
Abstract
Catalytic nonenzymatic kinetic resolution (KR) of racemates remains one of the most powerful tools to prepare enantiopure compounds, which dominantly relies on the manipulation of reactive functional groups. Moreover, catalytic KR of organic azides represents a formidable challenge due to the small size and instability of the azido group. Here, an effective KR of cyclic benzylic azides through site- and enantioselective C(sp3)–H oxidation is described. The manganese catalyzed oxidative KR reaction exhibits good functional group tolerance, and is applicable to a range of tetrahydroquinoline- and indoline-based organic azides with excellent site- and enantio-discrimination. Computational studies elucidate that the effective chiral recognition is derived from hydrogen bonding interaction between substrate and catalyst.
Similar content being viewed by others
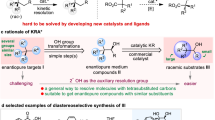
Introduction
Catalytic nonenzymatic kinetic resolution (KR) of racemates is one of the most powerful and practical tools to prepare valuable enantiopure targets, especially in cases where other methods are not possible or provide insufficient enantiocontrol1,2,3,4,5. Chiral organic azides are versatile synthetic precursors for a range of nitrogen-containing molecules and have found dramatically expanded utility in medicine, biology, and material science6,7,8,9,10,11,12,13,14,15. However, catalytic KR to provide optically pure azides has remained elusive, principally due to two essential features of the azido moiety: (1) the instability hampering the design of new reactivity with excellent chemoselectivity; (2) the small size hampering the achievement of effective chiral recognition16,17,18,19,20,21. Existing isolated examples always focused on manipulating the azido moiety through azide–alkyne cycloaddition (AAC)22,23,24,25,26 (Fig. 1A) or extra reactive functional groups preinstalled in substrates (Fig. 1B)27,28, which typically suffer from the use of excess azide substrates, poor chiral recognition, and narrow substrate scope. Developing an effective KR of organic azides relying on the reactivity of C(sp3)–H bonds would be highly desired29.

A KR of benzylic azides through azide–alkyne cycloaddition. B KR of allylic azides through asymmetric dihydroxylation of alkene. C KR of cyclic benzylic azides through asymmetric C(sp3)−H oxidation.
Nonenzymatic site- and enantioselective oxidation of ubiquitous C(sp3)−H bonds with a general scope and predictable selectivity represents a paradigm shift in the standard logic of organic synthesis30,31. However, such research topic has remained a formidable challenge, and current studies typically suffer from moderate enantioselectivity, low substrate conversion, and narrow substrate scope32,33,34,35,36,37,38,39,40,41,42,43,44. In particular, catalytic KR through C(sp3)–H oxidation dominantly focused on secondary alcohols45,46,47,48,49,50,51,52 and amines53,54 due to their high and well-known oxidized reactivity together with the presence of a strong interaction site with catalyst for efficient chiral recognition55,56. To our knowledge, selective oxidation of C(sp3)−H bond adjacent to azido moiety remains elusive. Moreover, organic azide lacks such an effective interaction site to direct substrate to an ideal location in the transition state. Therefore, chiral recognition of chemically similar C(sp3)−H bonds adjacent to azido group of two enantiomers would be difficult to accomplish.
Herein, we report the KR of organic azides through site- and enantioselective C(sp3)–H bond oxidation (Fig. 1C). First, given the significance of benzo-fused nitrogen-containing heterocycles in modern pharmacology, we choose a range of racemic benzylic azides bearing such skeletons as substrates. Second, we select the readily modifiable salen as the basal ligand to search for suitable base-metal catalyst. Third, varying the protecting group on the nitrogen moiety might also provide an opportunity to tune the chiral recognition. Based on these considerations, a range (36 examples) of cyclic benzylic azides participate in oxidative KR with good to excellent selectivity factors (s up to 95).
Results and discussion
Reaction condition optimization
The oxidative KR of tetrahydroquinoline (THQ) based organic azide rac-1a was selected as the model reaction for optimization (Table 1). In the presence of PhIO as the oxidant, no reaction was observed for either chiral Fe(salen) C1 or Co(salen) C2 (entry 1). Chiral Mn(salen) C3 exhibited good oxidation catalysis reactivity, though poor chiral recognition was obtained (entry 2). Oxidation proceeded with excellent site selectivity at the C4–H bond adjacent to azido moiety over C2–H bond α to amide motif, affording azine 2a as oxidized product. Mn(salen) C4 having cyclohexanediamine skeleton provided better results than C3 with 1,2-diphenyl-1,2-ethanediamine (entry 3). Careful examination of the substituent effects on the basal salen ligand revealed C12 with 2,4-difluorophenyl moieties at C3(3’) sites to be optimal (entries 4–10). Other oxidants such as H2O2 and NaClO afforded inferior oxidation reactivity (entry 11). Addition of PhIO as eight equal portions in 30 min intervals was beneficial for achieving an extremely high level of chiral discrimination, and (S)-1a was isolated in 48% yield with 98% ee (s = 91, entries 12 and 13).
Substrate scope
The scope of oxidative KR of THQ-based organic azides was explored (Fig. 2). In general, both electron-rich and -deficient THQ skeletons were well tolerated, as demonstrated by effective access to optically pure 1a-1m with good to excellent selectivity factors (s = 32–91). Resolution efficiency was not impaired for reaction on a 0.5 mmol scale. Common functional groups, including halide, acetate, silyl ether, benzoate, and triflate, were tolerated for further manipulation. Racemic THQ-based organic azides bearing two stereocenters were also suitable components with good enantio-discrimination. Oxidative KR of cis-2,4-disubstituted rac-1n proceeded, furnishing (2R,4S)-1n in 47% yield with 92% ee (The absolute configuration of recovered 1n was determined by X-ray diffraction analysis. See the Supporting Information for details). The reaction was not sensitive to the steric hindrance of C2-substituents, as demonstrated by access to respective enantiopure 1o-1u with good selectivity factors (s = 20–35).

Conditions: rac-1 (0.1 mmol), PhIO (0.08 mmol, addition as 8 portions in 30-min intervals over 3.5 h), and C12 (5 mol%) in ethyl acetate (1.0 ml) at rt for 4 h. aReaction with 0.5 mmol rac-1. bReaction with 1.0 gram of rac-1.
Indoline represents the other type of biologically important benzo-fused nitrogen-containing heterocycle. Accordingly, the applicability of the oxidative KR strategy in enantioselective access to indoline-based azides was next explored (Fig. 3). When Mn(salen) C7 was used as catalyst, oxidative KR of cyclic benzylic azide rac-3a proceeded, furnishing 2-aminobenzonitrile 4a as oxidized product together with recovered (S)-3a in 48% yield with 96% ee (s = 65) (The absolute configuration of recovered 3a was determined by X-ray diffraction analysis. See the Supporting Information for details). Notably, the reaction exhibited excellent site selectivity at the C3–H bond adjacent to azido moiety over C2–H bond α to amide motif. Indolines rac-3b-3h bearing electronically varied groups around the arene moiety were tolerated with a high level of chiral recognition (s = 36–95). Selectivities were not impaired for reaction on a 0.5 mmol scale. Substrates bearing other N-acyl groups, such as propanoyl (3i) and butyryl (3j), were also tolerated, though no reaction was observed for pivaloyl one (3k).

Conditions: rac-3 (0.1 mmol), PhIO (0.08 mmol, addition as 8 portions in 30-min intervals over 3.5 h), and C7 (5 mol%) in ethyl acetate (1.0 ml) at rt for 4 h. aReaction with 0.5 mmol rac-3.
Synthetic applications
Manipulating the azido moiety through copper-catalyzed AAC would allow facile integration of the other biologically important molecules into N-heterocycles for drug discovery. For example, vitamin E derivative (5) with potent antioxidation activity and estrone derivative (7) for treating abnormalities associated with menopause are efficiently installed into THQ skeleton using triazole as a linker, respectively (Fig. 4).

A CuSO4-catalyzed AAC of optically pure THQ-based organic azide and vitamin E derivative. B CuI-catalyzed AAC of optically pure THQ-based organic azide and estrone derivative.
Mechanistic and DFT studies
Control experiments were conducted to get a preliminary understanding of the reaction mechanism (Fig. 5). The relationship between ee values of Mn(salen) catalyst and recovered substrate was explored, showing that the enantioselectivity of recovered 3a is proportional to the ee of C7 (Fig. 5A). The absence of nonlinear effects indicated that the reaction might not involve heterochiral aglomerates57,58. No reaction was observed for stoichiometric Mn(salen) C12 mediated reaction in the absence of PhIO, suggesting that oxoMn(V) might be the species in charge of C–H oxidation (Fig. 5B). A competition deuterium kinetic isotope effect (KIE) study, using a mixture of rac-1a and [D]-rac-1a, revealed a KIE of 2.7 (Fig. 5C). The observation implied that C−H bond cleavage might be involved in the rate-determining step. The substituent effect of different acyl groups on THQ-based azides was explored (Fig. 5D). Several aspects of the data merit further comment. Firstly, no reaction was observed for N-acyl substituted 9a. Secondly, the oxidized reactivity was gradually enhanced as the increasing numbers of methyl groups at α-position of the carbonyl moiety (9a-9c and 1a). Thirdly, the oxidative reactivity was lost when placing an oxygen atom between the carbonyl and tBu groups. In general, more sterically hindered substrates should exhibit lower reactivity than that of less sterically hindered ones. We speculated that the opposite trend observed for THQ-based substrates in Fig. 5D might originate from the non-covalent interaction between the α-alkyl group of carbonyl moiety and Mn(salen) catalyst. The deuteration effect of the N-acyl moiety of indoline-based substrates was next evaluated (Fig. 5E). No oxidative conversion was observed for [D]-3a, indicating that the sp3 C–H bond at α-position of carbonyl motif is crucial to the reactivity of substrate 3a.

A Plot of enantiomeric excess of recovered 3a versus the enantiomeric excess of C7 at 20% conversion. The dotted line symbolized the linear correlation. B Stoichiometric Mn(salen) C12 mediated control experiment in the absence of PhIO. C The intermolecular kinetic isotope effect. D The N-acyl substituent effect for THQ-based azides. E Deuterated control experiment of indoline-based azide.
According to the generally accepted mechanism of manganese-catalyzed C(sp3)−H oxidation and the control experiments, a plausible mechanistic pathway for oxidative KR of benzylic azides was suggested (Fig. 6)59. Chiral MnIII catalyst is first oxidized by PhIO affording oxoMnV intermediate. THQ rac-1a underwent hydrogen atom transfer (HAT) to oxoMnV, giving benzylic radical 10 and MnIV−OH. Finally, azide 10 decomposed by losing molecular nitrogen to form iminyl radical 11, which immediately dimerized to provide 2a60. MnIV−OH dimerized by releasing H2O to generate μ-oxo bridged dimer MnIVOMnIV, which might undergo disproportionation reaction regenerating MnIII precursor and oxoMnV species for the catalytic cycle61,62,63. Based on the absolute configuration of recovered THQs, (R)-1a should be oxidized more preferentially than (S)-1a. With respect to the oxidative KR of indoline-based benzylic azides, rac-3a might proceed through a similar HAT process to oxoMnV species producing benzylic radical 12, which underwent azide collapse followed by C−C bond cleavage forming radical 14. Alkyl radical 14 underwent oxygen rebound with MnIV−OH followed by hemiaminal decomposition, generating 2-aminobenzonitrile 4a together with MnIII precursor for the catalytic cycle. Based on the absolute configuration of recovered indolines, (R)-3a should be oxidized more preferentially than (S)-3a.

The possible reaction pathway based on our studies and the previous literatures.
To elucidate the origin of the high level of chiral recognition of azide rac-1a, density functional theory (DFT) calculations were performed for the stereo-determining HAT process (Fig. 7). The Gibbs free energies of corresponding transition states follow the spin ordering of triplet < quintet < singlet, and the triplet state was determined to be the ground state (see Table S1 in the Supporting Information). 3TSR is 1.8 kcal/mol more favorable than 3TSS, which is consistent with experimentally observed stereoselectivity. The effective chiral recognition arises from additional CH···F hydrogen bonding interaction between tert-butyl group of (R)-1a and 2,4-difluorophenyl moiety of catalyst C12 in 3TSR. This CH···F hydrogen bonding interaction is further confirmed by independent gradient model analysis64.

Trivial hydrogen atoms are omitted for clarity. The isosurface of IGM analysis is 0.005. The bond distances are given in Å. All energies are given in kcal/mol.
In this work, the KR of organic azides through site- and enantioselective C(sp3)–H oxidation is described. The practical manganese catalyzed reaction exhibits good functional group tolerance, and is applicable to a variety of cyclic benzylic azides bearing pharmacologically significant nitrogen-containing heterocycle skeletons with extremely efficient site- and enantio-discrimination. The usefulness of products has also been demonstrated in synthetic applications. Detailed computational studies elucidate the origins of effective chiral recognition involving a hydrogen bonding interaction between substrate and catalyst. This strategically different approach would unlock opportunities for topologically straightforward synthetic planning for KR reactions relying on the reactivity of C(sp3)–H bonds.
Methods
General procedure
To a solution of rac-1a (0.1 mmol, 1.0 equiv) in ethyl acetate (1.0 ml) was added C12 (0.005 mmol, 0.05 equiv) at room temperature. Then PhIO (0.08 mmol, 0.8 equiv) was added as eight portions in 30-min intervals over 3.5 h. After that, the solvent was removed under vacuum and the residue was purified by flash chromatography on silica gel using ethyl acetate/petroleum ether as eluent to give the product (S)-1a.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files. Extra data are available from the corresponding author upon request. The X-ray crystallographic coordinates for structures reported in Supplementary Information have been deposited at the Cambridge Crystallographic Data Center (S1: CCDC 2009823, S2: CCDC 2009831). These data could be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif.
References
Keith, J. M., Larrow, J. F. & Jacobsen, E. N. Practical considerations in kinetic resolution reactions. Adv. Synth. Catal. 343, 5–26 (2001).
Breuer, M. et al. Industrial methods for the production of optically active intermediates. Angew. Chem. Int. Ed. 43, 788–824 (2004).
Vedejs, E. & Jure, M. Efficiency in nonenzymatic kinetic resolution. Angew. Chem. Int. Ed. 44, 3974–4001 (2005).
Pellissier, H. Catalytic non-enzymatic kinetic resolution. Adv. Synth. Catal. 353, 1613–1666 (2011).
Ren, J. et al. Kinetic and dynamic kinetic resolution of racemic tertiary bromides by pentanidium-catalyzed phase-transfer azidation. Angew. Chem. Int. Ed. 59, 9055–9058 (2020).
Bräse, S. & Banert, K. Organic Azides: Synthesis and Applications (Wiley, 2009).
Scriven, E. F. V. & Turnbull, K. Azides: their preparation and synthetic uses. Chem. Rev. 88, 297–368 (1988).
Jung, N. & Bräse, S. Vinyl and alkynyl azides: well-known intermediates in the focus of modern synthetic methods. Angew. Chem. Int. Ed. 51, 12169–12171 (2012).
Chiba, S. Application of organic azides for the synthesis of nitrogen-containing molecules. Synlett 1, 21–44 (2012).
Thirumurugan, P., Matosiuk, D. & Jozwiak, K. Click chemistry for drug development and diverse chemical–biology applications. Chem. Rev. 113, 4905–4979 (2013).
Grammel, M. & Huang, H. C. Chemical reporters for biological discovery. Nat. Chem. Biol. 9, 475–484 (2013).
Best, M. D. Click chemistry and bioorthogonal reactions: unprecedented selectivity in the labeling of biological molecules. Biochemistry 48, 6571–6584 (2009).
Pathak, T. Azidonucleosides: synthesis, reactions, and biological properties. Chem. Rev. 102, 1623–1668 (2002).
Binder, W. H. & Kluger, C. Azide/Alkyne-“Click” reactions: applications in material science and organic synthesis. Curr. Org. Chem. 10, 1791–1815 (2006).
Xi, W., Scott, T. F., Kloxin, C. J. & Bowman, C. N. Click chemistry in materials science. Adv. Funct. Mater. 24, 2572–2590 (2014).
Bräse, S., Gil, C., Knepper, K. & Zimmermann, V. Organic azides: an exploding diversity of a unique class of compounds. Angew. Chem. Int. Ed. 44, 5188–5240 (2005).
Ding, P., Hu, X., Zhou, F. & Zhou, J. Catalytic enantioselective synthesis of α-chiral azides. Org. Chem. Front. 5, 1542–1559 (2018).
Liu, W. et al. Iron-catalyzed enantioselective radical carboazidation and diazidation of α,β-unsaturated carbonyl compounds. J. Am. Chem. Soc. 143, 11856–11863 (2021).
Deng, Q.-H., Bleith, T., Wadepohl, H. & Gade, L. H. Enantioselective iron-catalyzed azidation of β‑keto esters and oxindoles. J. Am. Chem. Soc. 135, 5356–5359 (2013).
Martínez, L. E., Leighton, J. L., Carsten, D. H. & Jacobsen, E. N. Highly enantioselective ring opening of epoxides catalyzed by (salen)Cr(III) complexes. J. Am. Chem. Soc. 117, 5897–5898 (1995).
Seidl, F. J., Min, C., Lopez, J. A. & Burns, N. Z. Catalytic regio- and enantioselective haloazidation of allylic alcohols. J. Am. Chem. Soc. 140, 15646–15650 (2018).
Meng, J.-C., Fokin, V. V. & Finn, M. G. Kinetic resolution by copper-catalyzed azide–alkyne cycloaddition. Tetrahedron Lett. 46, 4543–4546 (2005).
Alexander, J. R., Ott, A. A., Liu, E.-C. & Topczewski, J. J. Kinetic resolution of cyclic secondary azides, using an enantioselective copper-catalyzed azide–alkyne cycloaddition. Org. Lett. 21, 4355–4358 (2019).
Zhou, F. et al. Asymmetric copper(I)-catalyzed azide−alkyne cycloaddition to quaternary oxindoles. J. Am. Chem. Soc. 135, 10994–10997 (2013).
Brittain, W. D. G., Buckley, B. R. & Fossey, J. S. Asymmetric copper-catalyzed azide−alkyne cycloadditions. ACS Catal. 6, 3629–3636 (2016).
Liu, E.-C. & Topczewski, J. J. Enantioselective copper catalyzed alkyne−azide cycloaddition by dynamic kinetic resolution. J. Am. Chem. Soc. 141, 5135–5138 (2019).
Ott, A. A., Goshey, C. S. & Topczewski, J. J. Dynamic kinetic resolution of allylic azides via asymmetric dihydroxylation. J. Am. Chem. Soc. 139, 7737–7740 (2017).
Yang, X. & Birman, V. B. Kinetic resolution of α-substituted alkanoic acids promoted by homobenzotetramisole. Chem. Eur. J. 17, 11296–11304 (2011).
Pramanik, S. & Ghorai, P. Synthesis and asymmetric resolution of α-azido-peroxides. Org. Lett. 15, 3832–3835 (2013).
Newhouse, T. & Baran, P. S. If C-H bonds could talk: selective C-H bond oxidation. Angew. Chem. Int. Ed. 50, 3362–3374 (2011).
Milan, M., Bietti, M. & Costas, M. Enantioselective aliphatic C–H bond oxidation catalyzed by bioinspired complexes. Chem. Commun. 54, 9559–9570 (2018).
Groves, J. T. & Viski, P. Asymmetric hydroxylation by a chiral iron porphyrin. J. Am. Chem. Soc. 111, 8537–8538 (1989).
Hamachi, K., Irie, R. & Katsuki, T. Asymmetric benzylic oxidation using a Mn-salen complex as catalyst. Tetrahedron Lett. 37, 4979–4982 (1996).
Zhang, R., Yu, W.-Y., Lai, T.-S. & Che, C.-M. Enantioselective hydroxylation of benzylic C–H bonds by D4-symmetric chiral oxoruthenium porphyrins. Chem. Commun. 1791–1792 (1999).
Srour, H., Maux, P. L. & Simonneaux, G. Enantioselective manganese-porphyrin-catalyzed epoxidation and C–H hydroxylation with hydrogen peroxide in water/methanol solutions. Inorg. Chem. 51, 5850–5856 (2012).
Talsi, E. P., Samsonenko, D. G., Ottenbacher, R. V. & Bryliakov, K. P. Highly enantioselective C−H oxidation of arylalkanes with H2O2 in the presence of chiral Mn-aminopyridine complexes. ChemCatChem 9, 4580–4586 (2017).
Burg, F., Gicquel, M., Breitenlechner, S., Pöthig, A. & Bach, T. Site- and enantioselective C−H oxygenation catalyzed by a chiral manganese porphyrin complex with a remote binding site. Angew. Chem. Int. Ed. 57, 2953–2957 (2018).
Abazid, A. H., Clamor, N. & Nachtsheim, B. J. An enantioconvergent benzylic hydroxylation using a chiral aryl iodide in a dual activation mode. ACS Catal. 10, 8042–8048 (2020).
Miyafuji, A. & Katsuki, T. Asymmetric desymmetrization of meso-tetrahydrofuran derivatives by highly enantiotopic selective C-H oxidation. Tetrahedron 54, 10339–10348 (1998).
Murahashi, S.-I., Noji, S., Hirabayashi, T. & Komiya, N. Manganese-catalyzed enantioselective oxidation of C–H bonds of alkanes and silyl ethers to optically active ketones. Tetrahedron. Asymmetry 16, 3527–3535 (2005).
Frost, J. R., Huber, S. M., Breitenlechner, S., Bannwarth, C. & Bach, T. Enantiotopos-selective C-H oxygenation catalyzed by a supramolecular ruthenium complex. Angew. Chem. Int. Ed. 54, 691–695 (2015).
Milan, M., Bietti, M. & Costas, M. Highly enantioselective oxidation of nonactivated aliphatic C–H bonds with hydrogen peroxide catalyzed by manganese complexes. ACS Cent. Sci. 3, 196–204 (2017).
Qiu, B. et al. Highly enantioselective oxidation of spirocyclic hydrocarbons by bioinspired manganese catalysts and hydrogen peroxide. ACS Catal. 8, 2479–2487 (2018).
Cianfanelli, M. et al. Enantioselective C–H lactonization of unactivated methylenes directed by carboxylic acids. J. Am. Chem. Soc. 142, 1584–1593 (2020).
Hashiguchi, S. et al. Kinetic resolution of racemic secondary alcohols by ruii-catalyzed hydrogen transfer. Angew. Chem. Int. Ed. 36, 288–290 (1997).
Jensen, D. R., Pugsley, J. S. & Sigman, M. S. Palladium-catalyzed enantioselective oxidations of alcohols using molecular oxygen. J. Am. Chem. Soc. 123, 7475–7476 (2001).
Ferreira, E. M. & Stoltz, B. M. The palladium-catalyzed oxidative kinetic resolution of secondary alcohols with molecular oxygen. J. Am. Chem. Soc. 123, 7725–7726 (2001).
Sun, W., Wang, H., Xia, C., Li, J. & Zhao, P. Chiral-Mn(Salen)-complex-catalyzed kinetic resolution of secondary alcohols in water. Angew. Chem. Int. Ed. 42, 1042–1044 (2003).
Radosevich, A. T., Musich, C. & Toste, F. D. Vanadium-catalyzed asymmetric oxidation of α-hydroxy esters using molecular oxygen as stoichiometric oxidant. J. Am. Chem. Soc. 127, 1090–1091 (2005).
Pawar, V. D., Bettigeri, S., Weng, S.-S., Kao, J.-Q. & Chen, C.-T. Highly enantioselective aerobic oxidation of α-hydroxyphosphonates catalyzed by chiral vanadyl(V) methoxides bearing N-salicylidene-α-aminocarboxylates. J. Am. Chem. Soc. 128, 6308–6309 (2006).
Arita, S., Koike, T., Kayaki, Y. & Ikariya, T. Aerobic oxidative kinetic resolution of racemic secondary alcohols with chiral bifunctional amido complexes. Angew. Chem. Int. Ed. 47, 2447–2449 (2008).
Murakami, K. et al. Highly enantioselective organocatalytic oxidative kinetic resolution of secondary alcohols using chiral alkoxyamines as precatalysts: catalyst structure, active species, and substrate scope. J. Am. Chem. Soc. 136, 17591–17600 (2014).
Saito, K., Shibata, Y., Yamanaka, M. & Akiyama, T. Chiral phosphoric acid-catalyzed oxidative kinetic resolution of indolines based on transfer hydrogenation to imines. J. Am. Chem. Soc. 135, 11740–11743 (2013).
Lu, R., Cao, L., Guan, H. & Liu, L. Iron-catalyzed aerobic dehydrogenative kinetic resolution of cyclic secondary amines. J. Am. Chem. Soc. 141, 6318–6324 (2019).
Larrow, J. F. & Jacobsen, E. N. Kinetic resolution of 1,2-dihydronaphthalene oxide and related epoxides via asymmetric C-H hydroxylation. J. Am. Chem. Soc. 116, 12129–12130 (1994).
Sun, S., Ma, Y., Liu, Z. & Liu, L. Oxidative kinetic resolution of cyclic benzylic ethers. Angew. Chem. Int. Ed. 60, 176–180 (2021).
Berrisford, D. J., Bolm, C. & Sharpless, K. B. Ligand-accelerated catalysis. Angew. Chem. Int. Ed. 34, 1059–1070 (1995).
Satyanarayana, T., Abraham, S. & Kagan, H. B. Nonlinear effects in asymmetric catalysis. Angew. Chem. Int. Ed. 48, 456–494 (2009).
Nam, W., Lee, Y.-M. & Fukuzumi, S. Hydrogen atom transfer reactions of mononuclear nonheme metal–oxygen intermediates. Acc. Chem. Res. 51, 2014–2022 (2018).
Wang, Y.-F., Lonca, G. H. & Chiba, S. PhI(OAc)2-mediated radical trifluoromethylation of vinyl azides with Me3SiCF3. Angew. Chem. Int. Ed. 53, 1067–1071 (2014).
Feichtinger, D. & Plattner, D. A. Direct proof for O=Mn”(salen) complexes. Angew. Chem. Int. Ed. 36, 1718–1719 (1997).
Feichtinger, D. & Plattner, D. A. Probing the reactivity of oxomanganese−salen complexes: an electrospray tandem mass spectrometric study of highly reactive intermediates. Chem. Eur. J. 7, 591–599 (2001).
Bryliakov, K. P., Babushkin, D. E. & Talsi, E. P. 1H NMR and EPR spectroscopic monitoring of the reactive intermediates of (Salen)MnIII catalyzed olefin epoxidation. J. Mol. Catal. A Chem. 158, 19–35 (2000).
Lefebvre, C. et al. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 19, 17928–17936 (2017).
Acknowledgements
We gratefully acknowledge the National Science Foundation of China (92156008, 22161142016, 21971148) and Youth Interdiscipline Innovative Research Group of Shandong University (2020QNQT009). The scientific calculations in this paper have been done on the HPC Cloud Platform of Shandong University.
Author information
Authors and Affiliations
Contributions
P.Y. conducted the asymmetric oxygenation experiments and mechanistic studies; A.F. and R.Z. performed the DFT calculations; L.W. prepared the substrates; M.C. initially developed the reaction; L.L. designed the experiments and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ye, P., Feng, A., Wang, L. et al. Kinetic resolution of cyclic benzylic azides enabled by site- and enantioselective C(sp3)–H oxidation. Nat Commun 13, 1621 (2022). https://doi.org/10.1038/s41467-022-29319-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-29319-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
