes at three additional locations using published proxy records22,23 (Figs. 1a; 3b–d). At all the investigated sites, our calculated δpCO2BIO suggests little-to-modest biological pump effects on SAZ surface-water pCO2 during times with large atmospheric pCO2 rises. For instance, except for a brief excursion at ~17 ka, δpCO2BIO at Site ODP 1090 (Fig. 3b) varied little and suggests minimal biological pump effects on surface-water pCO2 during the last deglaciation23. At site PS2498-1, despite an overall larger δpCO2BIO contribution to surface-water pCO2 compared to other sites, roughly stable average δpCO2BIO during ~14-11 ka contributed little to the observed surface-water pCO2 rise (Fig. 3c). At site TAN1106-28, δpCO2BIO showed a prominent peak at ~16 ka, but the general deglacial trend of δpCO2BIO is poorly defined by the low temporal resolution. In contrast to δpCO2BIO changes, our calculated δpCO2SOL at all studied sites supports that solubility pump changes consistently contributed to deglacial δpCO2TOTAL. From 18 to 10 ka, we observe well-defined δpCO2SOL increases of ~20 ppm at Site ODP 1090 from the South Atlantic. At site TAN1106-28 from the South Pacific, δpCO2SOL increased by ~50 ppm, in response to the deglacial SST change of ~7 °C in part caused by a possible frontal shift over this site23. Although the record at site PS2498-1 does not cover the entire last deglaciation, δpCO2SOL at this site shows a ~30-ppm increase during ~14–11 ka (Fig. 3c). These δpCO2SOL changes significantly contribute to, or even dominate, the surface-water pCO2 variations at these sites, strengthening our findings at site MD97-2106. Overall, our analyses of proxy data from different sectors of the Southern Ocean demonstrate that deglacial surface-water pCO2 changes in the SAZ are substantially affected by solubility pump changes, rather than solely by biological pump changes as previously assumed11,22,23.

a MD97-2106; b ODP 109011, 23, 46; c PS2498-122, 47; and d TAN1106-2823. Data in the four panels are arranged as follows. First row: surface-water pCO2 at investigated sites (red, orange, blue, and green curves) compared to atmospheric pCO2 recorded in Antarctic ice cores43,44 (gray curves). Second row: surface-water pCO2 change attributed to the solubility pumps (δpCO2SOL). Third row: surface-water pCO2 change attributed to the biological pump (δpCO2BIO). Fourth row: dust fluxes recorded in an Antarctic ice core45 (a), lithogenic (pale orange curve)22 and opal (orange curve with dots)46 fluxes at site ODP 1090 (b), lithogenic (pale blue curve) and opal (blue curve with dots) fluxes at site PS2498-147 (c). Fifth row: Foraminifera-bound δ15N (this study and11). Note that scales of δpCO2SOL and δpCO2BIO differ in d compared to a–c. Envelopes represent 15.9–84.1% uncertainties incorporating uncertainties from measurements, all individual parameters required for calculations, and age models. The vertical pale orange bars represent Heinrich Stadial 1 (HS1; ~18.0–14.6 ka BP) and the Younger Dryas (YD; ~12.8–11.7 ka BP). The vertical pale blue bar represents the Antarctic Cold Reversal (ACR; ~14.6–12.8 ka BP). The reference age for relative surface-water pCO2 change decomposition at these sites is set at 18 ka, except at site PS2498-1, where it is set at the oldest age of ~15.8 ka. For all sites examined, solubility pump changes consistently contribute ~20–50 ppm to the deglacial surface-water pCO2 changes (second row). Surface-water pCO2 changes attributed to the biological pump (third row) differ from dust and export production (fourth row).
We further scrutinize the role of the solubility pump in affecting deglacial SAZ surface-water pCO2 in a simulation by climate model LOVECLIM28. In this simulation, a 30-ppm atmospheric pCO2 increase is achieved during HS1 when the Southern Ocean overturning circulation and southern hemisphere westerly winds are intensified. The rising surface-water pCO2 in the Southern Ocean is diagnosed as a main CO2 source for the early deglacial atmospheric pCO2 increase28. Using the same method applied to proxy data, we quantify δpCO2BIO and δpCO2SOL changes between 19 ka and 15 ka in this simulation (“Methods”). Our decomposition (Fig. 4) reveals that δpCO2BIO changes are either small or tend to lower pCO2TOTAL in the SAZ. Because nutrient utilization forced by iron availability is not prescribed in the model, our calculated δpCO2BIO per se cannot be used to dismiss iron fertilization effect on deglacial SAZ surface-water pCO2 changes. By contrast, δpCO2SOL changed substantially due to strong surface warming, dominating surface-water δpCO2TOTAL rise in the SAZ (Fig. 4). Therefore, our model data analyses suggest that solubility pump changes are crucial for deglacial surface-water pCO2 and air-sea CO2 exchange in the SAZ, strengthening our findings based on above extensive proxy reconstructions.

a Total surface-water pCO2 change (δpCO2TOTAL). b surface-water pCO2 change attributed to the biological pump (δpCO2BIO). c, surface-water pCO2 change attributed to the solubility pump (δpCO2SOL). All anomalies (δ) represent changes between 19 ka and 15 ka. Note different δpCO2 scales for the three panels. The region between the dotted lines is the modern Subantarctic Zone (SAZ). The locations of MD97-2106 (red star) and other investigated sites (ODP 1090, orange square; PS2498-1, blue square; TAN1106-28 teal square) are shown. As can be seen, the total δpCO2TOTAL rise in the SAZ from 19 to 15 ka in this model simulation is mainly driven by solubility pump changes. The figure is made from recalculation based on data presented in Menviel et al.28.
Discussion
It is widely thought that declined biological pump efficiency, possibly owing to reducing nutrient utilization associated with dust-borne iron deposition, enhanced CO2 outgassing in the SAZ and hence atmospheric pCO2 rises during deglaciations1,10,11,24. During HS1 when atmospheric pCO2 raised substantially and dust deposition in Antarctica declined dramatically45 (Fig. 3), δpCO2BIO, together with independently measured δ15NFB, indicates little net biological pump change at site MD97-2106. Over the same period, δpCO2BIO shows little increase in response to the reduced iron fertilization inferred from dust deposition in Antarctica45 and other examined sites. At two of the examined sites (ODP 1090 and PS2498-1), where opal and lithogenic fluxes are available24,46,47,48, δpCO2BIO shows correlation with neither flux during HS124,46,47 (Fig. 3b, c). In addition to local nutrient utilization efficiency, δpCO2BIO and δ15NFB at a certain site in the SAZ may also be affected by nutrient supplies modulated by shifts of the Southern Ocean fronts11,21,39,41,42. Everything else being equal, poleward movements of the Subtropical and Subantarctic fronts and thus the SAZ during HS1 would reduce nutrient supply, with an effect to raise observed nutrient utilization efficiency, at any given location in the SAZ. During HS1, declining local nutrient utilization efficiency deduced from reduced iron fertilization and poleward front shifts would have opposing effects on SAZ δpCO2BIO, and their combined effects may result in minimal δpCO2BIO changes overall. Thus, despite the lack of any δpCO2BIO decline, our reconstructions imply a potential role of iron fertilization in affecting surface-water pCO2 through nutrient utilization in the SAZ during HS1.
Instead of a dominant biological contribution, our systematic investigations reveal persistent influences of the solubility pump on surface-water pCO2 fluctuations in the SAZ under both modern and past conditions. Modern hydrographical data shows that the solubility pump causes surface-water pCO2 to fluctuate by ~40 ppm seasonally (Fig. 1). New and published proxy data demonstrates a solubility effect that can modulate surface-water pCO2 by ~20–50 ppm on millennial timescales during the last deglaciation (Figs. 2 and 3). The strong solubility pump influence is likely widespread in the SAZ, as shown by >20 ppm surface-water pCO2 increase attributable to solubility pump changes during the early deglaciation in a model simulation28 (Fig. 4). Notably, in this model simulation, solubility pump changes in the SAZ lead to strong CO2 outgassing and, together with other Southern Ocean processes, contribute to a full-scale atmospheric pCO2 increase during HS1 without invoking nutrient utilization efficiency changes related to iron fertilization28. Based on our analyses on modern data, proxy reconstructions, and modeling outputs, we suggest that a weakened solubility pump, driven by warming, contributed critically to the rising surface-water pCO2 in the SAZ and thereby maintaining this region as a CO2 source to the atmosphere during the last deglaciation.
In sum, while our δpCO2BIO reconstructions, at face value, indicate little biological pump contributions to deglacial SAZ surface-water pCO2 variations at various SAZ sites locally, these reconstructions imply iron-related nutrient utilization effects on the deglacial SAZ surface-water pCO2 variabilities when the influence of frontal shift is considered. Nevertheless, such an iron-related biological effect appears to be smaller than previously thought. Therefore, the common view that the iron-regulated SAZ biological pump changes substantially contributed to the deglacial atmospheric pCO2 rise8,9,10,11 may need to be re-evaluated. In comparison to the widely recognized biological pump effect, the potential effect of the solubility pump in the SAZ has been previously overlooked when explaining the past atmospheric pCO2 changes. Our work demonstrates that the solubility pump plays an indispensable role in modulating SAZ surface-water pCO2 under both modern and past conditions. We suggest future works on quantifying the effect of SAZ solubility pump on past and possibly future atmospheric pCO2 changes, which would have important implications for our mechanistic understanding of the global carbon cycle.
Methods
Trace element and boron isotope analyses
About 30 to 40 shells of planktic foraminifera G. bulloides from the 300–355 µm size fraction and >5 mg of G. bulloides shells from the 250–355 µm size fraction were picked for trace element and δ11B analyses, respectively. These samples were cleaned following the “Mg-cleaning” procedure50,51,52,53. Measurements of B/Ca and Mg/Ca, along with Al/Ca and Mn/Ca for monitoring contaminants, were performed on an iCAP Inductively coupled plasma-mass spectrometry (ICP-MS) at the Australian National University (ANU), following an established method51.
Separation of boron from sample matrices and measurement of δ11B on a multi-collector-ICP-MS (MC-ICP-MS) generally follows the method of Foster31 with some modifications. The cleaned foraminifera shells were dissolved in 0.5 M HNO3, and buffered by 2 M NH4Ac, instead of NaAc-HAc mixture, to pH of ~5.5. We changed the buffering solution to eliminate potential matrix contamination of Na on the δ11B measurement. The buffered solution was gravitationally dripped into micro-columns, loaded with 20 µL ion exchange resin (Amberlite IRA-743, 63–125 µm size fraction), which was precleaned by 0.5 M HNO3 and then boron-free deionized water. These micro-columns were tested by processing reference materials (boric acid solutions, NIST SRM 951 and ERM AE-121, without and with addition of CaCO3 matrix; standard carbonates, NEP-3B) and generating values consistent with published values. After rinsing the resin eight times using Milli-Q water, boron was eluted by five aliquots of 90 µL 0.5 M HNO3. A sixth aliquot was also added and collected to check for complete boron recovery. Total procedure blanks for each batch of samples were monitored, and were between ~20 and 100 pg.
δ11B was measured on a MC-ICP-MS (Neptune Plus) at ANU using a standard bracketing method similar to Foster29. Following Farmer et al.54, we measured boron blanks before every bracketing standard (NIST SRM 951) and sample. We also introduce water aerosol into the spray chamber through a second nebulizer after every standard/sample measurement in addition to the routine rinse, to flush out boron in order to minimize the memory effect of boron. An analytical block is as follows: flush-blank-standard-flush-blank-sample-flush-blank-standard. With the additional water flushing in between, measured blanks for 11B can be kept <1.8% (an average for blanks of all the samples, external standards, and bracketing standards during 5 sessions) of the bracketing standard with 30 ppb of boron. Prior to and during analyses of these samples, repeating measurements of standard materials (NIST SRM 951, ERM AE-121, NEP-3B, and NIST RM 8301f) yield results consistent with their published values (Supplementary Table 2). The external reproducibility is estimated by repeating measurements of standard ERM AE-121 at 30-ppb boron concentration along with the samples (2σ = 0.17‰, n = 12). The boron concentration of the standard is chosen to match the expected median concentration of samples. Three of the foraminiferal samples were divided into two subsamples and processed separately from the cleaning step, and standard deviations of these replicated samples range from 0.08 to 0.27‰ (Supplementary Table 3).
During the late LGM and HS1, G. bulloides δ11B we measured at site MD97-2106 agrees with previous measurements by MC-ICP-MS in the Pacific and Atlantic SAZ23 and the Subtropical Southwest Pacific55, but is on average ~1‰ lower than those from the same site measured on a Negative Thermal Ionization Mass Spectrometry (N-TIMS) by a previous study56 (Supplementary Fig. 3). We tentatively attribute such offsets to potential analytical biases between MC-ICP-MS and N-TIMS that, as shown by a previous study, range from 0.5 to 2.7‰ and appear to enlarge for samples with low B/Ca values54. Despite that Moy et al.56 show different deglacial δ11B and thus surface-water pCO2 magnitudes, deglacial δpCO2SOL and δpCO2BIO calculated using their data show similar patterns to those based on our new data (Supplementary Fig. 3).
Carbonate chemistry system calculation
G. bulloides δ11B is converted into δ11B of seawater borate (δ11Bborate) using the calibration from Raitzsch, et al.30: δ11Bborate = (δ11BG. bulloides + 3.58 ± 11.77)/(1.09 ± 0.65). To estimate pH, SST and surface seawater salinity (SSS) are required. SST is estimated from G. bulloides Mg/Ca using the calibration of Elderfield and Ganssen35. SSS is estimated from the global sea-level change following Foster31. To calculate seawater pCO2, seawater alkalinity is estimated from the modern seawater SSS-alkalinity relation20. We then use the CO2sys script57 to calculate seawater pCO2 and other carbonate chemistry parameters including DIC. The 2.3–97.7% uncertainties of seawater pCO2 are propagated by a 10,000-iteration Monte-Carlo method incorporating uncertainties from δ11B (2σ = 0.17‰), SST (2σ = 1 °C), and SSS (2σ = 0.5), and alkalinity which is sourced from SSS and the modern SSS-alkalinity relation20. Using a different way to estimate alkalinity (Supplementary Fig. 7) does not substantially affect our calculated seawater pCO2 and its decompositions. For published δ11B records, surface-water pCO2 is recalculated using the same method as this study to be consistent with our methodology. Final uncertainties shown in Figs. 2 and 3 also incorporate age uncertainties. For site MD97-2106, age uncertainty (1σ ranging from 0.2 to 0.8 ka) is derived from the Undatable script58, and for published records, a uniform age uncertainty (1σ) of 0.5 ka is assigned.
We partition the total in-situ surface-water pCO2 changes (δpCO2TOTAL) into two components: solubility-driven (δpCO2SOL) and biology-driven (δpCO2BIO) components. In the main text, δpCO2SOL is derived first, and the different between δpCO2TOTAL and δpCO2SOL is defined as δpCO2BIO. Here, we provide an alternative method to derive δpCO2BIO first and subsequently δpCO2SOL. Firstly, δpCO2TOTAL is calculate the same way as described in the main text. Secondly, we use DIC and alkalinity values (the same as those used for in-situ pCO2 calculations), but constant SST and SSS values at 18 ka to calculate new surface-water pCO2. It is important to note that DIC and alkalinity are used as intermediate parameters for calculations, and their values do not need to be accurately quantified to yield well-quantified new pCO2 values. This is because DIC and alkalinity are inherently linked given constraint from pH (see Yu et al.59 for detailed discussions). Thirdly, δpCO2BIO is calculated by changes in the newly calculated surface-water pCO2 relative to 18 ka. Fourthly, δpCO2SOL is defined by differencing δpCO2TOTAL and δpCO2BIO. As can be seen from Supplementary Fig. 8, the two methods yield consistent results, strengthening reliability of our calculation. The small differences between these two methods are due to the non-linear responses of seawater pCO2 to temperature and DIC changes.
For δpCO2SOL, it may be further partitioned into temperature- and salinity-driven components (δpCO2T and δpCO2S, respectively). We first calculate new surface-water pCO2 at each age by using constant DIC, alkalinity, and SSS at 18 ka, but varying SST. Then, δpCO2T is derived as changes of the newly calculated pCO2 relative to 18 ka. Afterward, δpCO2S is defined as the difference between δpCO2SOL and δpCO2T. As can be seen from Supplementary Fig. 5, salinity changes tend to counter temperature effect on pCO2, but δpCO2S are limited to within 10 ppm at all four SAZ sites studied.
Regarding model outputs28, we first calculate δpCO2TOTAL between 15 ka and 19 ka, simply by differencing in-situ surface-water pCO2 values at these times (pCO2in-situ,15ka and pCO2in-situ,19ka, respectively). We re-calculate surface-water pCO2 at 15 ka (pCO2recalc,15ka) using DIC and alkalinity at 15 ka but using SST and SSS at 19 ka. Similar to proxy data, δpCO2BIO and δpCO2SOL are calculated by: δpCO2BIO = pCO2recalc,15ka − pCO2in-situ,19ka and δpCO2SOL = pCO2TOTAL − δpCO2BIO.
For modern hydrological data29, monthly δpCO2TOTAL, δpCO2BIO, and δpCO2SOL represent deviations from annual mean values. δpCO2TOTAL, δpCO2BIO, and δpCO2SOL are calculated similarly to those for model results described above. For example, monthly δpCO2BIO is calculated using monthly alkalinity and DIC but annual mean SST and SSS, while monthly δpCO2SOL is calculated using annual mean alkalinity and DIC but with monthly SST and SSS (Fig. S2).
Foraminifera-bound nitrogen isotope analyses
Sample preparation and measurements of δ15N follow protocols in Ren et al.60. For each sample, >3 mg of G. bulloides shells (250–355 µm size fraction) were picked and crushed under a dissecting microscope. Foraminiferal samples were sonicated in an ultrasonic bath for 5 min with 2% polyphosphate solution, treated in bicarbonate-buffered dithionite−citric acid in an 80 °C water bath for 1 h, and added with basic potassium persulfate solution and autoclaved at 121 °C for 1 h. After every cleaning step, samples were rinsed with deionized water. Cleaned samples were dried overnight at 55 °C. Each sample was weighed (~1.5–3.5 mg) into a combusted glass vial and then dissolved in 3 M HCl to release organic N from the calcite shell. Persulfate oxidation reagent (POR, 0.3 g of 3-time-recrystallized basic potassium persulfate and 0.7 g of NaOH dissolved in 100 mL of deionized water) were added to the dissolved samples which were then autoclaved at 121 °C for 1 h to convert organic N to nitrate. The nitrate concentrations of all POR-oxidized samples were measured to determine N contents after autoclaving using the chemiluminescence method61. Average N content of the cleaned calcite samples is 3.07 mmol N per gram. Nitrate concentration of POR and its δ15N were constrained by two organic standards (US Geological Survey (USGS) 40, δ15N = −4.5‰ vs. air; and a laboratory standard, mixture of 6-aminocaproic acid and glycine, δ15N = 5.4‰ vs. air) processed along with samples. Nitrate concentration of POR is 0.2 µM, representing 1–3% of the total N in samples.
The denitrifier method was applied to transform dissolved nitrate and nitrite into nitrous oxide (N2O) gas using a naturally occurring denitrifying bacterial strain, Pseudomonas chlororaphis, which lacks an active form of the enzyme N2O reductase. After degassing of the bacteria for 3 h, 1.5 mL of the bacterial concentrate was added with 5 nmol of samples acidified to pH of 3-7. Two nitrate reference materials (International Atomic Energy Agency NO3 reference (IAEA-N3), δ15N = 4.7‰ vs. air; and USGS 34, δ15N = −1.8‰ vs. air) were processed along with samples to monitor the bacterial conversion and were later repeatedly measured between samples to check the stability of the mass spectrometry.
δ15N of foraminiferal samples, together with bacterial blanks and organic standards, were determined by gas chromatography and isotope ratio mass spectrometry using a modified SigBench and MAT253 plus62. Due to the small sample size and low N content within foraminifera, no duplicates were made for these samples. Our IAEA-N3 and USGS 34 standards yielded standard deviation (1σ) of 0.06 and 0.07‰, respectively. The standard deviation (1σ) of the organic standards analyzed with these samples is 0.15‰, agreeing with the long-term variability of in-house carbonate standards using homogenized coral samples (±0.25‰). As a result, we assume that the analytical error for the δ15NFB is 0.25‰ in our new record.
Data availability
All data generated in this study have been deposited in the Zenodo database under access code: https://doi.org/10.5281/zenodo.6970032. All data generated in this study are also provided in the Supplementary Information.
Code availability
Codes used to produce Figs. 1 and 4 are available from Y.D. upon request. Mode output data used to produce Fig. 4 is available from https://doi.org/10.4225/41/5af39aae7960f28.
References
Sigman, D. M., Hain, M. P. & Haug, G. H. The polar ocean and glacial cycles in atmospheric CO2 concentration. Nature 466, 47–55 (2010).
Sigman, D. M. & Boyle, E. A. Glacial/interglacial variations in atmospheric carbon dioxide. Nature 407, 859–869 (2000).
Yu, J. et al. Millennial and centennial CO2 release from the Southern Ocean during the last deglaciation. Nat. Geosci. 15, 293–299 (2022).
Yu, J. et al. Loss of carbon from the deep sea since the Last Glacial Maximum. Science 330, 1084–1087 (2010).
Talley, L. D. Descriptive Physical Oceanography: An Introduction (Academic Press, 2011).
Rintoul, S. R. The global influence of localized dynamics in the Southern Ocean. Nature 558, 209–218 (2018).
Sarmiento, J., Gruber, N., Brzezinsld, M. & Dunne, J. High-latitude controls of thermocline nutrients and low lattitude biological productivity. Nature 427, 53–56 (2004).
Brovkin, V., Ganopolski, A., Archer, D. & Rahmstorf, S. Lowering of glacial atmospheric CO2 in response to changes in oceanic circulation and marine biogeochemistry. Paleoceanography 22, PA4202 (2007).
Hain, M. P., Sigman, D. M. & Haug, G. H. Carbon dioxide effects of Antarctic stratification, North Atlantic Intermediate Water formation, and subantarctic nutrient drawdown during the last ice age: diagnosis and synthesis in a geochemical box model. Glob. Biogeochem. Cycles 24, GB4023 (2010).
Jaccard, S. L. et al. Two modes of change in southern ocean productivity over the past million years. Science 339, 1419–1423 (2013).
Martínez-García, A. et al. Iron fertilization of the Subantarctic Ocean during the last ice age. Science 343, 1347–1350 (2014).
Takahashi, T. Global sea-air CO2 flux based on climatological surface ocean pCO2, and seasonal biological and temperature effects. Deep Sea Res. Part II 49, 1601–1622 (2002).
Takahashi, T. et al. Climatological mean and decadal change in surface ocean pCO2, and net sea–air CO2 flux over the global oceans. Deep Sea Res. Part II 56, 554–577 (2009).
Volk, T. & Hoffert, M. I. The Carbon Cycle and Atmospheric CO2: Natural Variations Archean to Present Vol. 32, 99–110 (the American Geophysical Union, 1985).
Hain, M., Sigman, D. & Haug, G. in Treatise on Geochemistry 2nd ed., Vol. 8, 485–517 (Elsevier, 2014).
Takahashi, T., olafsson, J., Goddard, J. G., Chipman, D. W. & Sutherland, S. C. Seasonal variation of CO2 and nutrients in the high‐latitude surface oceans: a comaprative study. Glob. Biogeochem. Cycles 7, 843–878 (1993).
Khatiwala, S., Schmittner, A. & Muglia, J. Air-sea disequilibrium enhances ocean carbon storage during glacial periods. Sci. Adv. 5, eaaw4981 (2019).
Köhler, P., Fischer, H., Munhoven, G. & Zeebe, R. E. Quantitative interpretation of atmospheric carbon records over the last glacial termination. Glob. Biogeochem. Cycles 19, GB4020 (2005).
Heinze, C., Maier-Reimer, E. & Winn, K. Glacial pCO2 reduction by the world ocean: experiments with the hamburg carbon cycle model. Paleoceanography 6, 395–430 (1991).
Sarmiento, J. L. & Gruber, N. Ocean Biogeochemical Dynamics (Princeton University Press, 2006).
Wang, X. T. et al. Deep-sea coral evidence for lower Southern Ocean surface nitrate concentrations during the last ice age. Proc. Natl Acad. Sci. USA 114, 3352–3357 (2017).
Martinez-Boti, M. A. et al. Boron isotope evidence for oceanic carbon dioxide leakage during the last deglaciation. Nature 518, 219–222 (2015).
Shuttleworth, R. et al. Early deglacial CO2 release from the Sub-Antarctic Atlantic and Pacific oceans. Earth Planet. Sci. Lett. 554, 116649 (2021).
Martinez-Garcia, A. et al. Southern Ocean dust-climate coupling over the past four million years. Nature 476, 312–315 (2011).
Pedro, J. B. et al. The spatial extent and dynamics of the Antarctic Cold Reversal. Nat. Geosci. 9, 51–55 (2015).
Jouzel, J. et al. Orbital and millennial Antarctic climate variability over the past 800,000 years. Science 317, 793–796 (2007).
Benz, V., Esper, O., Gersonde, R., Lamy, F. & Tiedemann, R. Last Glacial Maximum sea surface temperature and sea-ice extent in the Pacific sector of the Southern Ocean. Quat. Sci. Rev. 146, 216–237 (2016).
Menviel, L. et al. Southern Hemisphere westerlies as a driver of the early deglacial atmospheric CO2 rise. Nat. Commun. 9, 2503 (2018).
Gregor, L. & Gruber, N. OceanSODA-ETHZ: a global gridded data set of the surface ocean carbonate system for seasonal to decadal studies of ocean acidification. Earth Syst. Sci. Data 13, 777–808 (2021).
Raitzsch, M. et al. Boron isotope-based seasonal paleo-pH reconstruction for the Southeast Atlantic – a multispecies approach using habitat preference of planktonic foraminifera. Earth Planet. Sci. Lett. 487, 138–150 (2018).
Foster, G. L. Seawater pH, pCO2 and [CO32−] variations in the Caribbean Sea over the last 130 kyr: A boron isotope and B/Ca study of planktic foraminifera. Earth Planet. Sci. Lett. 271, 254–266 (2008).
Bostock, H. C., Hayward, B. W., Neil, H. L., Sabaa, A. T. & Scott, G. H. Changes in the position of the Subtropical Front south of New Zealand since the last glacial period. Paleoceanography 30, 824–844 (2015).
Sikes, E. L. et al. Southern Ocean seasonal temperature and Subtropical Front movement on the South Tasman Rise in the late Quaternary. Paleoceanography 24, PA2201 (2009).
Belkin, I. M. & Gordon, A. L. Southern Ocean fronts from the Greenwich meridian to Tasmania. J. Geophys. Res. Oceans 101, 3675–3696 (1996).
Elderfield, H. & Ganssen, G. Past temperature and δ18O of surface ocean waters inferred from foraminiferal Mg/Ca ratios. Nature 405, 442–445 (2000).
Dai, Y., Yu, J. & Rafter, P. A. Deglacial ventilation changes in the deep Southwest Pacific. Paleoceanogr. Paleoclimatol 36, e2020PA004172 (2021).
Ren, H. et al. Glacial-to-interglacial changes in nitrate supply and consumption in the subarctic North Pacific from microfossil-bound N isotopes at two trophic levels. Paleoceanography 30, 1217–1232 (2015).
Ren, H., Sigman, D. M., Thunell, R. C. & Prokopenko, M. G. Nitrogen isotopic composition of planktonic foraminifera from the modern ocean and recent sediments. Limnol. Oceanogr. 57, 1011–1024 (2012).
Studer, A. S. et al. Increased nutrient supply to the Southern Ocean during the Holocene and its implications for the pre-industrial atmospheric CO2 rise. Nat. Geosci. 11, 756–760 (2018).
Studer, A. S. et al. Antarctic Zone nutrient conditions during the last two glacial cycles. Paleoceanography 30, 845–862 (2015).
Ai, X. E. et al. Southern Ocean upwelling, Earth’s obliquity, and glacial-interglacial atmospheric CO2 change. Science 370, 1348–1352 (2020).
Li, T. et al. Rapid shifts in circulation and biogeochemistry of the Southern Ocean during deglacial carbon cycle events. Sci. Adv. 6, eabb3807 (2020).
Monnin, E. et al. Evidence for substantial accumulation rate variability in Antarctica during the Holocene, through synchronization of CO2 in the Taylor Dome, Dome C and DML ice cores. Earth Planet. Sci. Lett. 224, 45–54 (2004).
Marcott, S. A. et al. Centennial-scale changes in the global carbon cycle during the last deglaciation. Nature 514, 616–619 (2014).
Lambert, F. et al. Dust-climate couplings over the past 800,000 years from the EPICA Dome C ice core. Nature 452, 616–619 (2008).
Sachs, J. P. & Anderson, R. F. Fidelity of alkenone paleotemperatures in southern Cape Basin sediment drifts. Paleoceanography 18, 1082 (2003).
Anderson, R. F. et al. Biological response to millennial variability of dust and nutrient supply in the Subantarctic South Atlantic Ocean. Philos. Trans. A Math. Phys. Eng. Sci. 372, 20130054 (2014).
Garcia, H. E. et al. World Ocean Atlas 2018. Vol. 4: dissolved inorganic nutrients (phosphate, nitrate and nitrate+nitrite, silicate). A. Mishonov Technical Editor, NOAA Atlas NESDIS 84, 35pp. https://www.ncei.noaa.gov/sites/default/files/2020-04/woa18_vol4.pdf (2019).
Locarnini, R. A. et al. World Ocean Atlas 2018, Volume 1: Temperature. A. Mishonov, Technical Editor. NOAA Atlas NESDIS 81, 52pp. https://www.ncei.noaa.gov/sites/default/files/2020-04/woa18_vol1.pdf (2019).
Barker, S., Greaves, M. & Elderfield, H. A study of cleaning procedures used for foraminiferal Mg/Ca paleothermometry. Geochem. Geophys. Geosyst. 4, 8407 (2003).
Boyle, E. & Keigwin, L. Comparison of Atlantic and Pacific paleochemical records for the last 215,000 years: Changes in deep ocean circulation and chemical inventories. Earth Planet. Sci. Lett. 76, 135–150 (1985).
Yu, J., Elderfield, H., Greaves, M. & Day, J. Preferential dissolution of benthic foraminiferal calcite during laboratory reductive cleaning. Geochem. Geophys. Geosyst. 8, Q06016 (2007).
Yu, J., Day, J., Greaves, M. & Elderfield, H. Determination of multiple element/calcium ratios in foraminiferal calcite by quadrupole ICP-MS. Geochem. Geophys. Geosyst. 6, Q08P01 (2005).
Farmer, J. R., Hönisch, B. & Uchikawa, J. Single laboratory comparison of MC-ICP-MS and N-TIMS boron isotope analyses in marine carbonates. Chem. Geol. 447, 173–182 (2016).
Shao, J. et al. Atmosphere‐ocean CO2 exchange across the last deglaciation from the boron isotope proxy. Paleoceanogr. Paleoclimatol. 34, 1650–1670 (2019).
Moy, A. D. et al. Varied contribution of the Southern Ocean to deglacial atmospheric CO2 rise. Nat. Geosci. 12, 1006–1011 (2019).
Lewis, E., Wallace, D. & Allison, L. J. Program Developed for CO2 System Calculations (Carbon Dioxide Information Analysis Center, managed by Lockheed Martin Energy Research Corporation for the US Department of Energy Tennessee, 1998).
Lougheed, B. C. & Obrochta, S. P. A rapid, deterministic age-depth modeling routine for geological sequences with inherent depth uncertainty. Paleoceanogr. Paleoclimatol. 34, 122–133 (2019).
Yu, J. et al. More efficient North Atlantic carbon pump during the Last Glacial Maximum. Nat. Commun. 10, 2170 (2019).
Ren, H. et al. Impact of glacial/interglacial sea level change on the ocean nitrogen cycle. Proc. Natl Acad. Sci. USA 114, E6759–E6766 (2017).
Braman, R. S. & Hendrix, S. A. Nanogram nitrite and nitrate determination in environmental and biological materials by vanadium (III) reduction with chemiluminescence detection. Anal. Chem. 61, 2715–2718 (1989).
Weigand, M. A., Foriel, J., Barnett, B., Oleynik, S. & Sigman, D. M. Updates to instrumentation and protocols for isotopic analysis of nitrate by the denitrifier method. Rapid Commun. Mass Spectrom. 30, 1365–1383 (2016).
Acknowledgements
We thank Brad Opdyke and Will Howard for help with arranging core materials, and Laurie Menviel for providing model outputs. This work is supported by NSFC42076056 and ARC Discovery Project DP190100894 to J.Y., and Ministry of Science and Technology, Taiwan (111-2636-M-002-020) to H.R.
Funding
Open access funding provided by Lund University.
Author information
Authors and Affiliations
Contributions
J.Y. designed the project. Y.D. derived the pCO2 decomposition method. Y.D. made boron isotope measurements. X.J. made new trace element measurements. J.Y. supervised geochemical measurements made by Y.D. and X.J. H.R. made nitrogen isotope measurements. Y.D. performed data analyses and visualization. Y.D. wrote the first draft of the manuscript with significant inputs from J.Y. All authors contributed to the interpretation of the data and refinement of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Amy King for her contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dai, Y., Yu, J., Ren, H. et al. Deglacial Subantarctic CO2 outgassing driven by a weakened solubility pump. Nat Commun 13, 5193 (2022). https://doi.org/10.1038/s41467-022-32895-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-32895-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
