
Abstract
The radical cascade reaction is considered as one of the most powerful methods to build molecular complexity. However, highly stereoselective intermolecular radical cascade reactions that can produce complex cyclic compounds bearing multiple stereocenters via visible-light-induced photocatalysis have been challenging yet desirable. Herein we report a facile and efficient synthesis of multi-substituted trans-fused hexahydrocarbazoles via a stereoselective intermolecular radical cascade reaction of readily available tryptophans and acrylamides enabled by visible-light-induced photoredox catalysis. The trans-fused hexahydrocarbazoles with up to five stereocenters including two quaternary ones can be accessed in up to 82% yield, >20/1 diastereoselectivity, and 96% ee. Interestingly, the tetrahydrocarbazoles are favorably formed when the reaction is performed under air. Moreover, by simply switching the starting material from tryptophans to ɤ-alkenyl substituted α-amino acids, this protocol can be further applied to the stereoselective syntheses of 1,3,5-trisubstituted cyclohexanes which are otherwise challenging to access. Preliminary mechanistic studies suggest that the reaction goes through radical addition cascade and radical-polar crossover processes.
Similar content being viewed by others

Introduction
In the pharmaceutical industry, the growing demand for complex three-dimensional cyclic compounds urges the synthetic chemists to develop novel and efficient strategies to construct these motifs1,2. Radical cascade reactions are considered as one of the most powerful methods to meet this demand3,4,5,6. This protocol, which allows the rapid syntheses of valuable complex cyclic molecules, has been widely applied in the total syntheses of natural products7,8,9. However, many radical transformations involving alkyl radical species usually requires the use of organic halides as substrates, the addition of toxic stannanes as the halogen or hydrogen atom transfer agents, and the use of unstable initiators10,11. In recent years, photocatalysis using visible light has emerged as a promising strategy for the development of novel radical reactions due to its high efficiency and convenience in the generation of various reactive radical species under mild and environmentally friendly conditions12,13,14,15,16,17,18,19. Specifically, the visible-light-induced radical cascade reactions enable the facile construction of diverse cyclic compounds under sustainable conditions3,20,21. Despite these advances, plenty of radical cascade cyclization reactions still rely on either the carefully designed precursors or the intramolecular design, which tempers the appeal of their further synthetic applications. Therefore, it is highly desirable to develop stereoselective intermolecular radical cascade reactions that could produce complex cyclic compounds from readily available starting materials via visible-light-induced photocatalysis.
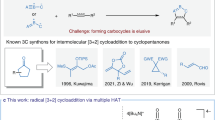
As a pivotal structure unit, hexahydrocarbazole motif widely occurs in many biologically active polycyclic indoline natural products22,23,24,25,26, such as kopsinine, tubifolidine, aspidospermidine, and vindoline (Fig. 1A). Consequently, great efforts have been devoted to the preparation of functionalized hexahydrocarbazoles27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43. Among those, direct catalytic dearomatization reaction of indole derivatives represents a straightforward and powerful strategy44,45,46,47,48,49,50. The cis-fused hexahydrocarbazoles are usually obtained through the dearomatization of substituted or pre-functionalized indole derivatives27,28,29,30,31,32,33. In contrast, the syntheses of the trans-fused hexahydrocarbazoles are less explored36,37,38,39,40,41,42,43. To date, methods to uncover the highly stereoselective synthesis of this structural motif have been limited to UV-light-induced intramolecular photocyclization of reactive enamine precursors36,37 (eq 1, Fig. 1B) as well as Pd-catalyzed intramolecular C–H activation/cyclization reaction (eq 2, Fig. 1B) 38,39,40,41,42,43.

A Natural products containing a hexahydrocarbazole ring. B Previous works. C Hypothesis. D Reaction of Boc-Trp and diethylethylidenemalonate. E Reaction of Boc-Trp and ɑ-CF3-alkene. F This work. PC photocatalyst, Trp tryptophan.
Tryptophan is a commercially available, non-toxic, abundant, and naturally occurring α-amino acid. Accordingly, we questioned whether this readily available material could be directly used to synthesize the trans-fused hexahydrocarbazole through a one-step visible-light-induced photocatalytic radical cascade reaction (Fig. 1C). In practice, tryptophan was often used as an α-amino alkyl radical precursor in radical addition reactions via visible-light-induced photocatalysis51,52,53,54,55,56,57,58. In pioneer work, MacMillan and coworkers reported an elegant radical Michael addition reaction of N-Boc-tryptophan and diethylethylidenemalonate via photocatalysis (Fig. 1D)54. In 2016, Zhou and coworkers described an interesting photocatalytic decarboxylative/defluorinative reaction of N-Boc-tryptophan and α-trifluoromethyl alkenes (Fig. 1E)56. Nevertheless, to the best of our knowledge, there is no general method for the generation of trans-fused hexahydrocarbazoles bearing multiple stereocenters through a one-step stereoselective radical cascade reaction of tryptophan via visible-light-induced photocatalysis. Herein, we report a facile and efficient synthesis of multi-substituted trans-fused hexahydrocarbazoles via a stereoselective intermolecular radical cascade reaction of readily available tryptophans and acrylamides enabled by visible-light-induced photoredox catalysis (eq 3, Fig. 1F). The trans-fused hexahydrocarbazoles with up to five stereocenters including two quaternary ones could be accessed in up to 82% yield, >20/1 diastereoselectivity, and 96% ee. Intriguingly, this protocol could further be applied to the stereoselective syntheses of 1,3,5-trisubstituted cyclohexanes, which are otherwise challenging to access in a single operation, by simply switching the starting material from tryptophans 1 to ɤ-alkenyl substituted amino acids 10 (eq 4, Fig. 1F).
Results
Reaction optimization
Our studies were initiated with an exploration of reaction conditions for the intermolecular coupling of commercially available N-(tert-butoxycarbonyl)tryptophan (1a) and N-phenyl-2-(trifluoromethyl)acrylamide (2a) (Table 1). After an extensive investigation of reaction conditions, it was found that the desired radical cascade cyclization product trans-fused hexahydro-1H-carbazole 3a was obtained in 75% yield and >20:1 dr when the reaction was performed with 1 mol% of Ir[dF(CF3)ppy]2(dtbbpy)PF6 as the photocatalyst, 3 equiv of Na2CO3 as the base and blue LEDs as the light source (entry 1, for the experimental setup, see Supplementary Fig. 1). Notably, the probably competitive radical addition product 3–1, radical addition/β-fluoride elimination product 3–2, and nucleophilic addition/cyclization product 3–3 were not detected under these photocatalytic conditions, which demonstrated the unique selectivity of the current reaction. The reaction could also proceed when the organic dye 4-CzIPN was used as the photocatalyst (entry 2). However, the use of a ruthenium-based photocatalyst led to low yield of the desired product (entry 3). The reaction also proceeded smoothly when different inorganic bases such as NaHCO3 and K2CO3 were used (entries 4–5). When the reaction was performed in the absence of base, the desired product was obtained with a significantly lower yield (entry 6). Control experiments demonstrated that the photocatalyst and light were indispensable for the product formation (entry 7). Interestingly, when the reaction was performed under air, 4% of the desired product was observed along with 61% of the tetrahydro-1H-carbazole 9a (entry 8, yields were determined by crude 1H NMR analysis. For details, see Fig. 4).
Evaluation of the substrate scope
With the optimized reaction conditions in hand, various substituted tryptophan derivatives 1 and N-aryl-2-(trifluoromethyl)acrylamides 2 were tested to establish the generality of the process (Fig. 2). Reactions of N-aryl-2-(trifluoromethyl)acrylamides 2 containing either electron-donating or electron-withdrawing groups on the aromatic ring (R4) attached to amide moiety all gave the corresponding products in moderate to good yields and good to excellent diastereoselectivities (3a − 3j). In addition, the tryptophan derivatives 1 containing either electron-donating or electron-withdrawing groups on the phenyl ring of the indole moiety (R1) were probed. The reactions occurred smoothly, affording the corresponding trans-fused hexahydrocarbazoles 3 in moderate to good yields and moderate to excellent diastereoselectivities (3k–3v). Moreover, substrates 1 containing different substituents on the nitrogen atom (R2) of the amino acid moiety were also tested. It was found that the carbamates, simple amides, and sulfonamide were all tolerated under these reaction conditions, affording the corresponding cyclization products in moderate to good yields and good to excellent diastereoselectivities (3w–3aa). Furthermore, reactions of ɑ, ɑ-disubstituted ɑ-amino acids also occurred, providing the trans-fused hexahydrocarbazoles 3ab–3ac with two quaternary carbon centers in moderate yields. Finally, the present method was applied to the late-stage functionalization of the derivatives of drug molecules, providing easy access to two hexahydrocarbazole-containing complex molecules (3ad–3ae) in moderate yields and excellent diastereoselectivities. The structure and relative configuration of both diastereomers of the hexahydrocarbazole products were assigned by X-ray crystallographic analyses of compounds 3k and 3ab (major diastereomers, see Supplementary Figs. 8 and 9), and compounds 3k' and 3ab' (minor diastereomers, see Supplementary Figs. 10 and 11) respectively.

a Reaction conditions: 1 mol% of Ir[dF(CF3)ppy]2(dtbbpy)PF6, 0.3 mmol of Na2CO3, 0.15 mmol of 1, 0.1 mmol of 2 in DMA (2.0 mL) for 12 h under blue LEDs irradiation at 30 °C. Isolated yields of major diastereomers are reported. The dr was determined by crude 1H NMR analysis. b The combined yield of both diastereomers. c The reaction was conducted at 40 °C.
Pleasingly, the present method was able to be applied to the diastereoselective syntheses of chiral trans-fused hexahydrocarbazoles with five contiguous stereogenic centers (Fig. 3). Reactions of chiral β-aryl-substituted tryptophan derivatives 7 containing either electron-withdrawing or electron-donating groups on the aromatic ring (R1) attached to β-carbon of the ɑ-amino acid moiety all gave the corresponding chiral trans-fused hexahydrocarbazoles bearing five contiguous stereocenters (8a–8f). In addition, reaction of the β-alkyl-substituted ɑ-amino acid also occurred smoothly, affording the desired product 8g in moderate yield and excellent diastereoselectivity. Notably, the enantiomeric purity of the starting material 6, which could be easily prepared via a simple Ag-catalyzed enantioselective reaction of glycine derivatives 5 with sulfonylindoles 459, was well-preserved under these photocatalytic conditions (8a–8g). The structure and absolute configuration of the major diastereomers of the hexahydrocarbazole products were assigned by an X-ray crystallographic analysis of compound 8a (Supplementary Fig. 12). The absolute configuration was determined as (1 S, 3 S, 4 R, 5 S, 6 S).

a Reaction conditions: 1 mol% of Ir[dF(CF3)ppy]2(dtbbpy)PF6, 0.1 mmol of 2, 0.15 mmol of 7 and 0.3 mmol of Na2CO3 in DMA (2.0 mL) for 12 h under blue LEDs irradiation at 30 °C. Combined isolated yields of the diastereomers are reported. b The dr was determined by crude 1H NMR analysis. c Enantiomeric excess (ee) of the major diastereomer was reported. The ee value was determined by HPLC analysis on a chiral stationary phase.
Interestingly, the reaction also proceeded when it was performed under air, affording tetrahydro-1H-carbazole 9a favorably (9a:3a = 14:1) with moderate yield and excellent diastereoselectivity (Fig. 4, for the experimental setup, see Supplementary Fig. 2). Several substituted tryptophan derivatives 1 and N-aryl-2-(trifluoromethyl)acrylamides 2 were tested to establish the generality of the process. Reactions of tryptophan derivatives 1 and N-aryl-2-(trifluoromethyl)acrylamides 2 containing either electron-donating or electron-withdrawing groups on the aromatic ring (R1 or R2) attached to the indole or amide moiety all led to the tetrahydro-1H-carbazoles 9 favorably in moderate yields and excellent diastereoselectivities (9b–9f). The structure and relative configuration of the tetrahydrocarbazole products were assigned by an X-ray crystallographic analysis of compound 9a (see Supplementary Fig. 13).

a Reaction conditions: 1 mol% of Ir[dF(CF3)ppy]2(dtbbpy)PF6, 0.15 mmol of 1, 0.1 mmol of 2, and 0.3 mmol of Na2CO3 in DMA (2.0 mL) for 6 h under blue LEDs irradiation at 30 °C. Isolated yields of compound 9 are reported. b The dr was determined by crude 1H NMR analysis. c The ratio was determined by crude 1H NMR analysis.
Mechanistic studies
To probe the reaction pathway, preliminary mechanistic studies were carried out (Figs. 5 and 6). It was found that the excited photocatalyst *Ir[dF(CF3)ppy]2(dtbbpy)PF6 could be quenched by acrylamide 2a, N-Boc-tryptophan 1a, and its sodium salt K1 respectively according to the Stern–Volmer luminescence quenching studies (Fig. 5, for experimental details, see Supplementary Fig. 7). However, the reductive quenching of *Ir[dF(CF3)ppy]2(dtbbpy)PF6 by either N-Boc-tryptophan 1a or its sodium salt K1 was more efficient than the oxidative quenching of the excited photocatalyst by substrate 2a, which indicates the former process is possibly the initiation step of the photoredox catalytic cycle (for the cyclic voltammetry measurement experiments of compounds 1a, 2a, and K1, see the Supplementary Figs. 3–6). In addition, when the enantiopure (R)-N-Boc-tryptophan (R)-1a was utilized under the reaction conditions, the racemic product 3a was obtained (Fig. 6A). These results suggest that the α-aminoalkyl radical is formed via photoinduced decarboxylation. Furthermore, 93% of deuterium incorporation at the C3-position of the hexahydrocarbazole product was observed when D2O was added to the reaction mixture (Fig. 6B). These results suggest that the carbon anion in the benzylic position of indoline moiety is plausibly generated via radical-polar crossover60,61. Interestingly, when the N-methyl substituted acrylamide 2' was used as a substrate for the reaction, the competitive radical addition/β-fluoride elimination product 3' was obtained instead of the hexahydrocarbazole56, which indicates that the N–H bond of the amide moiety is very crucial for this unique radical cascade cyclization reaction (Fig. 6C). The plausible hydrogen bonding effect between the amide N–H bond and the CF3 moiety might benefit the intramolecular cyclization. Moreover, in order to figure out whether the tetrahydrocarbazole 9a was formed via oxidation of trans-hexahydrocarbazole 3a, two control experiments were performed. Either low conversion or no reaction was observed when this transformation was carried out using the same conditions as shown in Fig. 4 or the conditions without photocatalyst (Fig. 6D). These results suggest that benzylic radical species I-3 might be involved as an intermediate during the oxidation process (for details, see Fig. 7) 62,63.

A solution of Ir[dF(CF3)ppy]2(dtbbpy)PF6 in N,N-dimethylacetamide was excited at 380 nm and the emission intensity at 481 nm was observed.

A Reaction of enantiopure (R)-N-Boc-tryptophan (R)-1a with substrate 2a. B Deuterium experiment. C Reaction of N-methyl substituted acrylamide 2' with substrate 1a. D Control experiments.

A plausible reaction pathway for the formation of products 3a and 9a was proposed.
Based on the above experimental observations, a plausible reaction pathway was proposed, as shown in Fig. 7. Firstly, the α-aminoalkyl radical I-1 generated by photoinduced decarboxylation adds to α,β-unsaturated amide 2a to form radical intermediate I-2. This tertiary alkyl radical intermediate would subsequently undergo a radical cyclization onto the indole ring to generate the tertiary benzylic radical intermediate I-3. Finally, the hexahydrocarbazole 3a is obtained via reduction of the benzylic radical intermediate I-3 by Ir(II) and subsequent protonation under inert atmosphere. Interestingly, when this benzylic radical intermediate I-3 is exposed to air under the reaction conditions, the tetrahydro-1H-carbazole 9a is formed favorably.
Rational expansion of the method
To further test the generality of the current method, this radical cascade cyclization strategy was applied to the synthesis of 1,3,5-trisubstituted cyclohexanes (Fig. 8). To our delight, by simply switching the starting material from tryptophans 1 to the alkenyl substituted amino acids 10, several 1,3,5-trisubstituted cyclohexanes bearing at least one quaternary carbon stereocenter were obtained in moderate yield and up to 3.5:1 dr under the photocatalytic conditions (11a–11h). Notably, the 1,3,5-trisubstituted cyclohexane with two quaternary carbon stereogenic centers (11h) could also be accessed via this one-step radical cascade cyclization reaction. This strategy provides a straightforward and efficient pathway for the construction of 1,3,5-trisubstituted cyclohexanes which are challenging to be synthesized in one step. The structure and relative configuration of both diastereomers of the 1,3,5-trisubstituted cyclohexane products were assigned by X-ray crystallographic analyses of the derivative of compound 11a (major diastereomer, see Supplementary Fig. 14) and compound 11d' (minor diastereomer, see Supplementary Fig. 15) respectively.

a Reaction conditions: 1 mol% of Ir[dF(CF3)ppy]2(dtbbpy)PF6, 0.1 mmol of 2, 0.15 mmol of 10 and 0.3 mmol of K2CO3 in DMA (2.0 mL) for 12 h under blue LEDs irradiation at 30 °C. Combined isolated yields of both diastereomers are reported. The dr was determined by crude 1H NMR analysis. b The reaction was performed with 2 mol% of Ir[dF(CF3)ppy]2(dtbbpy)PF6, and 2.0 equiv of 10 h for 18 h.
Synthetic applications
Finally, in order to demonstrate the synthetic utility of the present method, a 1.0 mmol-scale reaction and several transformations of the multi-substituted trans-fused hexahydrocarbazoles and cyclohexanes were carried out. Under the photocatalytic conditions, 1.0 mmol-scale reaction of substrates 1a and 2a occurred smoothly, providing product 3a in excellent diastereoselectivity and moderate yield (Fig. 9A). In addition, these radical cascade cyclization products are readily transformed to other valuable building blocks. Treatment of the trans-fused hexahydrocarbazole 3a with 2,2,2-trifluoroacetic acid and then 2,4,6-triphenylpyrylium tetrafluoroborate furnished Katritzky pyridinium salt K2 in excellent diastereoselectivity and moderate yield (path a, Fig. 9B)64,65,66. This pyridinium salt could be further transformed to other structural motifs bearing a trans-fused hexahydrocarbazole core through the mild, catalyst-free visible-light-induced deaminative functionalization reactions (paths b-d, Fig. 9B)67,68. Notably, the trans-fused hexahydrocarbazole product 12, bearing a Bpin group that could be readily applied in the following functionalization, could be easily accessed through this mild photoinduced deaminative borylation in excellent diastereoselectivity and moderate yield (path b, Fig. 9B)67. Interestingly, the bridged lactam-containing product 15 could be prepared in excellent diastereoselectivity and moderate yield through a simple two-step transformation of 1,3,5-trisubstituted cyclohexane 11d (path e, Fig. 9C).

A 1.0 mmol-scale reaction. B Synthetic transformations of 3a. C Transformation of 11d. Reaction conditions: [a] (i) CF3COOH, CH2Cl2, 0 °C → rt; (ii) 2,4,6-triphenylpyrylium tetrafluoroborate, EtOH, 140 °C, 68% yield over 2 steps, >20:1 dr. [b] (i) Bis(catecholato)diboron, DMA, blue LEDs, 30 °C; (ii) Et3N, pinacol, DMA, 30 °C, 57% yield over 2 steps, >20:1 dr. [c] Methyl 2-[(phenylsulfonyl)methyl]acrylate, diethyl 2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate, DMA, blue LEDs, 40 °C, 74% yield, 1.8:1 dr. [d] Methyl acrylate, diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate, DMA, blue LEDs, 40 °C, 50% yield, 3:1 dr. [e] (i) CF3COOH, CH2Cl2, 0 °C → rt; (ii) NaOH, MeOH/H2O, 100 °C, 69% yield over 2 steps, >20:1 dr.
Discussion
In this work, we have achieved the stereoselective intermolecular radical cascade reactions of readily available tryptophans and acrylamides via visible-light-induced photoredox catalysis, thus providing easy access to an array of trans-fused hexahydrocarbazoles with up to five stereogenic centers including two quaternary ones in up to 82% yield, >20/1 diastereoselectivity, and 96% ee. The reaction is distinguished by its broad substrate scope, its power in the rapid generation of molecular complexity from simple starting materials, environmentally friendly reaction conditions, and application to the late-stage functionalization. In addition, the reaction can be diverted to the syntheses of tetrahydrocarbazoles when it is performed under air. Moreover, this radical cascade protocol can further be applied to the stereoselective syntheses of 1,3,5-trisubstituted cyclohexanes by simply switching the starting material from tryptophans to ɤ-alkenyl substituted α-amino acids. This strategy allows the rapid preparation of trans-fused hexahydrocarbazoles and cyclohexanes with multiple stereocenters from simple amino acids and acrylamides. Further studies on the reaction mechanism and the development of other stereoselective intermolecular radical cascade reactions via visible-light-induced photoredox catalysis are ongoing in our laboratory.
Methods
Representative procedure for the synthesis of trans-fused hexahydrocarbazole 3a
To a Young Schlenk tube (10 mL) were added Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1.1 mg, 0.001 mmol, 1 mol%), 1a (45.7 mg, 0.15 mmol, 1.5 equiv), 2a (21.5 mg, 0.1 mmol, 1.0 equiv), Na2CO3 (31.8 mg, 0.3 mmol, 3.0 equiv), and N,N-dimethylacetamide (DMA, 2.0 mL). Subsequently, the reaction mixture was degassed through several freeze-pump-thaw cycles until no bubbles were released. The reaction mixture was stirred under argon at 30 °C, and irradiated by a 5 W blue LED lamp (λ = 450–460 nm, the tube was placed at ~2 cm away from the light source). After 12 h, the reaction mixture was passed through a short pad of celite and washed with ethyl acetate. The solvents were evaporated under reduced pressure to give the crude mixture, which was purified by flash column chromatography on silica gel (petroleum ether/ethyl acetate = 40:1 to 15:1, silica gel was soaked with a solution of petroleum ether and triethylamine (1000/1, v/v) before use) to afford the title compound 3a as a white solid (35.7 mg, 75% yield).
Data availability
The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 2108643 (3k), 2108649 (3ab), 2108644 (3k'), 2108650 (3ab'), 2108651 (8a), 2108652 (9a), 2108653 (the derivative of compound 11a), and 2108655 (11d'). The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif. Complete experimental procedures and compound characterization data are available in the Supplementary Information or from the corresponding author upon request.
References
Lovering, F., Bikker, J. & Humblet, C. Escape from Flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Taylor, R. D., MacCoss, M. & Lawson, A. D. G. Rings in drugs. J. Med. Chem. 57, 5845–5859 (2014).
Zhou, L., Hossain, M. L. & Xiao, T. Synthesis of N-containing heterocyclic compounds using visible-light photoredox catalysis. Chem. Rec. 16, 319–334 (2016).
Plesniak, M. P., Huang, H.-M. & Procter, D. J. Radical cascade reactions triggered by single electron transfer. Nat. Rev. Chem. 1, 0077 (2014).
Xuan, J. & Studer, A. Radical cascade cyclization of 1,n-enynes and diynes for the synthesis of carbocycles and heterocycles. Chem. Soc. Rev. 46, 4329–4346 (2017).
Huang, H.-M., Garduño-Castro, M. H., Morrill, C. & Procter, D. J. Catalytic cascade reactions by radical relay. Chem. Soc. Rev. 48, 4626–4638 (2019).
Hung, K., Hu, X. & Maimone, T. J. Total synthesis of complex terpenoids employing radical cascade processes. Nat. Prod. Rep. 35, 174–202 (2018).
Liu, X.-Y. & Qin, Y. Indole alkaloid synthesis facilitated by photoredox catalytic radical cascade reactions. Acc. Chem. Res. 52, 1877–1891 (2019).
Bonjoch, J. & Diaba, F. Radical reactions in alkaloid synthesis: a perspective from carbon radical precursors. Eur. J. Org. Chem. 5070−5100 (2020).
Khattab, M. A., Elgamal, M. A. & El-Batouti, M. Evaluation of thermal hazard of Azobisisobutyronitrile using accelerating rate calorimetry. Fire Mater. 20, 253–259 (1996).
O’Mahony, G. Triethylborane (Et3B). Synlett 572−573 (2004).
Narayanam, J. M. R. & Stephenson, C. R. J. Visible light photoredox catalysis: applications in organic synthesis. Chem. Soc. Rev. 40, 102–113 (2011).
Xuan, J. & Xiao, W.-J. Visible-light photoredox catalysis. Angew. Chem. Int. Ed. 51, 6828–6838 (2012).
Prier, C. K., Rankic, D. A. & MacMillan, D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 113, 5322–5363 (2013).
Schultz, D. M. & Yoon, T. P. Solar synthesis: prospects in visible light photocatalysis. Science 343, 985 (2014).
Xuan, J., Zhang, Z.-G. & Xiao, W.-J. Visible-light-induced decarboxylative functionalization of carboxylic acids and their derivatives. Angew. Chem. Int. Ed. 54, 15632–15641 (2015).
Romero, N. A. & Nicewicz, D. A. Organic photoredox catalysis. Chem. Rev. 116, 10075–10166 (2016).
Shaw, M. H., Twilton, J. & MacMillan, D. W. C. Photoredox catalysis in organic chemistry. J. Org. Chem. 81, 6898–6926 (2016).
Liu, Q. & Wu, L.-Z. Recent advances in visible-light-driven organic reactions. Natl Sci. Rev. 4, 359–380 (2017).
Zhao, Y., Lv, Y. & Xia, W. Synthesis of cyclic compounds via photoinduced radical cyclization cascade of C═C bonds. Chem. Rec. 19, 424–439 (2019).
Zhang, G., Liu, Y., Zhao, J., Li, Y. & Zhang, Q. Radical cascade reactions of unsaturated C–C bonds involving migration. Sci. China. Chem. 62, 1476–1491 (2019).
Zi, W., Zuo, Z. & Ma, D. Intramolecular dearomative oxidative coupling of indoles: a unified strategy for the total synthesis of indoline alkaloids. Acc. Chem. Res. 48, 702–711 (2015).
Li, L., Chen, Z., Zhang, X. & Jia, Y. Divergent strategy in natural product total synthesis. Chem. Rev. 118, 3752–3832 (2018).
Wang, Y., Xie, F., Lin, B., Cheng, M. & Liu, Y. Synthetic approaches to tetracyclic indolines as versatile building blocks of diverse indole Alkaloids. Chem. Eur. J. 24, 14302–14315 (2018).
Zheng, C. & You, S.-L. Catalytic Asymmetric Dearomatization (CADA) reaction-enabled total synthesis of indole-based natural products. Nat. Prod. Rep. 36, 1589–1605 (2019).
Thakur, A., Singh, A., Kaur, N., Ojha, R. & Nepali, K. Steering the antitumor drug discovery campaign towards structurally diverse indolines. Bioorg. Chem. 94, 103436 (2020).
Daniels, B. E., Ni, J. & Reisman, S. E. Synthesis of enantioenriched indolines by a conjugate addition/asymmetric protonation/Aza-Prins cascade reaction. Angew. Chem. Int. Ed. 55, 3398–3402 (2016).
Jiang, S.-Z. et al. Iridium-catalyzed enantioselective indole cyclization: application to the total synthesis and absolute stereochemical assignment of (−)-aspidophylline A. Angew. Chem. Int. Ed. 55, 4044–4048 (2016).
Zhu, J. et al. Highly efficient formal [2+2+2] strategy for the rapid construction of polycyclic spiroindolines: a concise synthesis of 11-demethoxy-16-epi-myrtoidine. Angew. Chem. Int. Ed. 55, 9224–9228 (2016).
Alpers, D., Gallhof, M., Witt, J., Hoffmann, F. & Brasholz, M. A photoredox-induced stereoselective dearomative radical (4 + 2)-cyclization/1,4-addition cascade for the synthesis of highly functionalized hexahydro-1H-carbazoles. Angew. Chem. Int. Ed. 56, 1402–1406 (2017).
Chen, H. et al. Access to hexahydrocarbazoles: the Thorpe–ingold effects of the ligand on enantioselectivity. Angew. Chem. Int. Ed. 56, 6942–6945 (2017).
Kuang, X.-K. et al. Synergetic tandem enantiomeric enrichment in catalytic Asymmetric Multi-Component Reactions (AMCRs): highly enantioselective construction of tetracyclic indolines with four continuous stereocenters. ACS Catal. 8, 4991–4995 (2018).
Wen, J. et al. Brønsted-acid-promoted Rh-catalyzed asymmetric hydrogenation of N-unprotected indoles: a cocatalysis of transition metal and anion binding. Org. Lett. 20, 2143–2147 (2018).
Dierks, A., Fliegel, L., Schmidtmann, M. & Christoffers, J. Preparation of hexahydrocarbazole derivatives by reductive indolization. Eur. J. Org. Chem. 7164–7175 (2020).
Ma, W.-Y. et al. Chiral phosphoric acid-catalyzed enantioselective construction of 2,3-disubstituted indolines. Org. Lett. 23, 442–448 (2021).
Chapman, O. L. & Eian, G. L. Photochemical synthesis of 2,3-dihydroindoles from N-aryl enamines. J. Am. Chem. Soc. 90, 5329–5330 (1968).
Chapman, O. L., Eian, G. L., Bloom, A. & Clardy, J. Nonoxidative photocyclization of N-aryl enamines. a facile synthetic entry to trans-hexahydrocarbazoles. J. Am. Chem. Soc. 93, 2918–2928 (1971).
Watanabe, T., Oishi, S., Fujii, N. & Ohno, H. Palladium-catalyzed Sp3 C–H activation of simple alkyl groups: direct preparation of indoline derivatives from N-alkyl-2-bromoanilines. Org. Lett. 10, 1759–1762 (2008).
Nakanishi, M., Katayev, D., Besnard, C. & Kündig, E. P. Fused indolines by palladium-catalyzed asymmetric C–C coupling involving an unactivated methylene group. Angew. Chem. Int. Ed. 50, 7438–7441 (2011).
Anas, S., Cordi, A. & Kagan, H. B. Enantioselective synthesis of 2-methyl indolines by palladium catalysed asymmetric C(sp3)–H activation/cyclisation. Chem. Commun. 47, 11483–11485 (2011).
Saget, T., Lemouzy, S. J. & Cramer, N. Chiral monodentate phosphines and bulky carboxylic acids: cooperative effects in palladium-catalyzed enantioselective C(Sp3)-H functionalization. Angew. Chem. Int. Ed. 51, 2238–2242 (2012).
Nakanishi, M., Katayev, D., Besnard, C. & Kündig, E. P. Palladium-NHC catalyzed enantioselective synthesis of fused indolines via Inert C(sp3)-H activation. Chimia 66, 241–243 (2012).
Larionov, E., Nakanishi, M., Katayev, D., Besnard, C. & Kündig, E. P. Scope and mechanism of asymmetric C(sp3)−H/C(Ar)−X coupling reactions: computational and experimental study. Chem. Sci. 4, 1995–2005 (2013).
Zhuo, C.-X., Zhang, W. & You, S.-L. Catalytic asymmetric dearomatization reactions. Angew. Chem. Int. Ed. 51, 12662–12686 (2012).
Zhuo, C.-X., Zheng, C. & You, S.-L. Transition-metal-catalyzed asymmetric allylic dearomatization reactions. Acc. Chem. Res. 47, 2558–2573 (2014).
Sheng, F. T., Wang, J. Y., Tan, W., Zhang, Y. C. & Shi, F. Progresses in organocatalytic asymmetric dearomatization reactions of indole derivatives. Org. Chem. Front. 7, 3967–3998 (2020).
Zheng, C. & You, S.-L. Advances in catalytic asymmetric dearomatization. ACS Cent. Sci. 7, 432–444 (2021).
Ma, J. et al. Gadolinium photocatalysis: dearomative [2 + 2] cycloaddition/ring-expansion sequence with indoles. Angew. Chem. Int. Ed. 59, 9639–9645 (2020).
Liang, R.-X. et al. Palladium-catalyzed enantioselective heteroarenyne cycloisomerization reaction. Angew. Chem. Int. Ed. 60, 7412–7417 (2021).
Jiang, R., Ding, L., Zheng, C. & You, S.-L. Iridium-catalyzed Z-retentive asymmetric allylic substitution reactions. Science 371, 380–386 (2021).
Liu, J.-Q., Shatskiy, A., Matsuura, B. S. & Kärkäs, M. D. Recent advances in photoredox catalysis enabled functionalization of α-amino acids and peptides: concepts, strategies and mechanisms. Synthesis 51, 2759–2791 (2019).
Rahman, M. et al. Recent advances on diverse decarboxylative reactions of amino acids. Adv. Synth. Catal. 361, 2161–2214 (2019).
Zuo, Z. & MacMillan, D. W. C. Decarboxylative arylation of α-amino acids via photoredox catalysis: a one-step conversion of biomass to drug pharmacophore. J. Am. Chem. Soc. 136, 5257–5260 (2014).
Chu, L., Ohta, C., Zuo, Z. & MacMillan, D. W. C. Carboxylic acids as a traceless activation group for conjugate additions: a three-step synthesis of (±)-Pregabalin. J. Am. Chem. Soc. 136, 10886–10889 (2014).
Noble, A., McCarver, S. J. & MacMillan, D. W. C. Merging photoredox and nickel catalysis: decarboxylative cross-coupling of carboxylic acids with vinyl halides. J. Am. Chem. Soc. 137, 624–627 (2015).
Xiao, T., Li, L. & Zhou, L. Synthesis of functionalized gem-difluoroalkenes via a photocatalytic decarboxylative/defluorinative reaction. J. Org. Chem. 81, 7908–7916 (2016).
Chen, Y., Lu, P. & Wang, Y. 3-Amino-fluorene-2,4-dicarbonitriles (AFDCs) as photocatalysts for the decarboxylative arylation of α-amino acids and α-oxy acids with arylnitriles. Org. Lett. 21, 2130–2133 (2019).
Ernouf, G., Chirkin, E., Rhyman, L., Ramasami, P. & Cintrat, J.-C. Photochemical strain-release-driven cyclobutylation of C(sp3)-centered radicals. Angew. Chem. Int. Ed. 59, 2618–2622 (2020).
Zheng, B.-H., Ding, C.-H., Hou, X.-L. & Dai, L.-X. Ag-Catalyzed diastereo- and enantioselective synthesis of β-substituted tryptophans from sulfonylindoles. Org. Lett. 12, 1688–1691 (2010).
Sim, B., Griller, D. & Wayner, D. Reduction potentials for substituted benzyl radicals: pKa values for the corresponding toluenes. J. Am. Chem. Soc. 111, 754–755 (1989).
Huang, H. et al. Chemo- and regioselective organo-photoredox catalyzed hydroformylation of styrenes via a radical pathway. J. Am. Chem. Soc. 139, 9799–9802 (2017).
Zhu, S. et al. Oxygen switch in visible-light photoredox catalysis: radical additions and cyclizations and unexpected C–C bond cleavage reactions. J. Am. Chem. Soc. 135, 1823–1829 (2013).
Shao, T. et al. Sequential photoredox catalysis for cascade aerobic decarboxylative povarov and oxidative dehydrogenation reactions of N-aryl α-amino acids. Adv. Synth. Catal. 360, 1754–1760 (2018).
Kong, D., Moon, P. J. & Lundgren, R. J. Radical coupling from alkyl amines. Nat. Catal. 2, 473–476 (2019).
He, F.-S., Ye, S. & Wu, J. Recent advances in pyridinium salts as radical reservoirs in organic synthesis. ACS Catal. 9, 8943–8960 (2019).
Roessler, S. L. et al. Pyridinium salts as redox-active functional group transfer reagents. Angew. Chem. Int. Ed. 59, 9264–9280 (2020).
Wu, J., He, L., Noble, A. & Aggarwal, V. K. Photoinduced deaminative borylation of alkylamines. J. Am. Chem. Soc. 140, 10700–10704 (2018).
Wu, J., Grant, P. S., Li, X., Noble, A. & Aggarwal, V. K. Catalyst-free deaminative functionalizations of primary amines by photoinduced single-electron transfer. Angew. Chem. Int. Ed. 58, 5697–5701 (2019).
Acknowledgements
We are grateful for financial support from the Recruitment Program of Global Experts and the startup funding from Xiamen University. We thank Mr. Zanbin Wei for solving the X-ray structure.
Author information
Authors and Affiliations
Contributions
J.-T. L., J.-N. L., J.-L. W., D.-K. W., and Y.-Z. Y. performed the experiments and analyzed the data. C.-X. Z. directed the project and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Liang-Qiu Lu the other anonymous reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, JT., Luo, JN., Wang, JL. et al. Stereoselective intermolecular radical cascade reactions of tryptophans or ɤ-alkenyl-α-amino acids with acrylamides via photoredox catalysis. Nat Commun 13, 1778 (2022). https://doi.org/10.1038/s41467-022-29464-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-29464-5
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
