
Abstract
Magnetic molecules known as molecular nanomagnets (MNMs) may be the key to ultra-high density data storage. Thus, novel strategies on how to design MNMs are desirable. Here, inspired by the hexagonal structure of the hardest intermetallic magnet SmCo5, we have synthesized a nanomagnetic molecule where the central lanthanide (Ln) ErIII is coordinated solely by three transition metal ions (TM) in a perfectly trigonal planar fashion. This intermetallic molecule [ErIII(ReICp2)3] (ErRe3) starts a family of molecular nanomagnets (MNM) with unsupported Ln-TM bonds and paves the way towards molecular intermetallics with strong direct magnetic exchange interactions—a promising route towards high-performance single-molecule magnets.
Similar content being viewed by others


Introduction
Rare-earth intermetallic magnets—samarium-cobalt (SmCo)1 and neodymium (NdFeB)2,3—are the strongest permanent magnets known to date with multiple applications in modern technology ranging from hard disk drives to electric vehicles and wind turbines4. Their commercial success results from the combination of the strong magnetic anisotropy of rare-earth ions and their direct magnetic coupling with transition metal ions. At the other far end of the current search for novel magnetic materials are magnetic molecules, called molecular nanomagnets5 (MNMs) or single-molecule magnets (SMMs). MNMs are molecule-sized objects with magnetic memory effects governed by quantum mechanics6,7,8,9,10. They are strongly believed to revolutionize magnetic information storage, but at the moment do not have any real-world applications, as their performance is limited by low blocking temperatures and quantum tunneling of magnetization (QTM)11. Current trends in the design of MNMs12,13 were ignited by the discovery of terbium double-decker [TbPc2]− (Pc = phthalocyanine dianion)14 and were fueled by the seminal perspective of Rinehart and Long15. Design principles focus on the precise control of the coordination sphere of a single lanthanide center to maximize the easy-axis magnetic anisotropy of the complex and to limit the spin-lattice relaxation rates16,17,18,19. While this approach led to several breakthroughs, including the observation of the magnetic hysteresis loop of molecular origin above the liquid nitrogen barrier for dysprosocenium6,7 and magnetic field control of the magnetization blocking barrier20, it appears to be reaching its limit as the proposed modifications were not as successful as expected21,22.
The original approach to MNMs was based on designing multinuclear systems with strong magnetic superexchange, where several paramagnetic centers are glued together by bridging ligands to form a high-spin molecule. If magnetic anisotropy is present in such a system, it shows MNM properties23,24. An MNM of this type stands behind the success story of the whole field as it was indeed started by the discovery of a dodecanuclear carboxylate-bridged cluster Mn12—a high-spin molecule (S = 10) comprising eight MnIII and four MnIV ions interacting magnetically via the bridging carboxylate ligands25,26,27. However, the indirect character of these magnetic interactions, based on a weak superexchange coupling mechanism, limits the performance of Mn12 to very low temperatures, below the boiling point of liquid helium. Attempts to adapt this particular strategy to dysprosocenium complexes suffer from a similar problem –weak superexchange interactions of the lanthanides with other metal centers through diamagnetic bridging ligands limit their influence on the slow magnetic relaxation28,29,30. Therefore, the route towards the high-performance MNMs based on superexchange interactions has been largely abandoned.
Overall, despite the huge progress in the field initiated by Mn12, its ultimate goal—the room temperature molecular nanomagnet (RT-MNM)—remains elusive. However, the aforementioned examples of the rare-earth intermetallics: SmCo51 and Nd2Fe14B2 provide clues for the possible direction in the design of RT-MNMs. Focusing the efforts on molecules comprising a rare-earth metal center coordinated solely by transition metal ions31,32,33,34, mimicking the first coordination sphere of the Sm center in the SmCo5 magnet (Fig. 1A–C) could enable the appearance of the direct exchange coupling between the highly anisotropic lanthanide central ion and the coordinated transition metals. This approach was recently predicted to efficiently suppress QTM in MNMs35 and demonstrated for the ultra-hard MNM (CpiPr5)2Dy2I3 where the direct exchange is operational36. Interestingly, even much weaker indirect interactions were reported to be quite efficient in this matter37.

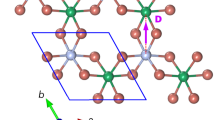
Graphical representation of the structural design transfer from the intermetallic magnet SmCo5 (A, B) to the molecular nanomagnet [ErIII(ReICp2)3] (C). The hexagonal planar SmCo6 coordination with unsupported coordination bonds between the center of the rare earth and six transition metals in SmCo5 (A, B) is mimicked by the trigonal planar coordination in ErRe3 (C). All three panels A–C are based on single-crystal structural models of SmCo51 and ErRe3. Sm-Co bonds in SmCo5: 2.888 Å; Er-Re bonds in ErRe3: 2.9004(5), 2.9124(5), 2.9172(5) Å. Panel D presents the synthesis of ErRe3 based on the work of Kempe et al.32. This approach utilizes the reaction of the weak Brønsted acid [Cp2ReH] with the strong Brønsted base btmsm− in [ErIII(btmsm)3] in benzene at room temperature (RT) resulting in (Me3Si)2CH2 elimination and the formation of [ErIII(ReICp2)3].
The concept of unsupported bonds between the lanthanide and the transition metal was first introduced and explored by Kempe et al.32,34. Later, it was proposed by Rinehart and Long15 as a possible strategy for molecular nanomagnets and put to use by Nippe et al. reporting MNMs with unsupported direct bonds between the dysprosium ion and 4d (Fe) or 4d (Ru) transition metal ions38. However, the magnetic memory effect (magnetic hysteresis) has not been observed, most probably due to the unfavorable ligand field geometry38 or the unfortunate choice of the rare-earth metal32. The introduction of p-block heavy metals directly into the coordination sphere of the lanthanide was also pursued resulting in interesting examples of MNMs39,40. Other systems involving direct bonds between actinides and d-metal elements were also reported, but slow magnetization dynamics were not studied41,42,43. Noteworthy, the concept of placing transition metal ions in the proximity of Ln-based MNMs employing suitable bridging ligands was pursued for tens of years with several examples showing enhancement of the slow relaxation of the magnetization due to the presence of the transition metal44,45,46. However, this strategy, while providing very good control of the connectivity of the target multi-metallic compounds, cannot warrant strong coupling between the two metal ions due to the weak overlap of the relevant orbitals of the lanthanide and the bridging moiety. Only the direct bonding of the transition metal to the lanthanide center could result in strong electronic and magnetic interactions.
Here, we apply the principles of easy-axis magnetic anisotropy design with the methodology of Kempe et al. that led previously to rare-earth metal complexes coordinated solely by transition metals32. We demonstrate an important step toward molecular intermetallic nanomagnets—a perfectly trigonal planar [ErIII(ReICp2)3] complex (ErRe3; Cp = cyclopentadienyl anion) with magnetic hysteresis up to 7.2 K (at 2.2 mT s−1 magnetic sweep rate) where ErIII is solely coordinated by three diamagnetic ReI ions.
Results and discussion
Synthetic strategy
ErRe3 is obtained following the literature procedure for [SmIII(ReICp2)3]32. The starting material [ErIII(btmsm)3] (obtained according to literature procedures47,48; btmsm = bis(trimethylsilyl)methyl anion) reacts with [Cp2ReH]49 in benzene (Fig. 1D). The btmsm− ligand coordinated to ErIII acts as a strong base capable of deprotonating [Cp2ReH]—a weak acid. Deprotonation of [Cp2ReH] leads to the elimination of the byproduct bis(trimethylsilyl)methane (“alkane elimination”34) and the formation of the anionic [Cp2ReI]- species that readily coordinate to ErIII resulting in the formation of a trigonal planar complex [ErIII(ReICp2)3].
Crystal structure
ErRe3 crystallizes slowly from benzene in the form of small yellow prism crystals which were characterized structurally by single-crystal X-ray diffraction (SCXRD; trigonal R-3; Supplementary Table 1). The SCXRD structural analysis confirmed the coordination of three ReI ions to the ErIII center with the formation of a nearly perfect trigonal planar neutral complex [ErIII(ReICp2)3] (Fig. 1C) with the Re-Er-Re angles very close to 120° (121.22(2), 119.15(2), and 119.62(2)°) and the Er atom lying only 0.013(1) Å above the Re3 plane. The cyclopentadienyl ligands are coordinated solely to ReI and arranged in a slightly tilted manner below and above the equatorial plane of the ErRe3 molecule (Fig. 1C). There are no other coordination bonds to ErIII. Other intramolecular contacts of ErIII involve Cp ligands with the shortest Er···C distances in the 2.829(8)–3.056(13) Å range—well beyond typical coordination bonds of lanthanide complexes. The Er-Re distances in ErRe3 (2.9004(5), 2.9124(5), and 2.9172(5) Å) are similar to those reported for [SmIII(ReICp2)3], [LaIII(ReICp2)3], and [LuIII(ReICp2)3]32 as well as other molecular compounds with unsupported rare earth—transition metal bonds34,38 and the intermetallic SmCo5 with Sm-Co distances of 2.888 Å1. The comparison of the Re-Cp distances in ErRe3 and the [Cp2ReH] starting material (SCXRD structural analysis of [Cp2ReH] was performed as part of this study Supplementary Table 1; CCDC 2027573) confirms the change of the valence state of the Re centers upon coordination to ErIII. The slightly unsymmetrical and tilted [Cp2ReH] with the average Re-Cpcentroid distance of 1.877(44) Å is similar to other compounds of this type50,51,52 and shows slight shortening of these contacts in ErRe3 to av. 1.864(8) Å (Supplementary Fig. 1). The [ErIII(ReICp2)3] molecules are stacked on top of each other in a hexagonal fashion along the c crystallographic direction, leading to the formation of channels filled with disordered benzene molecules (Supplementary Fig. 2). The shortest intermolecular distances between the ErIII centers can be found within the aforementioned stacks and amount to 7.6318(6) Å. There are six more nearest ErIII neighbors within the 10 Å radius with distances larger than 9 Å (Supplementary Fig. 3). This ensures magnetic isolation of the individual [ErIII(ReICp2)3] molecules that is sufficient for the observation of slow magnetization dynamics. Before attempting the magnetic characterization ErRe3 was subjected to a rigorous purity verification by performing a powder X-ray diffraction (PXRD) experiment for a sample loaded into a 0.3 mm glass capillary immersed in benzene and sealed using high vacuum grease (similar conditions were applied during the magnetic measurements discussed below). The experimental PXRD pattern shows very narrow peaks and matches almost perfectly the simulated one using 270 K SCXRD data (Supplementary Fig. 4).
Magnetic properties and calculations
The unusual coordination environment of ErIII in ErRe3 may cast doubts on the nature of its electronic ground state. However, magnetic measurements for ErRe3 confirm the valence states of the central ErIII ion and the diamagnetic ReI donor atoms. The χT (χ—molar magnetic susceptibility) value of 11.3 cm3 K mol−1 at 260 K is close to 11.48 cm3 K mol−1 expected for a single ErIII 4I15/2 ground multiplet (Supplementary Fig. 5). A similar agreement between the experiment and the expected χT was reported for [SmIII(ReICp2)3] (6H5∕2 multiplet)32. This is further confirmed by a very good agreement of the experimental χT and the ab initio calculations (red solid line in Supplementary Fig. 5 and supplemental information) using Molcas53,54. The χT(T) dependence shows an abrupt increase below 18 K, which can be ascribed to ferromagnetic intermolecular interactions and blocking of magnetization.
The M(H) (M—molar magnetization, H—magnetic field) measurements (magnetic field sweep rate of 2.2 mT s−1) revealed the presence of waist-restricted (pinched) magnetic hysteresis loops up to 7.2 K (Fig. 2A), originating from the slow relaxation of the magnetization of the trigonal ErRe3. This temperature matches ErIII sandwich and metallocene complexes55. Furthermore, the hysteresis loop in ErRe3 opens in the 0.05–1.5 T range at 1.8 K and is the widest among trigonal ErIII MNMs.

Magnetization (M) hysteresis loops were recorded with the 2.2 mT s−1 magnetic field (μ0H) sweep rate in the −2 to 2 T range (A). The energy of the calculated Kramers Doublets (KDs) arising from the splitting of the 4I15/2 multiplet of ErIII in ErRe3 with the most probable relaxation pathway represented by red arrows (B). In-phase (χ‘) and out-of-phase (χ“) AC molar magnetic susceptibility recorded at zero magnetic field in the 2.4–27 K temperature range, demonstrating the slow magnetization dynamics of ErRe3, solid lines are the best fits to the generalized Debye model with parameters presented in Supplementary Table 2 (C). Temperature dependence of the magnetization relaxation rate τ−1(T) at zero (D) and 0.15 mT (E) magnetic field obtained from the generalized Debye fitting of the respective AC molar magnetic susceptibility measurements (circles—experimental points, red line—best fits using Eq. 1 (D) and Eq. 2 (E), blue dashed line—Orbach relaxation, green dashed line—Raman relaxation, magenta dashed line—QTM relaxation). Standard deviations in A, C, D, and E are smaller than the size of the data points.
The slow magnetization dynamics were studied and confirmed by the alternating current (AC) magnetic susceptibility measurements performed in the 1–10000 Hz frequency range up to 27 K in the absence of the external magnetic field (Fig. 2C and Supplementary Table 2). However, the resulting temperature dependence of the magnetic relaxation rate τ−1(T) (Fig. 2D) contains contributions from three processes: quantum tunneling of magnetization (QTM), Raman and Orbach described by five parameters included in Eq. 1:
hence, all relevant parameters: A, C, n, τ0, and, most importantly, the effective energy barrier for the magnetization reversal Ueff arising from the Orbach relaxation, can be extracted only with limited confidence from AC data collected at zero DC field (best-fit parameters A = 179(4) s−1, C = 0.137(9) K−n s−1, n = 3.60(2), τ0 = 8.3(8)·10−10 s and Ueff = 285(3) K with R2 = 0.99994; Fig. 2D). To mitigate this issue, the AC magnetic studies were repeated under an applied DC magnetic field of 0.15 T (Supplementary Fig. 6 and Supplementary Table 3). The presence of a small applied magnetic field quenches the QTM with only a negligible direct process, which was accurately determined from the magnetic field dependence of the relaxation time (Supplementary Fig. 7 and Supplementary Table 4). Thus, the temperature dependence of the relaxation rate under HDC = 0.15 T could be reliably fitted including only the Raman and Orbach relaxation mechanisms using Eq. 2:
where τ−1 is the magnetic relaxation rate, C and n parameters describe the Raman-like process, and τ0 and Ueff are related to the Orbach process. The satisfactory fit could be obtained with C = 0.0035(4) K−n s−1, n = 4.74(3), τ0 = 3.0(5)·10−10 s, and Ueff = 314(5) K with R2 = 0.99987 (Fig. 2E). The relation between Ueff and τ0 falls perfectly in the statistically expected range for prolate Ln MNMs as analyzed by Gaita-Ariño et al. in their work covering 1405 examples of SIMs12. The observed low Raman exponent of 4.74 is most probably caused by spin-lattice coupling with optical phonons as summarized and discussed by Gu and Wu56.
The obtained effective energy barrier for the magnetization reversal Ueff = 314(5) K is significantly smaller than expected from the theoretical calculations (CASSCF-RASSI; OpenMolcas 19.1153; for details see Supplementary Information including Supplementary Tables 5 and 6 with the information on basis sets, spin-orbit energies and g-tensors of the lowest Kramers Doublets, respectively), predicting the fastest relaxation between the 5± Kramers Doublet lying 576 K above the highly axial ground state (Fig. 2B and Supplementary Table 6). The value 314(5) K is therefore underestimated. Additional fittings of the Raman and Orbach processes with fixed Ueff values corresponding to the calculated energies of the lower KDs were performed: 2± of 224 K, 3± of 363 K, and 4± of 475 K. The best fits (Supplementary Fig. 8A–C) are characterized by the following R2 values: 0.99702 for Ueff 2± = 224 K, 0.99960 for Ueff 3± = 363 K, and 0.99808 for Ueff 4± = 475 K. The best agreement is observed for Ueff 3± = 363 K, which corresponds to the energy of the third KD. This is also closest to the value obtained from the free fitting of the relaxation processes presented in Fig. 2E. An attempt to describe the magnetic relaxation of ErRe3 using only the Raman-like process leads to a very poor fit as depicted in Supplementary Fig. 8D. Thus we conclude, that the observed Orbach relaxation for ErRe3 proceeds predominantly through the third Kramers doublet, characterized by the estimated energy barrier Ueff approaching 363 K. This value is three times higher than that observed for the best trigonal48,57 or low-coordinate58,59 ErIII molecules and is comparable to the ErIII sandwich and metallocene complexes37,55,60,61,62,63,64. Interestingly, ErRe3 shows significantly slower relaxation as compared to [Er(N(SiMe3)2)3] characterized by Ueff = 122 K57 and surpasses the theoretical limit of 306 K (212.6 cm−1) for any trigonal planar molecule based on the theoretical analysis of a hypothetical ErIII(NH2)3 compound65. ErRe3 is a clear demonstration that direct bonding of transition metals to lanthanides might be one of the most promising directions towards room temperature MNMs, especially if paramagnetic donor atoms could be used.
Our findings regarding the magnetic dynamics of ErRe3 are consistent with the recent data analysis by Gaita-Ariño et al12. The analysis shows relatively high magnetic hysteresis temperatures for prolate systems such as equatorial ErIII SIMs (coordinated by rigid ligands) accompanied by the maxima of the out-of-phase magnetic susceptibility (χ“) at relatively low temperatures. ErRe3 is a perfect demonstration of this trend with the Thyst of 7.2 K and Ueff = 314 K.
In conclusion, the reported triangular molecule [ErIII(ReICp2)3] constitutes a molecular nanomagnet that mimics structurally the hardest commercial rare-earth magnet known SmCo5 and creates a connection between two incompatible fields: the rare-earth intermetallics and molecule-based compounds. ErRe3 is a rare example of a lanthanide single-molecule magnet coordinated solely by transition metals which enables the formulation of a promising strategy towards molecular intermetallic nanomagnets, where the lanthanide—the source of strong magnetic anisotropy—is directly coupled to the spins of softer transition metal ions playing the role of ligands.
Methods
General considerations
All manipulations were performed under an argon gas atmosphere inside the Inert PureLab HE glovebox (O2 < 0.1 ppm and H2O < 0.5 ppm). All solvents used in this study (except benzene) were of HPLC grade and were additionally dried under argon using the Inert PureSolv EN7 solvent purification system and stored over 3 Å molecular sieves for at least 24 h before use. Anhydrous benzene was purchased from commercial sources (Merck) and degassed by performing three freeze-pump-thaw cycles before use. Anhydrous ReCl5 (at least 99.9%) was purchased from Merck and used as received for the preparation of [Cp2ReH] according to the modified literature procedure49 described below. The obtained [Cp2ReH] was characterized by means of SCXRD and the purity was confirmed by performing PXRD measurements. [ErIII(btmsm)3] was also prepared following the modified literature procedures47,48—see below for the description of the preparation procedure. The purity of the obtained product was checked by PXRD. See Supplementary Information file for measurement details and ab initio calculations. Warning! Benzene is recognized as carcinogenic to humans and should be handled with caution using a well-ventilated hood or glovebox.
Preparation of [ErIII(btmsm)3]
[ErIII(btmsm)3] was prepared following the modified literature procedures47,48. Inside the Ar-filled glovebox bis(trimethylsilyl)methyllithium66 (Libtmsm; 0.91 g, 5.47 mmol) was slowly added in small portions to a stirred clear pink solution of [ErIII(BHT)3]48 (1.13 g, 1.37 mmol; BHT = 2,6-Di-tert-butyl-4-methylphenolate) in 17 ml of pentane. After the addition is finished the reaction mixture is still slightly turbid due to the undissolved Libtmsm, but becomes clear after ca. 20 min of stirring and then turbid again after ca. 1 h of stirring due to the precipitation of the byproduct LiBHT. The stirring is continued for 5 h at room temperature and then the thick suspension is vacuum filtered through a P4 fritted glass funnel in order to remove LiBHT. Please note that the funnel needs to be thoroughly clean, rinsed with THF, and dried at 140 °C prior to use. The product might start to crystallize in the filtrate during vacuum filtration. The pink filtrate is transferred into a tightly sealed glass vial and kept at −40 °C for 5 h for crystallization. The obtained pink needle crystals are filtered by vacuum filtration using a PTFE membrane, collected, and stored in the freezer. Yield: 350 mg (39%). Additional crop of the product (100 mg; 10%) is obtained by washing the LiBHT byproduct collected after the reaction with ca. 10 ml of pentane and keeping the washings at −40 °C for 24 h. The purity of both crops was confirmed by collecting PXRD patterns, which were identical to that simulated from the scXRD data.
Preparation of [Cp2ReH]
[Cp2ReH] was synthesized according to the modified literature procedure49. Inside the Ar-filled glovebox, a 100 mL 29/32 Schlenk flask was charged with a large magnetic stirrer, sodium cyclopentadienide (NaCp; 3.55 g, 0.04 mol), and 25 mL THF, and was cooled to −40 °C. The cold mixture was set on a fast stirring and ReCl5 (2.18 g, 0.006 mol) was added in small portions. The resulting mixture was left to heat to room temperature, then NaBH4 (0.57 g, 0.015 mol) was added, the flask was sealed with a glass stopper, and was removed from the glovebox. Using standard Schlenk line techniques, the mixture was refluxed under Ar for 2.5 h, then it was cooled to room temperature and the solvent was removed under vacuum. The resulting brown solid was dried under vacuum for 30 min at 70 °C, then the reaction flask was fitted with a 29/32 cold finger filled with dry ice/acetone mixture and the brown solid was sublimed under vacuum. The temperature during the sublimation was gradually increased up to 180 °C under vacuum (p ≈ 10−2 mbar) and continued until the product was deposited on the cold finger surface as a brownish-yellow powder, leaving most of the brown residue on the bottom of the reaction flask. The reaction flask was moved inside the glovebox, the yellow product was moved to another Schlenk flask and the vacuum sublimation was repeated at 120 °C (this time with ice-cooling of the cold finger) yielding Cp2ReH as yellow needles. Yield: 0.50 g (26%). The purity of the product was confirmed by collecting the PXRD pattern, which was identical to that simulated from the scXRD data.
Preparation of [ErIII(ReICp2)3]·0.5C6H6 (ErRe3)
All manipulations were performed in an argon-filled glovebox (Inert PureLab HE). [Cp2ReH] (242 mg, 0.76 mmol) was dissolved in anhydrous benzene (3.5 ml) and added to the benzene solution (2.5 ml) of [ErIII(btmsm)3] (160 mg, 0.25 mmol). The resulting orange solution was stirred using a glass rod for 5 min and then stored in a 20 ml scintillation vial that was left open for 5 h inside the glovebox. After this time yellow-orange crystals appeared which were collected by vacuum filtration using a 1-μm Teflon membrane (Whatman). Yield: 70 mg (0.061 mmol; 24%). The purity of the compound was checked by powder X-ray diffraction, with the experimental pattern (Supplementary Fig. 4) matching perfectly the simulated one from the SCXRD structural model obtained at 270 K (CCDC 2065531). IR, neat (cm−1): 796 s, 808 s, 816 s, 832 m, 850 m, 987 s, 1000 s, 1059 m, 1087 s, 1095vs, 1154vw, 1188vw, 1201vw, 1255 m, 1273 m, 1342 m, 1358 m, 1394 s, 1419 s, 1462 m, 1479 m, 1558 s, 1657s, 2001vw, 2034vw, 2854 s, 2929vs, 3036 m, 3063 s, 3089 s, 3550b (Supplementary Fig. 9).
Data availability
The data that support the findings of this study can be found in the manuscript or the accompanying supplementary information files. Source data for all Figures are provided with this paper as a single Excel file and are also available from the corresponding author: Dawid Pinkowicz (dawid.pinkowicz@uj.edu.pl). Crystallographic data generated during this study were deposited with the Cambridge Structural Database and can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif (ErRe3 at 100 K: CCDC 2065530, ErRe3 at 270 K: CCDC 2065531 and [Cp2ReH] at 100 K: CCDC 2027573). Source data are provided with this paper.
References
Buschow, K. H. J. & Van Der Goot, A. S. Intermetallic compounds in the system samarium-cobalt. J. Less Common Met. 14, 323–328 (1968).
Herbst, J. F., Croat, J. J., Pinkerton, F. E. & Yelon, W. B. Relationships between crystal structure and magnetic properties in Nd2Fe14B. Phys. Rev. B 29, 4176–4178 (1984).
Croat, J. J., Herbst, J. F., Lee, R. W. & Pinkerton, F. E. High‐energy product Nd‐Fe‐B permanent magnets. Appl. Phys. Lett. 44, 148–149 (1984).
Gutfleisch, O. et al. Magnetic materials and devices for the 21st century: stronger, lighter, and more energy efficient. Adv. Mater. 23, 821–842 (2011).
Gatteschi D., Sessoli R. & Villain J. Molecular Nanomagnets (Oxford Univ. Press, 2006).
Goodwin, C. A. P., Ortu, F., Reta, D., Chilton, N. F. & Mills, D. P. Molecular magnetic hysteresis at 60 kelvin in dysprosocenium. Nature 548, 439–442 (2017).
Guo, F.-S. et al. Magnetic hysteresis up to 80 kelvin in a dysprosium metallocene single-molecule magnet. Science 362, 1400–1403 (2018).
Gould, C. A. et al. Synthesis and magnetism of neutral, linear metallocene complexes of terbium(II) and dysprosium(II). J. Am. Chem. Soc. 141, 12967–12973 (2019).
Guo, F.-S. et al. A dysprosium metallocene single-molecule magnet functioning at the axial limit. Angew. Chem. Int. Ed. 56, 11445–11449 (2017).
Randall McClain, K. et al. High-temperature magnetic blocking and magneto-structural correlations in a series of dysprosium(iii) metallocenium single-molecule magnets. Chem. Sci. 9, 8492–8503 (2018).
Wu, S.-G. et al. Rectangle-like hysteresis in a dysprosium metallacrown magnet with linear F-Dy-F anisotropic moiety. Preprint at arXiv:2105.09586 (2021).
Duan Y. et al. Data mining, dashboards and statistics: a powerful framework for the chemical design of molecular nanomagnets. Preprint at arXiv:2103.03199 (2021).
Liu, J.-L., Chen, Y.-C. & Tong, M.-L. Symmetry strategies for high performance lanthanide-based single-molecule magnets. Chem. Soc. Rev. 47, 2431–2453 (2018).
Ishikawa, N., Sugita, M., Ishikawa, T., Koshihara, S.-Y. & Kaizu, Y. Lanthanide double-decker complexes functioning as magnets at the single-molecular level. J. Am. Chem. Soc. 125, 8694–8695 (2003).
Rinehart, J. D. & Long, J. R. Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chem. Sci. 2, 2078–2085 (2011).
Reta, D., Kragskow, J. G. C. & Chilton, N. F. Ab initio prediction of high-temperature magnetic relaxation rates in single-molecule magnets. J. Am. Chem. Soc. 143, 5943–5950 (2021).
Briganti, M. et al. A complete ab initio view of Orbach and Raman spin–lattice relaxation in a dysprosium coordination compound. J. Am. Chem. Soc. 143, 13633–13645 (2021).
Jackson, C. E., Moseley, I. P., Martinez, R., Sung, S. & Zadrozny, J. M. A reaction-coordinate perspective of magnetic relaxation. Chem. Soc. Rev. 50, 6684–6699 (2021).
Harriman, K. L. M. & Murugesu, M. An organolanthanide building block approach to single-molecule magnets. Acc. Chem. Res. 49, 1158–1167 (2016).
Wu, S.-G. et al. Field-induced oscillation of magnetization blocking barrier in a holmium metallacrown single-molecule magnet. Chem 7, 982–992 (2021).
Evans, P., Reta, D., Whitehead, G. F. S., Chilton, N. F. & Mills, D. P. Bis-monophospholyl dysprosium cation showing magnetic hysteresis at 48 K. J. Am. Chem. Soc. 141, 19935–19940 (2019).
Durrant, J. P., Tang, J., Mansikkamäki, A. & Layfield, R. A. Enhanced single-molecule magnetism in dysprosium complexes of a pristine cyclobutadienyl ligand. Chem. Commun. 56, 4708–4711 (2020).
Milios, C. J. et al. A record anisotropy barrier for a single-molecule magnet. J. Am. Chem. Soc. 129, 2754–2755 (2007).
Qian, K. et al. A single-molecule magnet based on heptacyanomolybdate with the highest energy barrier for a cyanide compound. J. Am. Chem. Soc. 135, 13302–13305 (2013).
Lis, T. Preparation, structure, and magnetic properties of a dodecanuclear mixed-valence manganese carboxylate. Acta Crystallogr. Sect. B 36, 2042–2046 (1980).
Sessoli, R. et al. High-spin molecules: [Mn12O12(O2CR)16(H2O)4]. J. Am. Chem. Soc. 115, 1804–1816 (1993).
Sessoli, R., Gatteschi, D., Caneschi, A. & Novak, M. A. Magnetic bistability in a metal-ion cluster. Nature 365, 141–143 (1993).
He, M., Guo, F.-S., Tang, J., Mansikkamäki, A. & Layfield, R. A. Fulvalene as a platform for the synthesis of a dimetallic dysprosocenium single-molecule magnet. Chem. Sci. 11, 5745–5752 (2020).
He, M., Guo, F.-S., Tang, J., Mansikkamäki, A. & Layfield, R. A. Synthesis and single-molecule magnet properties of a trimetallic dysprosium metallocene cation. Chem. Commun. 57, 6396–6399 (2021).
Evans, P. et al. A double-dysprosocenium single-molecule magnet bound together with neutral ligands. Chem. Commun. 56, 5677–5680 (2020).
Liu, J. et al. On-surface preparation of coordinated lanthanide-transition-metal clusters. Nat. Commun. 12, 1619 (2021).
Butovskii, M. V. et al. Molecules containing rare-earth atoms solely bonded by transition metals. Nat. Chem. 2, 741–744 (2010).
Shi, K. et al. Heterometallic clusters with multiple rare earth metal–transition metal bonding. J. Am. Chem. Soc. 143, 5998–6005 (2021).
Butovskii, M. V., Tok, O. L., Wagner, F. R. & Kempe, R. Bismetallocenes: lanthanoid–transition-metal bonds through alkane elimination. Angew. Chem. Int. Ed. 47, 6469–6472 (2008).
Swain, A., Sen, A. & Rajaraman, G. Are lanthanide-transition metal direct bonds a route to achieving new generation {3d–4f} SMMs? Dalton Trans. 50, 16099–16109 (2021).
Gould, C. A. et al. Ultrahard magnetism from mixed-valence dilanthanide complexes with metal–metal bonding. Science 375, 198–202 (2022).
Le Roy, J. J., Ungur, L., Korobkov, I., Chibotaru, L. F. & Murugesu, M. Coupling strategies to enhance single-molecule magnet properties of erbium–cyclooctatetraenyl complexes. J. Am. Chem. Soc. 136, 8003–8010 (2014).
Burns, C. P. et al. Structure and magnetization dynamics of Dy−Fe and Dy−Ru bonded complexes. Angew. Chem. Int. Ed. 57, 8144–8148 (2018).
Münzfeld, L. et al. Introduction of plumbole to f-element chemistry. Chem. Sci. 13, 945–954 (2022).
Zhang, P., Benner, F., Chilton, N. F. & Demir, S. Organometallic lanthanide bismuth cluster single-molecule magnets. Chem 8, 717–730 (2021).
Gardner, B. M., McMaster, J., Lewis, W. & Liddle, S. T. Synthesis and structure of [{N(CH2CH2NSiMe3)3}URe(η5-C5H5)2]: a heterobimetallic complex with an unsupported uranium–rhenium bond. Chem. Commun. 2851–2853 (2009).
Gardner, B. M. et al. An unsupported uranium–rhenium complex prepared by alkane elimination. Chem.—A Eur. J. 17, 6909–6912 (2011).
Ostrowski, J. P. A., Wooles, A. J. & Liddle, S. T. Synthesis and characterisation of molecular polarised-covalent thorium-rhenium and -ruthenium bonds. Inorganics 9, 30 (2021).
Darago, L. E. et al. Strong ferromagnetic exchange coupling and single-molecule magnetism in MoS43–-bridged dilanthanide complexes. J. Am. Chem. Soc. 143, 8465–8475 (2021).
Dey, A., Bag, P., Kalita, P. & Chandrasekhar, V. Heterometallic CuII–LnIII complexes: single molecule magnets and magnetic refrigerants. Coord. Chem. Rev. 432, 213707 (2021).
Andruh, M. Heterotrimetallic complexes in molecular magnetism. Chem. Commun. 54, 3559–3577 (2018).
Avent, A. G. et al. Synthetic and structural experiments on yttrium, cerium and magnesium trimethylsilylmethyls and their reaction products with nitriles; with a note on two cerium β-diketiminates. Dalton Trans. 1567–1577 (2004).
Zhang, H. et al. Low coordinated mononuclear erbium(iii) single-molecule magnets with C3v symmetry: a method for altering single-molecule magnet properties by incorporating hard and soft donors. Dalton Trans. 47, 302–305 (2018).
Green, M. L. H., Pratt, L. & Wilkinson, G. 795. Biscyclopentadienylrhenium hydride. J. Chem. Soc. (Resumed), 3916–3922 (1958).
Apostolidis, C., Kanellakopulos, B., Maier, R., Rebizant, J. & Ziegler, M. L. Bis(cyclopentadienyl)rhenium(III)chlorid: Kristallstruktur, Eigenschaften und Reaktivität; Darstellung und Charakterisierung von Cp3Re und Cp2ReH. J. Organomet. Chem. 409, 243–254 (1991).
Paciello, R. A., Kiprof, P., Herdtweck, E. & Herrmann, W. A. Synthesis and structural characterization of a mixed-ring rhenocene hydride complex, (.eta.5-cyclopentadienyl) (.eta.5-pentamethylcyclopentadienyl)hydridorhenium. Inorg. Chem. 28, 2890–2893 (1989).
Bandy, J. A. et al. Decamethylrhenocene, (.eta.5-C5Me5)2Re. J. Am. Chem. Soc. 110, 5039–5050 (1988).
Aquilante, F. et al. Modern quantum chemistry with [Open]Molcas. J. Chem. Phys. 152, 214117 (2020).
Chibotaru, L. F. & Ungur, L. Ab initio calculation of anisotropic magnetic properties of complexes. I. Unique definition of pseudospin Hamiltonians and their derivation. J. Chem. Phys. 137, 064112 (2012).
Chen, S.-M. et al. A soft phosphorus atom to “harden” an erbium(iii) single-ion magnet. Chem. Sci. 9, 7540–7545 (2018).
Gu, L. & Wu, R. Origin of the anomalously low Raman exponents in single molecule magnets. Phys. Rev. B 103, 014401 (2021).
Zhang, P. et al. Equatorially coordinated lanthanide single ion magnets. J. Am. Chem. Soc. 136, 4484–4487 (2014).
Brzozowska, M., Handzlik, G., Kurpiewska, K., Zychowicz, M. & Pinkowicz, D. Pseudo-tetrahedral vs. pseudo-octahedral ErIII single molecule magnets and the disruptive role of coordinated TEMPO radical. Inorg. Chem. Front. 8, 2817–2828 (2021).
Brown, A. J., Pinkowicz, D., Saber, M. R. & Dunbar, K. R. A trigonal-pyramidal erbium(III) single-molecule magnet. Angew. Chem. Int. Ed. 54, 5864–5868 (2015).
Münzfeld, L. et al. Synthesis, structures and magnetic properties of [(η9-C9H9)Ln(η8-C8H8)] super sandwich complexes. Nat. Commun. 10, 3135 (2019).
Jiang, S.-D., Wang, B.-W., Sun, H.-L., Wang, Z.-M. & Gao, S. An organometallic single-ion magnet. J. Am. Chem. Soc. 133, 4730–4733 (2011).
Meihaus, K. R. & Long, J. R. Magnetic blocking at 10 K and a dipolar-mediated avalanche in salts of the bis(η8-cyclooctatetraenide) complex [Er(COT)2]−. J. Am. Chem. Soc. 135, 17952–17957 (2013).
Le Roy, J. J., Korobkov, I. & Murugesu, M. A sandwich complex with axial symmetry for harnessing the anisotropy in a prolate erbium(iii) ion. Chem. Commun. 50, 1602–1604 (2014).
Meng, Y.-S. et al. Boratabenzene)(cyclooctatetraenyl) lanthanide complexes: a new type of organometallic single-ion magnet. Inorg. Chem. Front. 3, 828–835 (2016).
Lu, F., Ding, M.-M., Li, J.-X., Wang, B.-L. & Zhang, Y.-Q. Why lanthanide ErIII SIMs cannot possess huge energy barriers: a theoretical investigation. Dalton Trans. 49, 14576–14583 (2020).
Beswick, C. L. & Marks, T. J. Significant zirconium−alkyl group effects on ion pair formation thermodynamics and structural reorganization dynamics in zirconocenium alkyls. Organometallics 18, 2410–2412 (1999).
Acknowledgements
This work was financed by the Polish National Science Centre within the Sonata Bis 6 project no. 2016/22/E/ST5/00055 (D.P.). The research was partly carried out with the equipment purchased thanks to the financial support of the European Regional Development Fund in the framework of the Smart Growth Operational Program 2014-2020, contract no. POIR.04.02.00-00-D001/20-00 (D.P.). The open-access publication of this article was funded by the program “Excellence Initiative – Research University” at the Jagiellonian University in Krakow. The authors acknowledge Alejandro Gaita-Ariño for pointing out and formulating an important observation regarding the slow magnetic dynamics of the reported compound ErRe3 in comparison to known Ln-SIMs.
Author information
Authors and Affiliations
Contributions
M.M. prepared the starting material [Cp2ReH], participated in the preparation of the target compound, performed and analyzed the magnetic measurements, and wrote the relevant fragment of the manuscript; M.B. prepared the starting material [ErIII(btmsm)], participated in the preparation of the target compound, analyzed its structure and wrote a relevant fragment of the manuscript; S.B. participated in the AC magnetic measurements and their analysis. V.V. performed and analyzed the ab initio calculations and wrote the relevant fragment of the manuscript. D.P. acquired funding, designed the experiments, performed the synthesis of the target compound, coordinated and supervised the research, and wrote the manuscript. All authors reviewed and agreed to the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Thomas Albrecht-Schoenzart, Alejandro Gaita-Ariño and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Magott, M., Brzozowska, M., Baran, S. et al. An intermetallic molecular nanomagnet with the lanthanide coordinated only by transition metals. Nat Commun 13, 2014 (2022). https://doi.org/10.1038/s41467-022-29624-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-29624-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
