
Abstract
Introgression has been proposed as an essential source of adaptive genetic variation. However, a key barrier to adaptive introgression is that recombination can break down combinations of alleles that underpin many traits. This barrier might be overcome in supergene regions, where suppressed recombination leads to joint inheritance across many loci. Here, we study the evolution of a large supergene region that determines a major social and ecological trait in Solenopsis fire ants: whether colonies have one queen or multiple queens. Using coalescent-based phylogenies built from the genomes of 365 haploid fire ant males, we show that the supergene variant responsible for multiple-queen colonies evolved in one species and repeatedly spread to other species through introgressive hybridization. This finding highlights how supergene architecture can enable a complex adaptive phenotype to recurrently permeate species boundaries.
Similar content being viewed by others
Introduction
Alleles can be transferred between species by introgressive hybridization1,2. Is it possible for a complex trait involving alleles at many loci to evolve in one species and then introgress into another? One of the main barriers to this type of introgression is recombination, which breaks up stretches of DNA, producing haplotypes that combine alleles from donor and recipient species3. Genomic inversions can impede recombination to create supergene variants whereby entire genomic regions are inherited as units4,5. Recent studies have shown that supergene variants can spread, unhindered by recombination, not only among populations but also across species barriers6,7,8,9,10.
In this study, we test competing hypotheses regarding the origin and evolutionary history of a supergene that determines whether colonies of the fire ant Solenopsis invicta have one queen or multiple queens. Several behavioral, morphological, and physiological traits co-vary with queen number. For example, queens from single-queen colonies are better at dispersing to new habitats, while multiple-queen colonies are more competitive at high population densities11. The queen number social dimorphism is controlled by two variants of a supergene region, SB and Sb, on “social” chromosome 1612,13. In single-queen colonies, all individuals carry exclusively the SB variant, and workers kill any potential additional queens. In multiple-queen colonies, many workers carry the Sb variant, but kill queens that lack the Sb variant13. The supergene region contains more than 470 protein-coding genes, most of which are present in both supergene variants12. The duplication or absence of some genes in Sb14,15, and the divergence in sequence and expression of several others suggest that they contribute to the social phenotype16. In addition, Sb differs from SB by three large inversions, and Sb has expanded through the accumulation of repetitive elements17,18 (estimated sizes are 20.9 Mb for SB and 27.5 Mb for Sb).
Interestingly, close relatives of S. invicta are also socially dimorphic and carry the same social supergene system17,18,19. Although this supergene system could be a trans-species polymorphism that evolved in the common ancestor of these species17,18,19, or even a set of polymorphisms that originated independently in each species, it is possible that its spread17,18,20 was instead mediated by introgressive hybridization. Here, using coalescent-based phylogenies, we show that the supergene system originated in S. invicta and spread to different species through the introgression of the Sb variant of the supergene.
Results
Coalescent construction of a species phylogeny and a supergene phylogeny
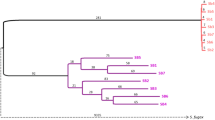
To understand the evolutionary history of the social supergene, we compared a phylogenetic tree representing the relationships among Solenopsis species with a phylogenetic tree representing the history of the supergene region (Fig. 1a–d illustrate potential scenarios). For this, we first obtained whole-genome sequences of 368 samples of Solenopsis. These include 261 samples published by ourselves and others12,14,16,17,18,21,22 and 107 samples that we additionally collected to expand taxonomic and geographical coverage (Supplementary Data 1 and Supplementary Fig. 1). Most of the samples are haploid males (365 out of 368). Using these genomes, we constructed a phylogenetic tree from each of 1728 informative single-copy genes23 (Supplementary Data 2). We then applied a coalescent-based method24 to reconstruct a species tree from 1631 single-copy genes mapping to chromosomes 1–15 and a supergene tree from 97 single-copy genes mapping to the supergene region of the social chromosome (Fig. 1e). In both trees, nodes critical for interpreting the histories of the species and the supergene are highly supported24 (bootstrap support 98–100%, local posterior probabilities 0.89–1.00).

a–d A simplified species tree (a), and hypothetical scenarios for the evolutionary history of the supergene (b–d). b SB and Sb supergene variants diverged (star) in the common ancestor of S. invicta and S. richteri; the supergene is thus a trans-species polymorphism. c Sb evolved twice from SB, representing independent origins (stars) after the separation of the two species. d Sb diverged in S. invicta and spread to S. richteri through introgression (arrows). e Empirical coalescent-based trees of 368 Solenopsis samples based on 1631 single-gene trees from chromosomes 1–15 (left; species tree) and 97 single-gene trees from the supergene region of chromosome 16 (right; supergene tree). Branches shorter than 0.05 were collapsed into polytomies. A tanglegram (middle) indicates the relative positions of each sample in both trees. Circled numbers highlight patterns consistent with introgression of Sb from S. invicta/macdonaghi into other species. On either side of the tanglegram black bars indicate where samples with the Sb genotype can be found in the two trees. Support values (ASTRAL bootstrap support | local posterior probability) are provided for key nodes of speciation and supergene differentiation.
The species tree (left side of Fig. 1e) shows that males from the socially dimorphic species S. invicta, S. macdonaghi and S. richteri are organized into two sister clades. One clade includes all S. richteri males. The second clade includes S. invicta and S. macdonaghi males in a paraphyletic relationship, suggesting that this clade may represent a single species. We hereafter refer to it as S. invicta/macdonaghi. All samples from other species, representing S. geminata, S. pusillignis, S. saevissima, S. interrupta, S. megergates and the undescribed species S. AdRX18,25, are organized into five clades branching off the base of the species tree.
The social supergene variant Sb evolved in one species and subsequently introgressed into others
Comparing the species tree with the supergene tree (Fig. 1e) allows us to infer the evolutionary history of the supergene. For males carrying the SB variant of the supergene and all but five males from other species (see below), the general topologies of the supergene tree and the species tree are congruent. However, all Sb males group into one clade that is sister to the SB S. invicta/macdonaghi clade. We can thus reject the hypothesis that the supergene is a trans-species polymorphism that originated before the speciation of S. richteri and S. invicta/macdonaghi. Indeed, if that were the case, each species would form one monophyletic group within the SB clade and another within the Sb clade (scenario in Fig. 1b). We can also reject the hypothesis that there were multiple origins of the supergene, whereby the S. richteri Sb samples would form a distinct sister group to the S. richteri SB samples (scenario in Fig. 1c). Instead, the grouping of Sb males as sister to the S. invicta/macdonaghi SB clade shows that the Sb supergene variant originated in chromosome 16 of S. invicta/macdonaghi after the divergence between this species and S. richteri. The presence of Sb sequences from other species within the S. invicta/macdonaghi Sb clade shows that Sb introgressed from S. invicta/macdonaghi into the other species (scenario in Fig. 1d). In addition, the hypothesis that the supergene arose through hybridization of two species with opposing orientations6 of the supergene-equivalent region on chromosome 16 is unparsimonious, as it would have required the introgression of Sb back into S. invicta/macdonaghi soon after its origin. Instead, the differentiation between Sb and SB suggests that the inversions that suppress recombination between Sb and SB12,17,18 in S. invicta/macdonaghi are conserved across species.
Recurring ancient and recent introgression of the supergene into different species
The Sb males from species other than S. invicta/macdonaghi are positioned in six Sb subclades within the supergene tree, suggesting that six independent introgression events occurred. The presence of two groups of S. richteri Sb samples within the Sb subtree (numbers 1 and 2 in Fig. 1e) could indicate that Sb introgressed twice from S. invicta/macdonaghi into S. richteri (see Discussion below). Strikingly, the other four cases of Sb introgression (numbered 3–6 in Fig. 1e) involve five Sb males positioned in early diverging clades of the species tree: one case involving S. interrupta (numbered 4; with two males), two cases involving S. megergates (numbered 3 and 5), and one case involving S. AdRX (numbered 6). All Sb haplotypes ultimately originate from the S. invicta/macdonaghi clade. However, in the supergene tree, one S. megergates Sb male (numbered 5) is adjacent to one of the S. richteri Sb clades (numbered 2), raising the possibility that Sb introgressed from one of these two species to the other secondarily.
A divergence time inference (Supplementary Note 1) suggests that the separation of SB and Sb occurred 0.97 million years ago (mya), after the split between S. richteri and S. invicta/macdonaghi (1.01 mya) and before the diversification of S. invicta/macdonaghi (0.81 mya) (Supplementary Figs. 2–4). All but one of the introgression events of the Sb supergene variant from S. invicta/macdonaghi into the other species occurred in the first quarter of its existence. The remaining introgression event is one of the two introgressions into S. megergates (numbered 3 in Fig. 1e); the age of the node representing this introgression event is within the age range of nodes separating individuals of the same species (Supplementary Note 1). Consistent with this, the putatively more ancient S. richteri Sb clades include long branch lengths in the supergene tree (clades 1 and 2 of Fig. 1e), and high nucleotide diversities (Supplementary Fig. 5 and Supplementary Note 2). In contrast, the younger Sb clade of S. megergates has a highly similar haplotype to those found in three S. invicta/macdonaghi Sb males we collected within 5.2 km of each other (Supplementary Note 3, Supplementary Fig. 6, and Supplementary Data 3). Altogether, our results suggest that introgression of the Sb supergene variant is a common mechanism for rapid adaptive evolution among fire ants and that this mechanism is not restricted to a particular Sb source haplotype or recipient species.
Discussion
Our findings contradict previous suggestions that the Sb supergene variant evolved in a common ancestor of S. invicta and S. richteri18 or within S. richteri20. By incorporating twice as many samples as the most recent large-scale study18, our work adds clades that had not yet been sampled. We focused on single-copy genes, which allowed us to avoid complex genealogies and potential genotyping artifacts caused by read mis-mapping of copy-number variants of genes and repetitive elements; such variation is particularly common in the degenerating Sb variant of the social chromosome14,17,21,26. Finally, rather than implying that all sequences share one evolutionary history, our coalescent-based tree reconstruction approach should be robust to gene tree discordance due to tree reconstruction errors and processes including incomplete lineage sorting and gene conversion24,27.
Nevertheless, we pursued five additional lines of analysis to challenge our finding of introgression. First, we produced a supergene tree and a species tree following the concatenated alignment approach similar to another recent study18, but after having excluded ambiguous regions of the genome (Supplementary Note 4 and Supplementary Fig. 7). The resulting phylogenies are consistent with those from the coalescent-based tree reconstructions (Fig. 1e) and not the previous studies, supporting the introgression of Sb from S. invicta/macdonaghi into each of the four Solenopsis species mentioned above. A notable difference between our two approaches is that the Sb samples of S. richteri named as group 1 in the coalescent-based tree are placed into one monophyletic group with the remaining S. richteri Sb samples in the concatenation-based tree, suggesting that Sb introgressed only once from S. invicta into this species. In addition, the Sb sample from S. AdRX (group 6 in the coalescent-based tree, Fig. 1e) is placed as a sister taxon to a relatively basal clade of Sb in the concatenation-based tree, suggesting that the introgression into S. AdRX occurred relatively early. Second, we created de novo genome assemblies using a subset of higher-coverage samples and derived an independent dataset of 2161 conserved single-copy ortholog sequences (Supplementary Note 1 and Supplementary Data 4). Phylogenetic inference yielded a dated species tree (Supplementary Fig. 2) with a topology that was fully consistent with the species tree from the coalescent-based tree reconstruction (Fig. 1e). Third, we quantified the support for introgression relative to incomplete lineage sorting using branch length distributions (QuIBL7; Supplementary Note 5) of trees built from single-copy genes from the supergene region. This analysis also supported the introgression of Sb between S. invicta/macdonaghi and the other four species (Supplementary Figs. 8–11). Fourth, if Sb originated in S. invicta/macdonaghi, any Sb haplotype should have more alleles in common with the SB variant of S. invicta/macdonaghi than with the SB variants of the other species. Indeed, in an analysis with equal number of samples for each species and supergene variant, we found that the Sb samples of S. invicta/macdonaghi and S. richteri shared 1921 alleles with the SB samples of S. invicta/macdonaghi, but only 994 alleles with the SB samples of S. richteri, a two-fold difference that is unlikely to be due to incomplete lineage sorting alone (exact binomial test, p < 10−12; Supplementary Note 6). We corroborated this result with an additional analysis of the remaining species, using one SB and one Sb sample per species (Supplementary Fig. 12). Finally, we used a phylogenetic weighting method (Twisst28; Supplementary Note 7) to quantify the support for alternative topologies in non-overlapping windows of four genes across the supergene. Introgression was the most highly supported topology, found in 2.5 times more windows than the topology in which the supergene is an ancestral trans-species polymorphism (Supplementary Figs. 13 and 14).
Outside the supergene region, the strong support for the species tree is in line with the absence of detectable contemporary gene flow between sympatric populations of S. invicta/macdonaghi and S. richteri in their native range29,30. Particular aspects of fire ant biology ensure at least occasional geographic contact between distinct lineages: flooding transports entire floating fire ant colonies along the major South American river systems31,32, while mating flights can reach altitudes of 300 m, with winds carrying young queens over great distances32. Importantly, S. invicta/macdonaghi and S. richteri do hybridize in their invasive North American range29,30. This shows that barriers to gene flow between these species are not yet irreversible and makes it plausible that such barriers were weaker in the past. Contact between lineages after allopatric speciation could therefore have facilitated the relatively old introgressions of Sb from S. invicta/macdonaghi to other species in our study. However, this explanation is unlikely for the more recent introgression into S. megergates, given the current sympatry between this species and S. invicta/macdonaghi. Instead, we hypothesize that, for this introgression and perhaps also some older introgression events, a hybrid Sb-carrying queen was accepted into an established multiple-queen colony of S. invicta/macdonaghi. This process could have been facilitated by the green-beard behavior of multiple-queen colonies whereby Sb-carrying workers accept Sb-carrying queens. The acceptance of Sb-carrying queens into existing multiple-queen colonies would have provided a crucial social buffer to any allelic incompatibilities of initial hybrid queens (see Supplementary Note 8).
Over long timescales, the retention of introgressed Sb supergene variants suggests that the costs of hybridization are outweighed by the combination of adaptive benefits of Sb in some environments, the green-beard behavior of Sb-carrying workers13 (in which they only accept Sb-carrying queens), and, potentially, meiotic drive33. Future studies of the evolution and function of the social chromosome supergene system will have to take the introgression of Sb into consideration. An important line of research will be to understand the dynamics of selection on Sb/Sb queens. Such queens are smaller and less fertile than SB/Sb or SB/SB queens34,35. Reproductive Sb/Sb queens are virtually absent in S. invicta populations, where the resulting lack of recombination is associated with an accumulation of deleterious mutations12,14,17,36. However, Sb/Sb queens are intriguingly present in at least some S. richteri populations37, thus enabling recombination between Sb haplotypes. Because of the differences in recombination regime and the bottlenecks associated with introgression, the Sb variants carried by each species may have different mutational loads or have different epistatic interactions with the rest of the genome. The accumulation of deleterious mutations in Sb likely creates a fitness cost that contributes to maintaining the system balanced in S. invicta. Indeed, this type of effect has been predicted by theoretical studies of supergene evolution38,39, and suggested to occur in the supergenes of species like the butterfly Heliconius numata40, the white-throated sparrow41, the ruff42 and the seaweed fly43. Further studying the dynamics of Sb degeneration will be essential to understand the evolution of the supergene system across all species.
Overall, our results clearly show that Sb evolved in an ancestor of S. invicta/macdonaghi that existed after divergence from the lineage leading to S. richteri. Through adaptive introgression, the Sb supergene variant spread to S. richteri, S. megergates, S. interrupta and S. AdRX. Supergene architecture can thus enable a complex social phenotype encoded by alleles at multiple genes to permeate species boundaries repeatedly. Our finding complements recently documented introgressions of smaller supergenes in other systems6,7,8,9. The introgression of the Sb supergene variant shows how a species can rapidly gain the genetic basis for a complex phenotype, and how selection for changes in a social organization can overcome the constraints of genomic architecture.
Methods
Collection and sequencing of fire ants from across their native range
We collected and flash-froze Solenopsis fire ants from 88 South American colonies (national collection and export permit from Uruguay N° 001/2015 to M.B, E.S., and Y.W., national and provincial collection and export permit for Argentina [Entre Rios, Santa Fe, Córdoba and Corrientes] N° 007/15, 282/2016, 20.911.387, 025052053-115, 433/02101-0014449-4 and 25253/16 to C.I.P., E.S. and Y.W., national collection and export permit for Brazil N° 14BR015531/DF to M.C.A.). After phenol-chloroform DNA extraction on a pool of ten workers from each colony, we performed a Restriction Fragment Length Polymorphism assay19 to determine whether both social chromosome variants are present (Supplementary Note 9 and Supplementary Data 1). From each of the 41 colonies that included only the SB variant, we retained one male. From 47 colonies that included both supergene variants, we either retained one SB and one Sb male (for 14 colonies), one Sb male (for 27 colonies), one SB male (for two colonies), two Sb males (for three colonies) or three Sb males (from one colony). We tentatively determined species identity through partial sequencing of the mitochondrial cytochrome oxidase I gene. Two colonies from S. pusillignis and S. saevissima lacked males; thus, we instead retained a pool of 10 workers. From each of the resulting 107 samples, we prepared an individually barcoded Illumina TruSeq PCR-Free library with 350 bp insert size, which we sequenced on Illumina HiSeq, providing 10.3-fold average genome coverage per sample. We additionally obtained sequence reads from 260 Solenopsis males and 1 pool of workers (S. geminata) from seven other studies: PRJNA39616114,17, PRJNA54260616, PRJNA42136718, PRJNA45075622, PRJNA18212714, SRR62111812, and SRX02192121. We performed optical duplicate filtering (minimum optical distance 2500), quality filtering and adapter trimming using clumpify (BBmap, v37.50) and skewer (v0.2.2, --mean-quality 20, --end-quality 15, minimum length of 80 for 100 bp reads and of 100 for 150 bp reads; removing degenerate reads). Supplementary Data 1 provides further details on sample collection and sequencing.
Genotyping through mapping reads to the reference genome
We aligned the filtered reads for each sample to the S. invicta reference genome assembly Si_gnGA17 and bait sequences including the S. invicta mitochondrion, phiX phage and Wolbachia sequences (NC_014672.1, NC_001422.1, AF243435.1, AF243436.1, CP001391.1, AM999887.1, GCA_003704235.1) using bwa-mem2 (v2.0pre2 with parameters -B 6 -E 2 -L25,25 -U 50 -T 50 -h 4,200 -a -V -Y -M). Sequence divergence within our dataset is sufficiently low for this approach as, for example, 98.95% of individual reads and 97.16% of pairs of reads from the outgroup species used (S. geminata; sequence divergence from S. invicta ~3.9 mya44), map directly to the S. invicta reference genome (Supplementary Data 1). For samples from PCR-based library preparation protocols, we marked duplicate reads with sambamba v0.7.1. We identified and subsequently excluded 23.05 Mb of regions that had coverage higher than median coverage plus 3 standard deviations of single-copy gene coverage across all samples because such high-coverage regions likely represent collapsed repeats (mosdepth v0.2.9, bedtools v2.27.1, bedops v2.4.30). We jointly called variants in all individuals using freebayes (v1.2.045, with parameters --min-alternate-count 4 --min-alternate-fraction 0.4 --min-coverage 4 --ploidy 2 --use-best-n-alleles 8 --min-mapping-quality 40 --min-base-quality 30 --use-reference-allele). Although male ants are normally haploid, the genotyping process considered that samples are diploid so that we could identify and remove diploid males and apparently heterozygous sites that could be due to copy-number variation or sequencing artifacts. After decomposing multi-nucleotide variants, we retained only single-nucleotide polymorphisms (SNPs) and excluded low-quality variants (Q < 30), low coverage variants represented by either only forward or only reverse reads, and variants overlapping high-coverage genomic regions or simple sequence repeats (trf v4.09, bcftools v1.10.2). After this first round of genotyping to identify high confidence variants, we performed relaxed targeted genotyping using freebayes --haplotype-basis-alleles of each sample with the following parameters: --ploidy 2 --haplotype-length −1 --use-best-n-alleles 4 --min-mapping-quality 30 --min-base-quality 28 --min-alternate-fraction 0.35 --min-alternate-total 2 --min-coverage 2 --use-reference-allele. After decomposition, individual genotypes where coverage was greater than median plus 3 standard deviations of coverage on single-copy genes or with an allelic balance between 0.25 and 0.75 were set to missing because such sites are not expected in haploids and thus are untrustworthy. For the three pools of workers, we created a haploid consensus by randomly selecting one allele at each heterozygous site. We had initially collected and sequenced 18 additional males (Supplementary Data 1, not included in the sample numbers used above), but for nine of these, more than 25% of variant sites could not be genotyped, and the nine others had particularly high numbers of heterozygous sites indicating that they are likely diploid. All 18 males were removed from further analysis. We retained variant sites for which more than 75% of the remaining 368 samples were genotyped. We analyzed sequences of the Gp-9 marker (Supplementary Data 1) and performed principal component analysis (Supplementary Fig. 15) to confirm supergene variants.
Coalescent-based phylogenetic inference
The S. invicta genome assembly (Annotation Release 100) contains exactly one copy of 5851 of the 5991 genes expected to be in a single copy across Hymenoptera (BUSCO v4.0.523). We made an initial consensus tree (which had the same topology as the species tree in Fig. 1e) based on genes mapped to chromosomes 1–15, rooted to S. geminata, which we used to label species of each sample according to Yan et al.18. Taxonomic assignment based on a mitochondrial phylogenetic tree was unreliable (Supplementary Note 10 and Supplementary Fig. 16). We retained the most informative 1881 out of 5851 genes for which the 342 males identified as S. invicta, S. macdonaghi and S. richteri (the expected closely related ingroup species in the phylogenetic tree of Solenopsis) had a per-gene average of at least ten single-nucleotide differences relative to the reference genome (this step removed non-informative genes that had few or mostly singleton SNPs or that were polymorphic only in the basally diverging species). We obtained a consensus sequence for each of these genes, including coding sequence and introns for each sample using bcftools consensus v1.10.2 from the VCF file of all genotypes. Modeltest-ng v0.1.646 identified the best substitution models for intronic sequences and for coding sequences (Supplementary Data 2). For each of the 1881 single-copy genes, we then used RAxML-NG v0.9.047 with 50 random and 50 parsimony starting trees to build maximum-likelihood trees with 100 bootstraps, using separate partitions for introns and exons. After removing the trees for the 153 genes that did not converge during the RAxML tree inference, we used ASTRAL-III v5.14.324 to create two coalescent-based trees, each supported by 100 bootstraps: one tree based on 97 single-copy genes in the supergene region of social chromosome 16, and one tree based on 1631 single-copy genes from chromosome 1–15. Trees were then rooted to the outgroup S. geminata, and extremely short branches (branch length < 0.05) were collapsed into polytomies. The topologies of these two trees were largely congruent with trees constructed from coding sequence alone, or constructed from the full set of 5851 genes with either a coalescent-based tree inference from gene trees (ASTRAL-III) or from concatenation (RAxML-NG, Supplementary Fig. 7 and Supplementary Note 4). To generate Fig. 1e, we used the R package dendextend48 and subsequently adjusted tree layouts, labels and background colors using Affinity Designer. We dated phylogenetic trees (Fig. 1) using calibrated node ages44,49 for major lineages in IQ-TREE v250 (Supplementary Note 1). The classic ABBA-BABA test51 is inappropriate for our data because of the topology of the supergene tree. Instead, we performed a similar test, examining the differences in allele sharing between the Sb samples and either the SB samples of S. invicta/macdonaghi or the SB of each of the other species (a “BBBAA-BBABA” test; Supplementary Note 6).
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All DNA sequences generated in this study have been deposited in the Sequence Read Archive database under accession code PRJNA685290 (individual accession numbers are detailed in Supplementary Data 1). The VCF genotype matrix and the de novo assemblies we generated are available at https://wurmlab.com/data/supergene_introgression. The source data used to produce each figure, including Newick tree files, are available on GitHub at https://github.com/wurmlab/2021-fire-ant-social-supergene-introgression, with additional information supplied as Supplementary Data.
Code availability
Code and analysis scripts are available on GitHub at https://github.com/wurmlab/2021-fire-ant-social-supergene-introgression, and at https://wurmlab.com/data/supergene_introgression.
References
Anderson, E. Introgressive Hybridization (J. Wiley, 1949).
Harrison, R. G. & Larson, E. L. Hybridization, introgression, and the nature of species boundaries. J. Hered. 105, 795–809 (2014).
Yeaman, S. & Whitlock, M. C. The genetic architecture of adaptation under migration-selection balance. Evolution 65, 1897–1911 (2011).
Thompson, M. J. & Jiggins, C. D. Supergenes and their role in evolution. Heredity 113, 1–8 (2014).
Kirkpatrick, M. How and why chromosome inversions evolve. PLoS Biol. 8, e1000501 (2010).
Jay, P. et al. Supergene evolution triggered by the introgression of a chromosomal inversion. Curr. Biol. 28, 1839–1845.e3 (2018).
Edelman, N. B. et al. Genomic architecture and introgression shape a butterfly radiation. Science 366, 594–599 (2019).
Norris, L. C. et al. Adaptive introgression in an African malaria mosquito coincident with the increased usage of insecticide-treated bed nets. Proc. Natl Acad. Sci. USA. 112, 815–820 (2015).
Brion, C., Caradec, C., Pflieger, D., Friedrich, A. & Schacherer, J. Pervasive phenotypic impact of a large nonrecombining introgressed region in yeast. Mol. Biol. Evol. 37, 2520–2530 (2020).
Dixon, G., Kitano, J. & Kirkpatrick, M. The origin of a new sex chromosome by introgression between two stickleback fishes. Mol. Biol. Evol. 36, 28–38 (2019).
Ross, K. G. & Keller, L. Ecology and evolution of social organisation: insights from fire ants and other highly eusocial insects. Annu. Rev. Ecol. Syst. 26, 631–656 (1995).
Wang, J. et al. A Y-like social chromosome causes alternative colony organization in fire ants. Nature 493, 664–668 (2013).
Keller, L. & Ross, K. G. Selfish genes: a green beard in the red fire ant. Nature 394, 573–575 (1998).
Pracana, R. et al. Fire ant social chromosomes: differences in number, sequence and expression of odorant binding proteins. Evol. Lett. 1, 199–210 (2017).
Cohanim, A. B., Amsalem, E., Saad, R., Shoemaker, D. & Privman, E. Evolution of olfactory functions on the fire ant social chromosome. Genome Biol. Evol. 10, 2947–2960 (2018).
Martinez-Ruiz, C. et al. Genomic architecture and evolutionary antagonism drive allelic expression bias in the social supergene of red fire ants. eLife 9, e55862 (2020).
Stolle, E. et al. Degenerative expansion of a young supergene. Mol. Biol. Evol. 36, 553–561 (2019).
Yan, Z. et al. Evolution of a supergene that regulates a trans-species social polymorphism. Nat. Ecol. Evol. 4, 240–249 (2020).
Krieger, M. J. B. & Ross, K. G. Identification of a major gene regulating complex social behavior. Science 295, 328–332 (2002).
Cohen, P. & Privman, E. The social supergene dates back to the speciation time of two Solenopsis fire ant species. Sci. Rep. 10, 11538 (2020).
Wurm, Y. et al. The genome of the fire ant Solenopsis invicta. Proc. Natl. Acad. Sci. USA. 108, 5679–5684 (2011).
Privman, E. et al. Positive selection on sociobiological traits in invasive fire ants. Mol. Ecol. 27, 3116–3130 (2018).
Seppey, M., Manni, M. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness. Methods Mol. Biol. 1962, 227–245 (2019).
Zhang, C., Rabiee, M., Sayyari, E. & Mirarab, S. ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 19, 153 (2018).
Shoemaker, D. D., Ahrens, M. E. & Ross, K. G. Molecular phylogeny of fire ants of the Solenopsis saevissima species-group based on mtDNA sequences. Mol. Phylogenet. Evol. 38, 200–215 (2006).
Fontana, S. et al. The fire ant social supergene is characterized by extensive gene and transposable element copy number variation. Mol. Ecol. 29, 105–120 (2020).
Kubatko, L. S. & Degnan, J. H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 56, 17–24 (2007).
Martin, S. H. & Van Belleghem, S. M. Exploring evolutionary relationships across the genome using topology weighting. Genetics 206, 429–438 (2017).
Ross, K. G. & Trager, J. C. Systematics and population genetics of fire ants (Solenopsis saevissima complex) from Argentina. Evolution 44, 2113–2134 (1990).
Cohen, P. & Privman, E. Speciation and hybridization in invasive fire ants. BMC Evol. Biol. 19, 111 (2019).
Adams, B. J., Hooper-Bùi, L. M., Strecker, R. M. & O’Brien, D. M. Raft formation by the red imported fire ant, Solenopsis invicta. J. Insect Sci. 11, 171 (2011).
Tschinkel, W. R. The Fire Ants (Harvard Univ. Press, 2006).
Ross, K. G. & Shoemaker, D. Unexpected patterns of segregation distortion at a selfish supergene in the fire ant Solenopsis invicta. BMC Genet. 19, 101 (2018).
DeHeer, C. J., Goodisman, M. A. D. & Ross, K. G. Queen dispersal strategies in the multiple-queen form of the fire ant Solenopsis invicta. Am. Nat. 153, 660–675 (1999).
DeHeer, C. J. A comparison of the colony-founding potential of queens from single- and multiple-queen colonies of the fire ant Solenopsis invicta. Anim. Behav. 64, 655–661 (2002).
Pracana, R., Priyam, A., Levantis, I., Nichols, R. A. & Wurm, Y. The fire ant social chromosome supergene variant Sb shows low diversity but high divergence from SB. Mol. Ecol. 26, 2864–2879 (2017).
Hallar, B. L., Krieger, M. J. B. & Ross, K. G. Potential cause of lethality of an allele implicated in social evolution in fire ants. Genetica 131, 69–79 (2007).
Kirkpatrick, M. & Barton, N. Chromosome inversions, local adaptation and speciation. Genetics 173, 419–434 (2006).
Berdan, E. L., Blanckaert, A., Butlin, R. K. & Bank, C. Deleterious mutation accumulation and the long-term fate of chromosomal inversions. PLoS Genet. 17, e1009411 (2021).
Jay, P. et al. Mutation load at a mimicry supergene sheds new light on the evolution of inversion polymorphisms. Nat. Genet. 53, 288–293 (2021).
Tuttle, E. M. et al. Divergence and functional degradation of a sex chromosome-like supergene. Curr. Biol. 26, 344–350 (2016).
Küpper, C. et al. A supergene determines highly divergent male reproductive morphs in the ruff. Nat. Genet. 48, 79–83 (2016).
Butlin, R. K. & Day, T. H. Genic and karyotypic selection on an inversion polymorphism in the seaweed fly, Coelopa frigida. Heredity 54, 267–274 (1985).
Moreau, C. S. & Bell, C. D. Testing the museum versus cradle tropical biological diversity hypothesis: phylogeny, diversification, and ancestral biogeographic range evolution of the ants. Evolution 67, 2240–2257 (2013).
Garrison, E. & Marth, G. Haplotype-based variant detection from short-read sequencing. Preprint at arXiv [q-bio.GN] (2012).
Darriba, D. et al. ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294 (2020).
Kozlov, A. M., Darriba, D., Flouri, T., Morel, B. & Stamatakis, A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35, 4453–4455 (2019).
Galili, T. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics 31, 3718–3720 (2015).
Ward, P. S., Brady, S. G., Fisher, B. L. & Schultz, T. R. The evolution of myrmicine ants: phylogeny and biogeography of a hyperdiverse ant clade (Hymenoptera: Formicidae). Syst. Entomol. 40, 61–81 (2015).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Green, R. E. et al. A draft sequence of the Neandertal genome. Science 328, 710–722 (2010).
Acknowledgements
This work was supported by the European Union (FP7 Marie-Curie-Fellowship PIEF-GA-2013-623713 to E.S. and Y.W.), the German Academic Exchange (DAAD 570704 83 to E.S.), the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP BEPE grant 2014/04943-0 to M.C.A.), COLFUTURO (to G.L.H.), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (Ciência sem Fronteiras grant 248391/2013-5 to Y.W.), the Biotechnology and Biological Sciences Research Council (grants BB/K004204/1, BB/T015683/1, and BB/S507556/1 to Y.W.) and the Natural Environment Research Council (grant NE/L00626X/1 to Y.W.). We thank S. Coelho, D. Pereira Nogueira Da Silva, N. Curi, N. Souza Araujo (Universidade de São Paulo), R. Jaffé (Instituto Tecnológico Vale), Pablo Álvarez Osorio (Buenos Aires), E. Boné (Universidad de Buenos Aires), Y. Guillij (Dir. de Usos Sust. de los Recursos Naturales, Entre Ríos), Dir. de Conservación de la Biodiversidad Argentina, C. Durrant, M. dos Reis, L. Rodrigues Santiago, C. Martínez-Ruiz, R. Nichols, and C. Eizaguirre (Queen Mary University of London), for their help with organization, sampling, permits, preparation, sequencing or analysis, useful discussions, and comments on the manuscript.
Author information
Authors and Affiliations
Contributions
Initial conceptualization: E.S., Y.W. Experimental design: R.P., E.S., F.L.-O., Y.W. Sample collection, identification, and processing: E.S., C.C.-C., M.C.A., C.I.P., M.B., Y.W. DNA extraction and sequence library construction: E.S., C.C.-C.. Initial data analysis: E.S., R.P., A.P. Main data analyses: E.S., R.P., Y.W. Additional data analyses and visualization: E.S., R.P., M.K.P., G.L.H., A.P., F.L.-O. Supervision: Y.W. Writing—original draft: E.S., R.P., Y.W. Writing—review and editing: R.P., E.S., M.K.P., G.L.H., A.P., F.L.-O., Y.W.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stolle, E., Pracana, R., López-Osorio, F. et al. Recurring adaptive introgression of a supergene variant that determines social organization. Nat Commun 13, 1180 (2022). https://doi.org/10.1038/s41467-022-28806-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28806-7
This article is cited by
-
Integrative characterization of genetic and phenotypic differentiation in an ant species complex with strong hierarchical population structure and low dispersal abilities
Heredity (2023)
-
Functional properties of ant queen pheromones as revealed by behavioral experiments
Behavioral Ecology and Sociobiology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
