
Abstract
Dopant segregation, frequently observed in ionic oxides, is useful for engineering materials and devices. However, due to the poor driving force for ion migration and/or the presence of substantial grain boundaries, dopants are mostly confined within a nanoscale region. Herein, we demonstrate that core–shell heterostructures are formed by oriented self-segregation using one-step thermal annealing of metal-doped hematite mesocrystals at relatively low temperatures in air. The sintering of highly ordered interfaces between the nanocrystal subunits inside the mesocrystal eliminates grain boundaries, leaving numerous oxygen vacancies in the bulk. This results in the efficient segregation of dopants (~90%) on the external surface, which forms their oxide overlayers. The optimized photoanode based on hematite mesocrystals with oxide overlayers containing Sn and Ti dopants realises high activity (~0.8 μmol min−1 cm−2) and selectivity (~90%) for photoelectrochemical H2O2 production, which provides a wide range of application for the proposed concept.
Similar content being viewed by others


Introduction
Ionic oxide heterostructures have attracted significant attention in diverse fields ranging from catalysis to (magneto) optoelectronics owing to their tunable optical, electrical, and magnetic properties by precisely controlling the concentration and location of elements1,2,3. These heterostructures are mostly fabricated by vacuum technologies, such as atomic layer deposition4 and chemical vapor deposition5. They rely on high-precision equipment and specialized precursor reagents, which limit their large-scale application for production in industries.
Dopant segregation is another approach. It usually occurs in ionic solids containing aliovalent dopant ions and is driven by elastic and/or electrostatic interactions (Fig. 1a)6,7,8. However, in many cases, only small amounts of dopants can reach the external surface of polycrystalline or nanocrystalline materials under elevated temperatures (i.e., 1300 °C for Sn-doped hematite9) owing to the limited ion migration that results from poor driving forces or grain boundaries (GBs) (Fig. 1b)10. Extrinsic or intrinsic defects (e.g., vacancies and interstitial atoms) in the crystals yield space charge regions that modify a local electrostatic potential, but often lead to inhomogeneous properties and inevitably degrade their performance11. Thus, it is challenging to build the heterostructures by dopant segregation. External segregation may be promoted by removing the GBs from doped materials and adding excess space charges; however, these actions are incompatible. The concept of mesocrystal (MC)12,13, which is an ordered assembly of nanocrystals via oriented attachment, provides a solution to this problem. We recently discovered that thermal treatment at relatively lower temperatures (e.g., 700 °C for hematite (α-Fe2O3) MCs) induces the sintering of an interface (i.e., GB elimination) and creates numerous interfacial oxygen vacancies (VO)14,15, which facilitate the oriented migration of dopant ions as well as photogenerated charges (Fig. 1b).

a Distribution of dopants and electrostatic potential (donor-doping case) in ionic oxides based on the space charge theory (Supplementary Fig. 1 for acceptor-doping case). Strong segregation of dopants in the space charge layers is induced to compensate the excess charges on the crystal surfaces and bulk GBs (Supplementary Fig. 2). See Supplementary Note 1 for more details on the space charge theory. b Schematic illustration of the dopant segregation in different types of ionic oxide crystals: (I) Crystal with numerous GBs: dopants tend to segregate at the surface and GBs. (II) Disordered nanocrystals: a small number of dopants tend to segregate on the surface due to the lack of driving force even at high temperatures. (III) Ordered nanocrystals with highly aligned interfaces: a large number of dopants segregate on the outer surface because of interface sintering (GB elimination), creating numerous interfacial VO and narrowing the space charge layer to drive the charge migration.
Charge transfer efficiency and catalytic activity of semiconducting materials are highly influenced by their bulk electronic and surface structures. For example, hematite MCs with thin rutile TiO2 overlayers exhibit excellent performance in photoelectrochemical (PEC) water oxidation to obtain O2 owing to the suppressed surface recombination of photogenerated electrons and holes14,15. H2O2, which is another product of the water oxidation, exceeds O2 owing to its usefulness as a green oxidant for industrial chemistry and environmental purification, as well as a clean energy source for fuel cells16,17. The PEC H2O2 production has been mostly realized by a two-electron pathway from water oxidation using BiVO4-based photoanodes18,19,20,21; however, these photoanodes are still unstable for practical use due to the dissolution of V5+ that arises from anodic photocorrosion22.
Herein, we present MC-based dopant segregation to easily and effectively modify the surface of hematite MCs for highly efficient and selective solar-driven H2O2 production. Hematite (α-Fe2O3) is naturally abundant with good stability and suitable bandgap (~2.1 eV) for sunlight absorption and has been extensively studied for solar-driven water oxidation to obtain O223,24. To the best of our knowledge, there are no reports on hematite-based photoanodes for water oxidation to obtain H2O2, probably due to the unfavorable surface properties required for H2O2 generation25,26,27. By controlling the type and concentration of the dopants, as well as the annealing conditions, the hierarchical structures and catalytic activities of hematite MCs can be rationally optimized.
Results
Structures of hematite-based MCs
The as-synthesized Sn, Ti-codoped Fe2O3 (SnTi–Fe2O3) MCs (Supplementary Fig. 3), which cause self-segregation during heating, exhibit uniform cuboid-like morphology (length = ~150 nm, width = ~110 nm, and height = ~90 nm) assembled with closely stacked nanocrystal subunits (~20 nm), as observed from the field-emission scanning electron microscopy (FE-SEM) (Supplementary Fig. 4) and transmission electron microscopy (TEM) images (Fig. 2a, Supplementary Fig. 5). A coherent crystal lattice of hematite (104) is seen with adjacent nanocrystals and intimate interfaces (Fig. 2b and Supplementary Fig. 5c), indicating that the as-synthesized particle is composed of crystallographically aligned nanocrystals. The specific structure of the MC is further featured by single crystal-like diffraction spots obtained from the adjacent nanocrystals (Supplementary Fig. 5d)28,29. After annealing it at 700 °C in air for 20 min, GBs in the bulk region almost disappeared, while disordered overlayers with a thickness of 1–7 nm were formed on the outer surface and inner mesopores were created inside the crystal (Fig. 2c, d). The interfacial sintering also creates numerous interfacial VO, which appear as deficient regions in the crystal (white dotted line in Fig. 2c), resulting in an exceedingly high carrier density (1020–1021 cm−3) and ultrathin space charge layers (Supplementary Note 2 and Supplementary Table 1)14,15. The energy-dispersive X-ray (EDX) maps (Fig. 2d) and corresponding EDX spectra (Supplementary Fig. 6) suggest that the concentrations of Ti and Sn are higher at the surface region than in the bulk. We obtained hematite MCs with dopant oxide overlayers (SnO2 or TiO2) by the same procedure (Supplementary Fig. 7), confirming the viability and validity of our approach.

a TEM and b HRTEM images of as-synthesized SnTi–Fe2O3 MCs. c HRTEM image of annealed SnTi–Fe2O3 MC. Insets indicate inverse fast Fourier transform (FT) images of the selected regions with the same colors as indicated by the dashed frames. d HAADF-STEM images and corresponding EDX chemical composition maps of annealed SnTi–Fe2O3 MCs. e HAADF-STEM image (left) and corresponding EELS composition maps of Ti (451.7–469.7 eV) (middle) and Sn (507.5–525.5 eV) (right) signals of a typical annealed SnTi–Fe2O3 MC. f XPS depth analysis of the as-synthesized and annealed SnTi–Fe2O3 samples. g Sn 3d XPS spectra of annealed SnTi–Fe2O3 and Sn–Fe2O3 MCs. h Ex situ Sn K-edge XANES spectra of the annealed Sn-containing samples measured in CEY mode. i The corresponding Sn K-edge FT-EXAFS spectra of the samples.
We then employed high-angle annular dark-field scanning TEM (HAADF-STEM) combined with electron energy loss spectroscopy (EELS) to derive information about the local compositions, structures, and chemical states of the annealed SnTi–Fe2O3 MCs. Ti and Sn species are present with the same spatial distribution at the outer surface and near the edge of pores (as specified by the white dotted circles) (Fig. 2e), implying a single phase of the Sn–Ti complex. This speculation was partly supported by the fact that aligned high-loss EEL spectra obtained from the surface region show much higher peak intensities of Ti–L2,3 and Sn–M4,5 signals (Supplementary Fig. 8). The thermal accelerated dopant segregation at the crystal surface was further suggested by the significantly increased concentrations of the dopants at the surface region based on the X-ray photoelectron spectroscopy (XPS) depth profiles (Fig. 2f). In addition, the XPS analysis revealed that the valance state of Sn ions for the annealed SnTi–Fe2O3 is lower than that (Sn4+) of the annealed Sn–Fe2O3 (Fig. 2g). This was further confirmed by the Sn K-edge X-ray absorption near edge structure (XANES) spectrum of the annealed SnTi–Fe2O3 measured in a conversion electron yield (CEY) mode, an effective method to analyze the surface compositions of materials, which indicates a clear shift to lower energy compared with Sn–Fe2O3 and SnO2 (Fig. 2i). The corresponding Sn K-edge extended X-ray absorption fine structure FT spectrum of SnTi–Fe2O3 exhibits the main first shell of Sn–O at 1.38 Å and second Sn–Ti shell at 2.76 Å30,31, in addition to the weak coordination of Sn–Sn as those of SnO2 and Sn–Fe2O3 at ~3.28 Å. The above results indicate the formation of SnTiOx hetero-overlayer with the possibility of a small amount of SnO2 at the outer surface, as suggested by the Sn 3d XPS depth profile analysis (Supplementary Fig. 8d).
Heterostructures formed by dopant segregation
All doped hematite MCs are composed of a hematite phase, as deduced from the powder X-ray diffraction (XRD) patterns (Supplementary Fig. 9). However, owing to the lattice expansion via the replacement of smaller Fe3+ ions (0.55 Å) with larger Sn2+ (0.999 Å) and/or Ti4+ (0.605 Å) ions32,33, the peak positions of the hematite diffraction lines shifted towards smaller angles (solid lines in Fig. 3a). The corresponding lattice d(104) space values increased from 2.709 to 2.730 Å with an increase in total dopant concentration from 0 to 30 mol% (Supplementary Fig. 10), confirming the uniform incorporation of Sn and/or Ti ions in the hematite lattice owing to the replacement with Fe3+ ions34. After thermal treatment, the diffraction peak positions (dashed lines in Fig. 3a) and d values of the doped samples exhibited the same level of undoped Fe2O3 (Fig. 3b), suggesting that most of the doped Sn2+ and/or Ti4+ ions (~90%) were diffused out from the hematite lattice. We observed a broad diffraction peak located at 27.0° (and 26.5°) for the annealed Ti–Fe2O3 (and Sn–Fe2O3) sample (Fig. 3c), supporting the formation of the (110) phase of the TiO2 (and SnO2) overlayer. For the annealed SnTi–Fe2O3 sample, no clear diffraction peak was detected, probably due to the disordered structure of the overlayer, as indicated by the HRTEM image (Fig. 2c).

a Powder XRD patterns of the samples measured for as-synthesized (solid lines) and annealed (dashed lines) samples with a scanning rate of 10° min−1. b Lattice spacing d104 values of the samples before and after annealing. c Powder XRD patterns of annealed samples measured with a scanning rate of 1.0° min−1. d PDF analyses of the samples. The peak at approximately 2 Å is composed of shorter Fe–O (1.94 Å) and longer Fe–O (2.12 Å) distances. The peak at approximately 3 Å is composed of the first neighbor edge-sharing (Fe–Fee, 2.95 Å) and face-sharing (Fe–Fef, 2.90 Å) Fe–Fe distances. The first and second neighbor corner-sharing Fe–Fe pairs are at 3.39 Å (Fe–Fec1) and 3.72 Å (Fe–Fec2), respectively62. e Schematic illustration of the driving force for oriented dopant segregation in hematite during thermal treatment. The self-segregation is driven by the space charges induced by the dopants and VO, RF between the dopant cations and positively charged VO, and EE induced by the size mismatch between the dopant and the host cations.
The synchrotron-based X-ray total scattering measurements and pair distribution function (PDF) analysis are powerful methods to characterize disordered or amorphous structures35. The as-synthesized SnTi–Fe2O3 sample shows a peak shift, along with the broadening of the peaks, towards a larger interatomic distance (r) compared to the undoped Fe2O3 (Fig. 3d and Supplementary Fig. 11), suggesting an expansion of the hematite lattice by the replacement of Fe3+ sites with larger-sized Sn2+ and Ti4+ ions. The thermal treatment leads to a lattice rearrangement in the bulk as indicated by the peak shifts toward the r values of the undoped Fe2O3. Considering that the length of corner-sharing Ti–Ti (and Sn–Sn) bond of rutile TiO2 (and SnO2) is 3.55 Å36 (and 3.68 Å37) along with the corresponding PDFs (Supplementary Fig. 12), an increase in amplitude at 3.5–3.7 Å implies the formation of the dopant oxide overlayers. A new peak with a Q value of 1.872 Å−1 (1.916 Å−1) for the annealed Sn–Fe2O3 (Ti–Fe2O3) is assigned to SnO2 (rutile TiO2) (Supplementary Fig. 12)37,38. Meanwhile, only a weak and broad scattering peak located between that of SnO2 and TiO2 was detected for the annealed SnTi–Fe2O3. This result suggests the formation of SnTiOx phases, not the simple mixture state of TiO2 and SnO2.
Based on these results, we propose dopant segregation, as illustrated in Fig. 3e. Owing to the sintering of nanocrystal interfaces (GB elimination) by annealing, interfacial VO with a positive charge(s) (i.e., \({{{{{{\rm{V}}}}}}}_{{{{{{\rm{o}}}}}}}^{{{{{{\rm{\bullet }}}}}}{{{{{\rm{\bullet }}}}}}}\) in the Kröger–Vink notation) are formed, which can create the space charge regions (Supplementary Fig. 13 and Supplementary Table 1) and yield a repulsive force against the dopant cations. This effectively drives the oriented self-segregation of larger-sized dopants to the external surface in addition to the elastic energy (EE) induced by the size mismatch between the dopant and host ions without any significant dopant accumulation in the GB regions (see Supplementary Note 1 for details).
Formation of composite oxide overlayers
To elucidate the local structures of the overlayers, X-ray absorption spectroscopy (XAS) at Fe, Ti, and Sn K-edges was performed. The normalized Fe K-edge XANES spectra (Supplementary Fig. 14) of all the hematite-based samples are close to the standard α-Fe2O3, indicating that the doping and annealing treatments have negligibly affected the valence and coordination states of the iron cation. The normalized Ti K-edge XANES spectra of the Ti-containing samples are shown in Fig. 4a. For transition metals in oxides, the threshold energy position of the spectra is very sensitive to their oxidation states, while the shapes of the peaks give information about the local structural environments of the absorbing elements39. The peak shapes of the annealed Ti–Fe2O3 MCs in pre- and post-edge regions are similar to those of rutile TiO2, but the line position is located between those of rutile and FeTiO3. These results are consistent with the fact that the rutile overlayer with the FeTiOx intermediate layer is seen in the HRTEM image (Supplementary Fig. 7). For the annealed SnTi–Fe2O3 sample, the characteristic peak of rutile in the pre-edge (indicated by the black arrow) did not appear. Moreover, the peak shape and position are different from those of reference TiO2 and annealed Ti–Fe2O3, indicating that no rutile TiO2 phase was formed as the main product in this sample. The annealed SnTi–Fe2O3 sample also has a similar strongest absorption peak with FeTiO3 at the post-edge region; thus, the oxidation state of Ti ions is similar to that of FeTiO3. Besides, a much stronger first post-edge peak may indicate the Sn–Ti coordination. This was further supported by the comparison of the ex situ Ti K-edge XAS profile measured in transmission and CEY modes (Supplementary Fig. 15).

a Ex situ Ti K-edge XANES spectra of annealed Ti-containing samples and reference samples. b In situ Ti K-edge XANES spectra of as-synthesized SnTi–Fe2O3 measured with the similar heating procedure used for electrodes preparation. The spectra are shifted along the y-axis for the sake of better clarity. c In situ FT-EXAFS spectra of as-synthesized SnTi–Fe2O3 measured with the similar heating procedure used for electrodes preparation. d Schematic illustration of the binary dopant segregation enabling the heterostructures during thermal treatment of as-synthesized SnTi–Fe2O3 MC.
Figure 4b depicts in situ Ti K-edge XANES spectra of as-synthesized SnTi–Fe2O3 MCs. When the temperature increased from 40 to 700 °C, a negligible change in the peak shape and position was observed. No characteristic peak of the rutile phase was observed during heating, whereas the two post-edge peaks shifted to the higher energy and became smooth with an increase in the first post-edge peak at 700 °C due to the formation of binary Sn–Ti oxides. For the Ti–Fe2O3 sample, the rutile phase was formed at 700 °C (Supplementary Fig. 16).
Ex and in situ Sn K-edge FT-EXAFS measurements enhanced the investigation of the dynamics of dopant segregation (Figs. 2i and 4c, and Supplementary Figs. 17 and 18). The peak position of the Sn–Sn bond for annealed Sn–Fe2O3 sample is close to that of the reference SnO2 (Fig. 2i and Supplementary Fig. 17), proving the formation of SnO2 overlayers. For the annealed SnTi–Fe2O3 sample, the second main shell has a shorter radial distance than that of SnO2 owing to the formation of Sn–Ti coordination (i.e., SnTiOx)30,31. The peak intensity of the Sn–Fe coordination shell gradually decreases as the temperature increases from 48 to 700 °C (Fig. 4c) because the Sn-coordination becomes disordered due to the diffusion of Sn ions from the hematite lattice. A second Sn–Sn coordination peak is formed and grows when the temperature rises to ~450 °C (Supplementary Fig. 19). Meanwhile, the Sn–Ti coordination peak appears at 700 °C and becomes stronger during the heat preservation and cooling stages, which agrees with the Ti K-edge XANES result (Fig. 4b). The growth of the Sn–Sn and Sn–Ti coordination peaks during the cooling stage indicates ordered Sn-coordinations by suppressed oscillation or diffusion of the elements at lower temperatures. For Sn–Fe2O3 sample, the SnO2 phase is formed, as indicated by the increase in the Sn–Sn coordination at ~550 °C (Supplementary Fig. 18). Based on these results, it is concluded that Sn2+ ions migrate from the hematite lattice before Ti4+ ions owing to their larger radius (i.e., larger EE), and segregate on the surface to form the SnO2 (or SnO) phase at ~450 °C. The deficient SnTiOx overlayers are then formed when Ti4+ ions segregate at the surface and react with SnO2 at 700 °C (Fig. 4d). In addition, a very small amount of SnO2 remained at the outer surface during the annealing treatment at 700 °C.
Performance of PEC H2O2 synthesis
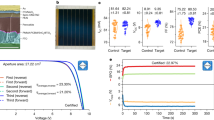
Next, we demonstrate an outstanding ability of the hematite-based heterostructure as a photocatalyst for PEC H2O2 synthesis (Fig. 5a). Figure 5b shows the current density–voltage curves of photoanodes, which were prepared by spin-coating a suspension of as-synthesized MCs followed by the same thermal treatment as mentioned earlier, operated in the dark or under simulated sunlight illumination. The photocurrent density obtained for the optimized SnTi–Fe2O3 photoanode at 1.23 V vs. RHE was approximately 1.1 mA cm−2, which is much higher than those of Fe2O3 (0.37 mA cm−2), Ti–Fe2O3 (0.83 mA cm−2), and Sn–Fe2O3 (0.23 mA cm−2). This improved performance may result from the reduced electron transfer resistances in the bulk and at the interfaces of hematite/FTO and hematite/electrolyte by the surface passivation with overlayers and n-type conductivity arising from the interfacial VO in addition to interparticle sintering and necking14,15, as suggested by the electrochemical impedance spectroscopy (EIS) (Supplementary Fig. 20) and Mott–Schottky plots (Supplementary Fig. 13 and Supplementary Table 1).

a Illustration of PEC water splitting system using a hematite-based photoanode. b Current density–voltage curves of Fe2O3, Sn–Fe2O3, Ti–Fe2O3, and SnTi–Fe2O3 photoanodes in 1.0 M NaHCO3 under dark and back illumination with AM 1.5 G simulated sunlight. c Amounts of H2O2 generated from the photoanodes at 1.23 V vs. RHE with an increase in illumination time. The error bars represent the standard deviation. d The photocurrent densities and the FEs of H2O2 obtained by different electrodes with back illumination at 1.23 V vs. RHE. The error bars represent the standard deviation. e Structural models of hematite (110) (left) and SnO2−x (110) (right) for DFT calculations. f Activity volcano plots based on calculated limiting potentials as a function of ΔGOH*. The calculated values are summarized in Supplementary Table 3.
We quantitatively analyzed the H2O2 generation by the N,N-diethyl-1,4-phenylenediamine (DPD) method (Supplementary Fig. 21)40. The number of H2O2 produced from the SnTi-Fe2O3 photoanode increased almost linearly with the illumination time, which is much more active than Ti–Fe2O3, Sn–Fe2O3, and Fe2O3 (Fig. 5c). No H2O2 was generated without light irradiation or from the Pt cathode under the operating conditions (Supplementary Fig. 22a). The Sn–Fe2O3 photoanode (i.e., the SnO2 overlayer) showed a much lower photocurrent but a higher Faradaic efficiency (FE) of H2O2, while the Ti–Fe2O3 photoanode (i.e., the TiO2 overlayer) exhibited an opposite trend (Fig. 5d). No gaseous oxygen was detected by the gas chromatography (GC) analysis, possibly due to the low concentration of evolved O2, whereas the amount of H2 (with FE over 90%) linearly increased with the illumination time (Supplementary Fig. 22b, c).
Furthermore, the co-doping of Sn and Ti significantly enhanced the generation of both photocurrent (i.e., H2) without any notable decrease over 90 min and H2O2 with high FEs (>90%) in the range of 1.0–1.8 V vs. RHE (Supplementary Fig. 23). The optimized SnTi–Fe2O3 photoanode realized an H2O2 generation rate of ~0.8 μmol min−1 cm−2, which is comparable to those of the active BiVO4-based photoanodes (Supplementary Fig. 24). To the best of our knowledge, this is the first example of a hematite-based PEC water-splitting system that achieves a high FE for H2O2 generation with a good H2/H2O2 co-production capability. At higher voltages (>1.8 V vs. RHE), dark currents are more significant owing to the EC oxidation of water for obtaining O2 (Fig. 5b)20, leading to the decreased FE of H2O2 down to ~65% (Supplementary Fig. 23c). Here, we notice that SnTi–Fe2O3 photoanode exhibits the lowest activity for EC oxidation among the doped samples, implying that the hetero-overlayer has a specific structure suitable for H2O2 generation.
DFT calculations of adsorption energies
To identify reaction active sites, we calculated the adsorption energies of relevant intermediates O* and OH* for various structures, including pure hematite (Fig. 5e), using density functional theory (DFT) based on the computational hydrogen electrode model41,42. We then calculated free energy changes of OH* and O* (ΔGOH* and ΔGO*) to construct activity volcano plots for two-electron water oxidation to H2O2.
As shown in Fig. 5f, bare hematite (110) surface is not suitable for H2O2 evolution, but it is for O2 evolution (Supplementary Figs. 25 and 26, Supplementary Table 2, and Supplementary Note 3), judging from the calculated ΔGOH* (1.33 eV) and ΔGO* (3.45 eV), which are deviated from the preferred range for H2O2 evolution (1.6 eV < ΔGOH* < 2.4 eV, ΔGO* < 3.5 eV)19,43. We also point out that a simple doping treatment cannot improve the reaction selectivity, according to the fact that the ΔGOH* values calculated for two local structures, Sn4+-doped TiO2 (Sn-TiO2) and Ti4+-doped SnO2 (Ti–SnO2), where the dopants are considered as the surface active sites, are comparable to those of SnO2 and TiO2, respectively (Fig. 5f, Supplementary Fig. 27, and Supplementary Table 3).
Recently, Diehl et al. reported an ilmenite-type SnTiO3 structure where each Sn2+ possesses a lone pair, forming layers separated by a van der Waals gap44,45. This finding inspired us to explore local structures of SnTiOx overlayers, but our calculations revealed that the ideal SnTiO3 (0001) surface has a ΔGOH* of 1.24 eV (ΔGO* of 3.04 eV), which is not suitable for H2O2 evolution (Fig. 5f, Supplementary Fig. 28, and Supplementary Table 3). They also reported that an oxidized passivation layer (~2 nm thickness) that resembles SnO2 formed at the top surface of SnTiO3. We thus modeled various rutile SnO2 structures possessing VO, where subsurface VO mimics the Sn2+ support from SnTiO3 and examined their possibility as a catalytic site on the annealed SnTi-Fe2O3. As demonstrated in Fig. 5e and f, when VO was introduced near the surface of SnO2 (site #1), the OH adsorption was significantly enhanced, leading to a poor H2O2 evolution activity. The two electrons are left behind when a neutral VO is formed and these electrons are delocalized over neighboring Sn4+ 46,47. An increase in electron density on the Sn ions alters their coordination from 6- to 5-fold coordination. This local under-coordination increases the electronegativity of Sn and reduces the reorganization energy required to distort Sn–O bonds for OH adsorption. Meanwhile, when VO is present in deeper positions, the ΔGOH* value shifts toward the volcano peak where the catalyst is optimal for H2O2 production according to Sabatier’s principle (Fig. 5e, f, Supplementary Fig. 29, and Supplementary Table 4)48. We also found a similar tendency for rutile-type Sn0.5Ti0.5O2−x with VO (Supplementary Fig. 30 and Supplementary Table 5) 30.
Discussion
The activity of heterogeneous photocatalysts is strongly influenced by their ability to exhibit chemisorption on reactants and intermediates. Since the surface Sn2+ ions are probably oxidized to Sn4+ during the annealing treatment in the air (Supplementary Fig. 8d), partially amorphized SnO2 (or Sn0.5Ti0.5O2) (below 2 nm thickness) could form at the outer surface of disordered SnTiOx overlayers. Such a heterostructure could be realized by successive binary dopant segregation through nanoparticle networks in the MCs (Fig. 4d). Among the structures utilized in the DFT calculations, the prospective ones are SnO2−x or Sn0.5Ti0.5O2−x with VO at depths of 1.2–1.7 nm (e.g., site #5 in Fig. 5e, f, and Supplementary Fig. 29), which are structurally analogous to the Sn4+ species on the disordered SnTiOx overlayers. Our calculations further point out the importance of the VO location in controlling the selectivity of the water oxidation reaction (Fig. 5f). It was reported that surface VO can lower the H2O2 evolution activity by promoting water dissociation to form intermediates for O2 evolution on BiVO449, but the VO-position dependence of ΔGOH* has been overlooked so far.
For PEC H2O2 synthesis, several mechanisms have been proposed (Supplementary Note 4)50. In particular, the presence of HCO3− is significant to accelerate the water oxidation to H2O2. Baek et al. also demonstrated that the tuning of ΔGOH* by doping of Gd ions to BiVO4 significantly improves the activity and selectivity of PEC H2O2 synthesis in 2 M KHCO3 electrolyte. In our system, the oxidized layers supported on SnTiOx serve as a bifunctional catalyst to adequately adsorb water molecules (i.e., OH*) and facilitate HCO3−-mediated interfacial transfer51 of photo-holes from excited hematite core for efficient H2O2 evolution. In the future, detailed analyses of reaction intermediates using operando spectroscopic methods will be important to refine the mechanism of H2O2 formation.
In summary, we developed MC-based binary dopant segregation to construct heterostructures with preferential properties for solar H2O2 synthesis. The oxidized surface Sn4+ species on the disordered SnTiOx overlayers are potential active sites for efficient H2O2 generation. The composite overlayers on the hematite can be modified to further improve the PEC performance for practical use and fit specific other sustainable reactions like CO2 reduction52. Moreover, other types of overlayers, such as nitrides and hydrides53,54, may be fabricated by varying the synthesis conditions (e.g., annealing in N2 or H2 atmosphere) for emerging functionalities.
Methods
Synthesis of hematite-based MCs
The hematite MCs containing Sn2+ (6.5 mol%) and Ti4+ (6.5 mol%) dopants (SnTi–Fe2O3) were synthesized via a simple surfactant-free solvothermal method (Supplementary Fig. 3)14. Briefly, a mixed metal precursor of 1 mmol of Fe(NO3)3·9H2O (FUJIFILM Wako Pure Chemical, 99.9%), 0.075 mmol of TiF4 (Sigma-Aldrich), and 0.075 mmol of SnCl2 (FUJIFILM Wako Pure Chemical, 97.0 + %) were dissolved in a mixed solvent of 40 mL of N,N-dimethylformamide (FUJIFILM Wako Pure Chemical, 99.0 + %) and 10 mL of methanol (FUJIFILM Wako Pure Chemical, 99.8 + %). The above solution was then treated at 180 °C for 24 h in a 100 mL Teflon-lined autoclave reactor. After naturally cooling, the resulting solid product was thoroughly washed with water and ethanol and dried at 60 °C (8 h). The hematite MCs with individual dopants of Sn2+ (Sn–Fe2O3) and Ti4+ (Ti–Fe2O3), and undoped hematite (Fe2O3) MCs were synthesized via the same method by varying the dopant precursors. The doping levels of Sn2+ and Ti4+ were controlled by varying the amounts of Sn2+ and Ti4+ in the precursor solution.
Fabrication of hematite MC-based photoanodes
The hematite MC-based films were prepared by multiple spin-coating (3000 rpm) of an ethanol solution containing the highly dispersed hematite-based MCs (10 mg mL−1) on a piece of cleaned fluorine-doped tin oxide (FTO)-coated glass (2.5 × 1.7 cm). To obtain the stable films, the above-prepared electrodes were annealed in a furnace at 700 °C for 20 min in the air with a heating rate of 20 °C min−1 and collected for further use after naturally cooling. This annealing treatment is also a key step for the formation of dopant oxide overlayers by accelerating the dopant segregation to the hematite surface.
Characterizations
Atomic force microscopy (AFM) and Kelvin probe force microscopy (KPFM) measurements were performed on a Dimension Icon (Bruker) using a silicon nitride probe (Bruker, PFQNE-AL). FE-SEM observations were performed on JSM-7100F (JEOL). TEM observations were performed on JEM-2100F (JEOL) operated at 200 kV. EDX mapping and HAADF-STEM images combined with EELS analysis were collected on a JEM-ARM200F Cold FEG (JEOL) microscope operated at 200 kV. The powder XRD patterns were recorded on a Rigaku Ultima IV diffractometer with Cu Kα radiation (λ = 1.5418 Å) at a voltage of 40 kV and a current of 40 mA. The XPS measurements were performed on a PHI X-tool (ULVAC-PHI). The spectra were calibrated by the reference of the C1s peak at 284.8 eV. Synchrotron-based X-ray total scattering measurements with PDF analysis were performed with the incident X-ray energy of 61.4 keV at BL04B2 beamline in SPring-8, Japan. The data were collected using the hybrid detectors of Ge and CdTe. The reduced PDF G(r) was obtained by the conventional Fourier transform of the Faber–Ziman structure factor S(Q)55 extracted from the collected date56. Ex and in situ X-ray absorption spectral measurements were performed at BL01B1 beamline in SPring-8, Japan. The samples were prepared by pelletizing the uniform mixtures of hematite-based MCs or reference samples with dehydrated boron nitride powders. The collected data were processed using the IFEFFIT software package. The filtered k3 weighted χ spectra were Fourier transformed into r space (k range: 3.0–16.0 Å−1 for Fe K, 1.0–6.8 Å−1 for Ti K, and 1.0–9.0 Å−1 for Sn K).
PEC measurements
The PEC measurements were conducted in a typical three-electrode system in 1.0 M NaHCO3 aqueous solution (pH = 8.3) with the fabricated hematite MC-based film as working electrode, Pt wire as the counter electrode, and Ag/AgCl electrode with saturated KCl solution as the reference electrode. The reactors with photoanode and cathode were separated with a membrane film (Sigma-Aldrich, Nafion® 117). All the electrochemical data were recorded and analyzed on an electrochemical workstation (ALS, model 608E). A xenon light source (Asahi Spectra, LAX-C100) equipped with an AM 1.5 filter with the light intensity of 100 mW cm−2 (calibrated by a silicon photodiode detector (Asahi Spectra, CS-30)) was used to irradiate the photoanodes from the backside (through the FTO glass) with a working area of 0.72 cm2. The current–voltage curves were obtained by cyclic voltammetry at a scan rate of 20 mV s−1. All the applied potentials have been converted into the potential vs. RHE via the Nernst equation (ERHE = EAg/AgCl + 0.059 × pH + E°Ag/AgCl). The EIS measurements were performed at 1.23 V vs. RHE with frequencies between 0.5 Hz and 10 kHz under back illumination with the simulated sunlight. The Mott–Schottky plots were measured in the dark at a frequency of 10 kHz.
Product analysis
The evolved gas from the PEC cell was analyzed by a GC (Shimadzu, GC-8A) equipped with an MS-5A column and a thermal conductivity detector. To remove air in the reactor, the electrolyte solutions in well-sealed working and counter electrode cells that were separated by the Nafion membrane were bubbled with Ar gas for at least 30 min before the measurements. To minimize the influence of air leaks, the analysis of gas products was carried out independently using the same electrode under the same conditions. A relatively large amount of gas sample (50 μL) from the working electrode cell was measured every time for O2 detection. 20 μL of gas sample from the counter electrode cell was collected and measured for H2 analysis. The GC was calibrated by injecting exact volumes of pure O2 and H2 gases (Supplementary Fig. 32). H2O2 evolution was evaluated using N,N-diethyl-1,4-phenylenediamine (DPD) sulfate. Typically, 200 μL sample aliquots collected with a syringe during irradiation were mixed with 200 μL potassium phosphate buffer solution (pH = 7), 2 mL water, 20 μL DPD solution (0.1 g DPD in a 10 mL 0.05 M H2SO4 solution), and 20 μL freshly prepared peroxidase (POD) solution (3 mg POD in 3 mL deionized water), and the mixtures were shaken for 120 s. The above solutions were analyzed by UV-vis spectroscopy (JASCO, V-700). The H2O2 concentrations were calibrated with standard H2O2 solutions. The concentration of dissolved O2 in the electrolyte was evaluated using a free radical analyzer (World Precision Instruments, TBR4100), which is based on an electrochemical (amperometric) detection, with an ISO-OXY-2 oxygen sensor (World Precision Instruments). The detected currents were converted to O2 concentrations using the obtained calibration curve.
DFT calculations
All the calculations reported in this work were carried out using the revised version of Perdew-Burke-Ernzerhof exchange-correlation functional57 and the projector-augmented wave method as implemented in Vienna Ab Initio Simulation Package58,59. The spin-polarization effect and dipole correction were both taken into account. Convergence was considered to be achieved when the energy difference was less than 10−6 eV and the force was less than 10−2 eV per Å for geometry optimization. The (110) slab model of Fe2O3 was prepared by first performing the geometry optimization of the anti-ferromagnetic bulk system with a cut-off energy of 520 eV and Monkhorst–Pack k-point meshes of 4 × 4 × 4 to determine the lattice constants, and then by using 4 Fe-layers with a vacuum of 15 Å and 4 × 4 × 1 Monkhorst–Pack k-points. For the calculations of OH* and O*, several coverage patterns have been considered, from 1/12 to 0.5, corresponding to 1 and 6 adsorbates per cell. For Fe2O3, a Hubbard U value of 4.3 eV was employed60. The computational hydrogen electrode was employed, for which the Gibbs free energy of 1/2 H2 ⇌ H+ + e− is set to zero. The free energy change was computed as ΔG = ΔE + ΔZPE − TΔS, where previously reported values are used for the zero-point energy and entropy to compute ΔZPE − TΔS at standard conditions61, since they are known to be very similar between different oxides. For the calculations of SnO2−x, the same procedure as Fe2O3 was employed, except that the cut-off energy used was 400 eV and the (110) slab model consisted of 1 × 2 super-cell with 8 Sn-layers. The adsorption energies were computed with one adsorbate for each cell. The VO dependence of the volcano plot was investigated with changing vacancy sites (Supplementary Fig. 28).
Data availability
Source data are provided with this paper.
References
Vurgaftman, I., Meyer, J. R. & Ram-Mohan, L. R. Band parameters for III–V compound semiconductors and their alloys. J. Appl. Phys. 89, 5815–5875 (2001).
Ahn, C. H. Ferroelectricity at the nanoscale: local polarization in oxide thin films and heterostructures. Science 303, 488–491 (2004).
Zhou, P., Yu, J. & Jaroniec, M. All-solid-state Z-scheme photocatalytic systems. Adv. Mater. 26, 4920–4935 (2014).
Marichy, C., Bechelany, M. & Pinna, N. Atomic layer deposition of nanostructured materials for energy and environmental applications. Adv. Mater. 24, 1017–1032 (2012).
Gong, Y. et al. Vertical and in-plane heterostructures from WS2/MoS2 monolayers. Nat. Mater. 13, 1135–1142 (2014).
Kliewer, K. L. & Koehler, J. S. Space charge in ionic crystals. I. General approach with application to NaCl. Phys. Rev. 140, A1226–A1240 (1965).
Chung, S.-Y., Choi, S.-Y., Yoon, H.-I., Kim, H.-S. & Bae, H. B. Subsurface space-charge dopant segregation to compensate surface excess charge in a perovskite oxide. Angew. Chem. Int. Ed. 55, 9680–9684 (2016).
Yoon, H. I. et al. Probing dopant segregation in distinct cation sites at perovskite oxide polycrystal interfaces. Nat. Commun. 8, 1417 (2017).
Soares, M. R. S. et al. Unraveling the role of Sn segregation in the electronic transport of polycrystalline hematite: raising the electronic conductivity by lowering the grain‐boundary blocking effect. Adv. Electron. Mater. 5, 1900065 (2019).
Hwang, S.-L. & Chen, I.-W. Grain size control of tetragonal zirconia polycrystals using the space charge concept. J. Am. Ceram. Soc. 73, 3269–3277 (1990).
Li, W., Shi, J., Zhang, K. H. L. & Macmanus-Driscoll, J. L. Defects in complex oxide thin films for electronics and energy applications: challenges and opportunities. Mater. Horiz. 7, 2832–2859 (2020).
Cölfen, H. & Antonietti, M. Mesocrystals: inorganic superstructures made by highly parallel crystallization and controlled alignment. Angew. Chem. Int. Ed. 44, 5576–5591 (2005).
Tachikawa, T. & Majima, T. Metal oxide mesocrystals with tailored structures and properties for energy conversion and storage applications. NPG Asia Mater. 6, 1–11 (2014).
Zhang, Z. et al. Interfacial oxygen vacancies yielding long-lived holes in hematite mesocrystal-based photoanodes. Nat. Commun. 10, 4832 (2019).
Zhang, Z., Nagashima, H. & Tachikawa, T. Ultra-narrow depletion layers in a hematite mesocrystal-based photoanode for boosting multihole water oxidation. Angew. Chem. Int. Ed. 59, 9047–9054 (2020).
Campos-Martin, J. M., Blanco-Brieva, G. & Fierro, J. L. G. Hydrogen peroxide synthesis: an outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 45, 6962–6984 (2006).
Sayama, K. Production of high-value-added chemicals on oxide semiconductor photoanodes under visible light for solar chemical-conversion processes. ACS Energy Lett. 3, 1093–1101 (2018).
Fuku, K. & Sayama, K. Efficient oxidative hydrogen peroxide production and accumulation in photoelectrochemical water splitting using a tungsten trioxide/bismuth vanadate photoanode. Chem. Commun. 52, 5406–5409 (2016).
Shi, X. et al. Understanding activity trends in electrochemical water oxidation to form hydrogen peroxide. Nat. Commun. 8, 701 (2017).
Baek, J. H. et al. Selective and efficient Gd-doped BiVO4 photoanode for two-electron water oxidation to H2O2. ACS Energy Lett. 4, 720–728 (2019).
Zhang, K. et al. Near-complete suppression of oxygen evolution for photoelectrochemical H2O oxidative H2O2 synthesis. J. Am. Chem. Soc. 142, 8641–8648 (2020).
Hermans, Y., Murcia-López, S., Klein, A. & Jaegermann, W. BiVO4 surface reduction upon water exposure. ACS Energy Lett. 4, 2522–2528 (2019).
Sivula, K., Le Formal, F. & Grätzel, M. Solar water splitting: progress using hematite (α-Fe2O3) photoelectrodes. ChemSusChem 4, 432–449 (2011).
Shen, S., Lindley, S. A., Chen, X. & Zhang, J. Z. Hematite heterostructures for photoelectrochemical water splitting: rational materials design and charge carrier dynamics. Energy Environ. Sci. 9, 2744–2775 (2016).
Kormann, C., Bahnemann, D. W. & Hoffmann, M. R. Environmental photochemistry: is iron oxide (hematite) an active photocatalyst? A comparative study: α-Fe2O3, ZnO, TiO2. J. Photochem. Photobiol. A Chem. 48, 161–169 (1989).
Dotan, H., Sivula, K., Grätzel, M., Rothschild, A. & Warren, S. C. Probing the photoelectrochemical properties of hematite (α-Fe2O3) electrodes using hydrogen peroxide as a hole scavenger. Energy Environ. Sci. 4, 958–964 (2011).
Avital, Y. Y. et al. Two-site H2O2 photo-oxidation on haematite photoanodes. Nat. Commun. 9, 4060 (2018).
Bian, Z., Tachikawa, T., Zhang, P., Fujitsuka, M. & Majima, T. A nanocomposite superstructure of metal oxides with effective charge transfer interfaces. Nat. Commun. 5, 3038 (2014).
Zhang, P. et al. Topotactic epitaxy of SrTiO3 mesocrystal superstructures with anisotropic construction for efficient overall water splitting. Angew. Chem. Int. Ed. 56, 5299–5303 (2017).
Chen, Y. C. et al. Rutile-type (Ti,Sn)O2 nanorods as efficient anode materials toward its lithium storage capabilities. Nanoscale 5, 2254–2258 (2013).
Cao, N. et al. Electrochemical nitrogen fixation via bimetallic Sn-Ti sites on defective titanium oxide catalysts. J. Colloid Interface Sci. 588, 242–247 (2021).
Shannon, R. D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 32, 751–767 (1976).
Baloch, A. A. B. et al. Extending Shannon’s ionic radii database using machine learning. Phys. Rev. Mater. 5, 043804 (2021).
Wang, Y., Yang, Y., Zhang, X., Liu, X. & Nakamura, A. Optical investigation on cadmium-doped zinc oxide nanoparticles synthesized by using a sonochemical method. CrystEngComm 14, 240–245 (2012).
Bordet, P. Application of the pair distribution function analysis for the study of cultural heritage materials. Comptes Rendus Phys. 19, 561–574 (2018).
Jiang, Z. et al. Filling metal–organic framework mesopores with TiO2 for CO2 photoreduction. Nature 586, 549–554 (2020).
Jensen, K. M. Ø. et al. Revealing the mechanisms behind SnO2 nanoparticle formation and growth during hydrothermal synthesis: an in situ total scattering study. J. Am. Chem. Soc. 134, 6785–6792 (2012).
Ferri, F., Bertolotti, F., Guagliardi, A. & Masciocchi, N. Nanoparticle size distribution from inversion of wide angle X-ray total scattering data. Sci. Rep. 10, 12759 (2020).
Liu, F., He, H. & Xie, L. XAFS study on the specific deoxidation behavior of iron titanate catalyst for the selective catalytic reduction of NOx with NH3. ChemCatChem 5, 3760–3769 (2013).
Bader, H., Sturzenegger, V. & Hoigné, J. Photometric method for the determination of low concentrations of hydrogen peroxide by the peroxidase catalyzed oxidation of N,N-diethyl-p-phenylenediamine (DPD). Water Res. 22, 1109–1115 (1988).
Nørskov, J. K. et al. Origin of the overpotential for oxygen reduction at a fuel-cell cathode. J. Phys. Chem. B 108, 17886–17892 (2004).
Shi, X. et al. Understanding activity trends in electrochemical water oxidation to form hydrogen peroxide. Nat. Commun. 8, 701 (2017).
Siahrostami, S., Li, G.-L., Viswanathan, V. & Nørskov, J. K. One- or two-electron water oxidation, hydroxyl radical, or H2O2 evolution. J. Phys. Chem. Lett. 8, 1157–1160 (2017).
Diehl, L. et al. Structure-directing lone pairs: synthesis and structural characterization of SnTiO3. Chem. Mater. 30, 8932–8938 (2018).
Diehl, L. et al. Interplay between valence band tuning and redox stability in SnTiO3: implications for directed design of photocatalysts. Chem. Mater. 33, 2824–2836 (2021).
Themlin, J. M. et al. Resonant-photoemission study of SnO2: cationic origin of the defect band-gap states. Phys. Rev. B 42, 11914–11925 (1990).
Godinho, K. G., Walsh, A. & Watson, G. W. Energetic and electronic structure analysis of intrinsic defects in SnO2. J. Phys. Chem. C 113, 439–448 (2009).
Che, M. Nobel Prize in chemistry 1912 to Sabatier: organic chemistry or catalysis? Catal. Today 218–219, 162–171 (2013).
Wang, L. et al. Suppressing water dissociation via control of intrinsic oxygen defects for awakening solar H2O‐to‐H2O2 generation. Small 17, 2100400 (2021).
Liu, J., Zou, Y., Jin, B., Zhang, K. & Park, J. H. Hydrogen peroxide production from solar water oxidation. ACS Energy Lett. 4, 3018–3027 (2019).
Patra, S. G., Mizrahi, A. & Meyerstein, D. The role of carbonate in catalytic oxidations. Acc. Chem. Res. 53, 2189–2200 (2020).
Li, X., Yu, J., Jaroniec, M. & Chen, X. Cocatalysts for selective photoreduction of CO2 into solar fuels. Chem. Rev. 119, 3962–4179 (2019).
Sun, W. et al. A map of the inorganic ternary metal nitrides. Nat. Mater. 18, 732–739 (2019).
Mohtadi, R. & Orimo, S. The renaissance of hydrides as energy materials. Nat. Rev. Mater. 2, 16091 (2017).
Faber, T. E. & Ziman, J. M. A theory of the electrical properties of liquid metals. Philos. Mag. 11, 153–173 (1965).
Ohara, K. et al. Time-resolved pair distribution function analysis of disordered materials on beamlines BL04B2 and BL08W at SPring-8. J. Synchrotron Radiat. 25, 1627–1633 (2018).
Hammer, B., Hansen, L. B. & Nørskov, J. K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 59, 7413–7421 (1999).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Zhang, X., Klaver, P., van Santen, R., van de Sanden, M. C. M. & Bieberle-Hütter, A. Oxygen evolution at hematite surfaces: the impact of structure and oxygen vacancies on lowering the overpotential. J. Phys. Chem. C 120, 18201–18208 (2016).
Man, I. C. et al. Universality in oxygen evolution electrocatalysis on oxide surfaces. ChemCatChem 3, 1159–1165 (2011).
Sanson, A., Mathon, O. & Pascarelli, S. Local vibrational dynamics of hematite (α-Fe2O3) studied by extended X-ray absorption fine structure and molecular dynamics. J. Chem. Phys. 140, 1–8 (2014).
Acknowledgements
Mr. Hideo Maruyama (KANEKA TECHNO RESEARCH CORPORATION) is acknowledged for AFM/KPFM measurements. The X-ray total scattering and absorption measurements were carried out at SPring-8 under the proposals 2020A1208 and 2021A1114, and 2020A1209 and 2021A1113, respectively. This work was partially supported by Nagoya University microstructural characterization platform as a program of “Nanotechnology Platform” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, MEXT as “Program for Promoting Researches on the Supercomputer Fugaku” (Realization of innovative light energy conversion materials, Grant Number JPMXP1020210317), JST A-STEP Grant Number JPMJTR20TD, JSPS KAKENHI Grant Numbers JP18H01944, JP20H04673, JP21H02049, and others. KANEKA CORPORATION is acknowledged for financial support. Zhujun Zhang thanks to the financial support from Marubun Research Promotion Foundation.
Author information
Authors and Affiliations
Contributions
Z.Z. and T. Tachikawa wrote the paper with contributions from all co-authors. Z.Z. synthesized the materials and performed most of the experiments. Z.Z. and T. Tachikawa performed the data analysis. T. Tsuchimochi and S.L.T. performed the DFT calculations. T.I. performed the X-ray absorption measurements. Y.K. performed parts of the PEC experiments and data analysis. S.M. performed the STEM-EELS measurements. K.O. and H.Y. performed the X-ray total scattering measurements. T. Tachikawa conceived and supervised the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiao-Ming Cao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Z., Tsuchimochi, T., Ina, T. et al. Binary dopant segregation enables hematite-based heterostructures for highly efficient solar H2O2 synthesis. Nat Commun 13, 1499 (2022). https://doi.org/10.1038/s41467-022-28944-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28944-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
