
Abstract
Artificial photosynthesis of H2O2 using earth-abundant water and oxygen is a promising approach to achieve scalable and cost-effective solar fuel production. Recent studies on this topic have made significant progress, yet are mainly focused on using organic polymers. This set of photocatalysts is susceptible to potent oxidants (e.g. hydroxyl radical) that are inevitably formed during H2O2 generation. Here, we report an inorganic Mo-doped faceted BiVO4 (Mo:BiVO4) system that is resistant to radical oxidation and exhibits a high overall H2O2 photosynthesis efficiency among inorganic photocatalysts, with an apparent quantum yield of 1.2% and a solar-to-chemical conversion efficiency of 0.29% at full spectrum, as well as an apparent quantum yield of 5.8% at 420 nm. The surface-reaction kinetics and selectivity of Mo:BiVO4 were tuned by precisely loading CoOx and Pd on {110} and {010} facets, respectively. Time-resolved spectroscopic investigations of photocarriers suggest that depositing select cocatalysts on distinct facet tailored the interfacial energetics between {110} and {010} facets and enhanced charge separation in Mo:BiVO4, therefore overcoming a key challenge in developing efficient inorganic photocatalysts. The promising H2O2 generation efficiency achieved by delicate design of catalyst spatial and electronic structures sheds light on applying robust inorganic particulate photocatalysts to artificial photosynthesis of H2O2.
Similar content being viewed by others


Introduction
Harvesting solar fuels by artificial photosynthesis has great values in the global missions on tackling climate change and environmental pollutions1,2,3. Among various artificial photosynthetic reactions, solar-driven water splitting for hydrogen generation has attracted the most attention in the past half century. Yet, its practical application is challenged by the low-energy density, storability, and transportability of hydrogen gas4,5. To this end, photosynthesis of H2O2, an emerging liquid fuel and also a green oxidant, is attracting growing interests6. Among primary photosynthetic systems, including photovoltaic-assisted electrolysis7, photoelectrochemical catalysis8,9, and particulate photocatalysis (PC)10, PC is the most cost-effective because of its simplicity and scalability11. With regard to reaction processes, PC systems are advantageous for the mass transport of reagents and products, which greatly reduces the concentration overpotential and pH gradient during reactions12. For these reasons, it is desirable to develop efficient PC systems for H2O2 generation.
Recently, various PC systems based on organic-polymer semiconductors have been developed for photocatalytic H2O2 generation with a recording solar-to-H2O2 (STH) conversion efficiency of 0.61%13,14,15. Nevertheless, these semiconductors have a potential concern of their stability since photocatalytic H2O2 generation is inevitably accompanied by hydroxyl radical (•OH) generation (H2O2 + hν → 2•OH or H2O2 + e− + H+ → •OH + H2O) and such a potent oxidant (E0 (•OH/H2O) = 2.18 V vs. NHE at pH 7.0) is damaging to organic structures16. For instance, after 24-h incubation under •OH-rich conditions, graphitic carbon nitride (C3N4, one of the most widely studied organic photocatalysts for H2O2 photosynthesis) lost over 60% activity for H2O2 generation (Fig. S1). In contrast, inorganic semiconductors (e.g., BiVO4) are resistant to •OH-mediated oxidation, so they are more favored by long-term reactions. Yet, inorganic semiconductors remain inefficient for photocatalytic H2O2 generation (<150 µM/h, see Table S1) due to high-charge recombination13,17. For an efficient inorganic photocatalyst, it needs to exhibit (i) a suitable band structure for O2 reduction and H2O oxidation coupled with a narrow band-gap, (ii) efficient charge separation, and (iii) high surface-reaction kinetics and selectivity.
Here, we report a faceted Mo-doped BiVO4 (Mo:BiVO4) with dual cocatalysts selectively loaded on its reduction and oxidation facets (Fig. 1a). BiVO4 is a photocatalyst with a suitable band structure and relatively narrow band-gap (2.4 eV) for H2O2 photosynthesis, yet the reported efficiency remains unsatisfying (<12 µM/h, see Table S1) due to severe charge recombination, even in the presence of sacrificial agents18,19. We synthesized monoclinic Mo:BiVO4 and anchored CoOx onto the oxidative {110} facet via photooxidation, which served to promote the water oxidation kinetics. In the meantime, Pd was anchored onto the reductive {010} facet via photoreduction and served to steer the oxygen-reduction pathway from four-electron processes for H2O formation to two-electron processes for H2O2 generation. In-depth time-resolved spectroscopic investigations of photocarriers demonstrates that CoOx and Pd depositions tailored the energetics of the respective facet for improved charge separation, a key obstacle limiting the performance of inorganic photocatalysts. Without using any sacrificial reagent, the reasonably designed CoOx/Mo:BiVO4/Pd produced H2O2 at a rate of 1425 μM/h, an apparent quantum yield (AQY) of 1.2% over the full spectrum of sunlight, and a STH of 0.29%, surpassing other inorganic photocatalysts by one order of magnitude (Table S1). With comparable efficiency with organic ones in photocatalytic H2O2 generation, our work demonstrates the feasibility of applying robust inorganic particulate photocatalysts to efficient photocatalytic H2O2 generation through delicate design of catalyst spatial and electronic structures.

a Schematic deposition processes of CoOx and Pd on Mo:BiVO4 and the corresponding SEM images of b Mo:BiVO4, c CoOx/Mo:BiVO4, and d CoOx/Mo:BiVO4/Pd. e, f Energy-dispersive X-ray spectroscopy (EDS) elemental mapping and line profile along with the white arrow of CoOx/Mo:BiVO4/Pd.
Results and discussion
Synthesis and characterization of CoOx/Mo:BiVO4/Pd
We first prepared faceted Mo:BiVO4 particles using a solid-liquid-reaction method19,20. Mo was doped to the V site to increase the bulk conductivity. Mo doping amount was optimized to be 0.025 mol% based on the activity of photocatalytic H2O2 generation over CoOx/Mo:BiVO4/Pd (Fig. S2). The X-ray diffraction (XRD) pattern of Mo:BiVO4 as well as CoOx/Mo:BiVO4, Mo:BiVO4/Pd and CoOx/Mo:BiVO4/Pd particles matched well with that of monoclinic BiVO4, with {010} and {110} facet peaks located at 30.6° and 18.7°, respectively (Fig. S3). The Brunner−Emmet−Teller (BET) tests show Mo:BiVO4 as well as CoOx/Mo:BiVO4, Mo:BiVO4/Pd and CoOx/Mo:BiVO4/Pd particles exhibit similar surface areas (1.43–1.71 m2/g, Table S2). The Mo:BiVO4 particles exhibit a decahedron structure with clear facets as shown in scanning electron microscope (SEM) images (Fig. 1a, b). The selected area electron diffraction (SEAD, Fig. S4) pattern confirms the Millier index of top and side facets are {010} reduction facet and {110} oxidation facet, respectively21. The Mo doping amount (Mo/V) is 0.023 mol% tested by inductively coupled plasma mass spectrometry (ICP-MS).
Secondly, we selectively loaded cocatalysts onto different facets of Mo:BiVO4 particles via stepwise photodeposition (Fig. 1a). CoOx as a cocatalyst for water oxidation reaction (WOR) was selectively deposited onto the {110} facet of Mo:BiVO4 via photooxidation of Co2+ ions. The loading amount of Co was optimized to be 0.2 wt% based on the activities of photocatalytic H2O2 generation over CoOx/Mo:BiVO4/Pd (Fig. S5). Prominent Co 2p X-ray photoelectron spectroscopy (XPS) peaks demonstrate the successful loading of Co species (Fig. S6a). The Co 2p3/2 peak can be deconvoluted to a Co2+ peak at 781.6 eV and a Co3+ peak at 780.6 eV, suggesting that the valence of Co was in-between +2 and +3, therefore the cocatalyst is denoted as CoOx. The SEM image (Fig. 1c) shows that CoOx particles are uniformly distributed across the {110} facet of Mo:BiVO4. Consistent with the SEM results, energy-dispersive X-ray spectroscopy (EDS) elemental mapping and line profile (Fig. 1e, f) show 4.1-fold stronger Co signal on the {110} facet compared to that on the {010} facet. Further, SEM (Fig. S7) and TEM (Fig. S8) line profiles of CoOx/Mo:BiVO4 indicate that Co signal on {110} facet is much higher than that on {010} facet. These results demonstrate the selective deposition of CoOx on the {110} facet of Mo:BiVO4.
Loading CoOx cocatalyst significantly enhanced the WOR surface kinetics of Mo:BiVO4. The photocatalytic O2 evolution activity of CoOx/Mo:BiVO4 was more than twice as much as that of Mo:BiVO4 (Fig. S9a). The enhancement water oxidation was further verified by the improved photoelectrochemical performance of CoOx/Mo:BiVO4 electrode compared to that of Mo:BiVO4 electrode. The onset potential of the CoOx/Mo:BiVO4 photoanode was ~0.1 V more negative than that of Mo:BiVO4 (Fig. S9b). At a given potential, the photoanodic current density of CoOx/Mo:BiVO4 was much higher than that of Mo:BiVO4 (Fig. S9b). Such stark contrasts between O2 production and photoelectrochemical performance clearly demonstrate the improved WOR activity of Mo:BiVO4 upon CoOx deposition.
Following the photodeposition of CoOx, Pd as an oxygen-reduction reaction (ORR) cocatalyst was selectively deposited on the {010} facet of Mo:BiVO4 via photoreduction of PdCl42− (Fig. 1a). The loading amount of Pd was optimized to be 0.4 wt% based on the activity of photocatalytic H2O2 generation over CoOx/Mo:BiVO4/Pd (Fig. S10). The distinct Pd 3d XPS peaks indicate the successful loading of Pd species (Fig. S6b). The Pd 3d5/2 peak can be deconvoluted to a main Pd0 peak at 335.1 eV and a minor Pd2+ peak at 337.0 eV, attributing to the metallic Pd from photoreduction and PdO from partial oxidation of Pd in air, respectively. SEM images (Fig. 1d and S11) show that Pd particles were uniformly and selectively distributed across the {010} facets of Mo:BiVO4. The facet-selective loading of Pd was further demonstrated by the stark contrast between distinctive Pd signal on the {010} facet and negligible Pd signal on the {110} facet from EDS elemental mapping and line profile of Mo:BiVO4 (Fig. 1e, f and S12). SEM (Fig. S13) and TEM (Fig. S14) line profile of Mo:BiVO4/Pd also suggest that Pd signal on {010} facet is much higher than that on {110} facet.
Loading Pd cocatalyst significantly enhanced the selectivity of H2O2 generation from 13% by pristine Mo:BiVO4 to 89% (Fig. S15; H2O2 generation selectivity is defined as the ratio of electrons utilized for H2O2 synthesis to the total number of electrons consumed). These results indicate that Pd, as a verified catalyst for selective H2O2 synthesis22,23,24, steered the ORR on Mo:BiVO4 from four-electron (O2 + 4H+ + 4e− → 2H2O) to two-electron (O2 + 2H+ + 2e− → H2O2) processes. Moreover, the enhanced H2O2 production selectivity with Pd loading was slightly perturbed by the presence of CoOx, likely attributed to improved H2O2 decomposition (Fig. S15). Note that the two-electron H2 evolution (2H+ + 2e− → H2), another major side reaction likely limiting the selectivity for H2O2 generation19, was prohibited because the conduction band of Mo:BiVO4 is too deep to evolve H2.
Photocatalytic performance
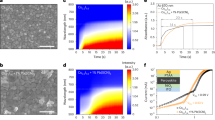
The photocatalytic H2O2 generation performance of the particulate photocatalyst was evaluated under simulated sunlight irradiation without any sacrificial reagent (Fig. 2a). Bare Mo:BiVO4 exhibits minimal performance for 60-min H2O2 generation (4.1 μM). Loading CoOx cocatalyst enhanced the photocatalytic H2O2 generation performance by a factor of 1.8 (7.5 μM, Fig. 2a), attributing to promoted water oxidation and consequentially reduced detrimental charge recombination (Fig. 2c). In the meanwhile, loading Pd cocatalyst onto Mo:BiVO4 improved H2O2 generation selectivity (Fig. 2c), resulting in a 53.7-fold enhancement of 60-min H2O2 generation (220.0 μM, Fig. 2a).

a Time courses of photocatalytic H2O2 generation over CoOx/Mo:BiVO4/Pd, Mo:BiVO4-CoOx-Pd, CoOx/Mo:BiVO4, Mo:BiVO4/Pd, and Mo:BiVO4. Reaction conditions: photocatalyst amount, 2 mg; reactant solution, 12 mL PBS aqueous solution (pH = 7.4) saturated with O2; light source, xenon lamp solar simulator, 100 mW/cm2, AM 1.5 G. We note the data series for CoOx/Mo:BiVO4 (7.5 μM at the reaction time of 60 min) overlap with those of Mo:BiVO4 (4.1 μM at the reaction time of 60 min). b Apparent quantum yield (AQY) of photocatalytic H2O2 generation over CoOx/Mo:BiVO4/Pd as a function of the incident light wavelength. Reaction conditions: photocatalyst amount, 2 mg; reactant solution, 5 mL PBS aqueous solution (pH = 7.4) saturated with O2; light source, monochromatic LED light. c Schematic of photocatalytic H2O2 generation over CoOx/Mo:BiVO4/Pd. CB and VB are short for conduction band and valence band, respectively.
Simultaneous loading of CoOx and Pd cocatalysts enhanced H2O2 generation by 347.6-fold compared to pristine Mo:BiVO4 (Fig. 2a). Without any sacrificial reagent, CoOx/Mo:BiVO4/Pd generated 1425 µM H2O2 after one-hour reaction. When the suspension was N2-purged, the H2O2 production was inhibited by over 99% (Fig. S16), confirming that H2O2 generation proceeded mainly through ORR. The wavelength-dependent AQYs measured by light-emitting diode (LED) light irradiation agree well with the absorption spectrum of CoOx/Mo:BiVO4/Pd (Fig. 2b), suggesting that H2O2 was generated following its band-gap excitation. The AQY at 420 nm was determined to be 5.8%, the highest reported for inorganic semiconductors to the best of our knowledge (see Table S1). Furthermore, the AQY of CoOx/Mo:BiVO4/Pd reached 1.2% over the full spectrum, and its STH reached 0.29%. Such an efficiency surpasses other inorganic semiconductor photocatalysts by one order of magnitude (Table S1) and indicates that inorganic photocatalysts are competent for efficient photocatalytic H2O2 generation. Most importantly, in stark contrast to the high •OH-susceptibility of organic photocatalyst, after 24-h incubation under •OH-rich conditions, CoOx/Mo:BiVO4/Pd exhibits nominal change in H2O2 productions (Fig. S1b) and chemical compositions (Fig. S17), demonstrating its high resistance to •OH-mediated oxidation.
The photocatalytic H2O2 generation activity of CoOx/Mo:BiVO4/Pd decreased gradually as shown in Fig. 2a. The reaction rate in the fourth 15 min corresponds to 47% of that in the first one. To clarify such decay, CoOx/Mo:BiVO4/Pd was tested with cycles of reaction. The photocatalytic H2O2 generation rate in the second cycle was 420 μM/h, 29% of that in the first cycle (Fig. S18a). It is supposed that the decay is related to the gradual transformation of CoOx to CoPi in PO43- solution (applied as H2O2 stabilizer25). Although CoPi has been widely applied to photoanodes a cocatalyst, its roles in photocatalysis are controversial mostly because of inconsistent sample conditions26. Here we believe that CoPi solely facilitated the surface kinetics for O2 evolution and hardly affected the energetics for charge separation27. Since the photocatalytic H2O2 generation activity is determined by the charge-separation efficiency, CoPi behaved inferior to CoOx in our system. The transformation of CoOx to CoPi was confirmed by characterizing CoOx/Mo:BiVO4/Pd before and after the reaction, where a prominent peak at 133.6 eV attributing to Co-Pi bond was observed in the post-catalysis P 2p3/2 XPS spectra (Fig. S19). Furthermore, the photocatalytic H2O2 generation activity of freshly prepared CoPi/Mo:BiVO4/Pd was 21% of that of CoOx/Mo:BiVO4/Pd and was similar to that of spent CoOx/Mo:BiVO4/Pd (Fig. S20). These results confirm that the transformation of CoOx to CoPi undermined CoOx/Mo:BiVO4/Pd for photocatalytic H2O2 generation. In order to avoid the deterioration of CoOx/Mo:BiVO4/Pd in PO43− solution, photocatalytic H2O2 generation was conducted in pure water as shown in Fig. S18b. As expected, CoOx/Mo:BiVO4/Pd was highly stable over five cycles of reaction. To further demonstrate the stability of CoOx/Mo:BiVO4/Pd, we test the performance of CoOx/Mo:BiVO4/Pd in seawater, which is a desirable solution condition for artificial photosynthesis28. No deactivation was observed over five-round repetitive use (Fig. S21). Yet the cumulative production of H2O2 was lower than that in phosphate solution owing to H2O2 decomposition, consistent with the previous report29. In following studies, the cumulative production of H2O2 in pure water will be improved with rapid H2O2 diffusion by a large-scale photosynthesis setup where the CoOx/Mo:BiVO4/Pd photocatalyst will be immobilized on a support using drop-casting or screen printing technologies and integrated in a flow cell photolysis system19,30.
Charge separation
We note that the enhancement of H2O2 production by the synergistic effect of coloading Pd and CoOx is even higher than the multiplication of the enhancements by loading Pd and CoOx individually, i.e., 347.6-fold for coloading Pd and CoOx, 53.7-fold for solely loading Pd, and 1.8-fold for solely loading CoOx. In the meantime, when CoOx and Pd were randomly deposited on Mo:BiVO4 (denoted as Mo:BiVO4-CoOx-Pd with a SEM image in Fig. S22), it was only 32% as active as CoOx/Mo:BiVO4/Pd, though these two catalysts had similar surface kinetics and selectivity. Further, the photocatalytic O2 evolution activity of CoOx/Mo:BiVO4/Pd was 3.2-fold higher than that of CoOx/Mo:BiVO4 (Fig. S9a). These results suggest that selective coloading of CoOx and Pd not only improved surface kinetics for O2 evolution and selectivity for H2O2 production as introduced above, but also tuned other critical processes like charge separation31. Furthermore, selectively coloading dual cocatalysts has been applied to enhance charge separation for sacrificial photocatalytic O2 evolution21,32,33. To this end, charge separation processes in Mo:BiVO4, CoOx/Mo:BiVO4, Pd/Mo:BiVO4, CoOx/Mo:BiVO4/Pd were thoroughly studied by transient absorption spectroscopy (TAS).
The TA spectra of photogenerated charge carriers in Mo:BiVO4 was examined upon band-gap excitation as shown in Fig. S23. The spectra exhibited strong absorption in 20,000–17,000 cm−1, a broad absorption in 170,000–5000 cm−1, and a weak absorption <5000 cm−1. These absorptions are attributed to trapped holes, deeply trapped electrons and free/shallowly trapped electrons, respectively (see supplementary discussions in Figs. S23 for detailed peak assignments)34,35,36. The dynamics of these photocarriers were compared in the ultrafast region (picosecond time-scale) as shown in Fig. 3. The photocarriers probed at 781 nm (~1.59 eV, deeply trapped electrons) and 505 nm (~2.45 eV, trapped holes) exhibited comparably slow decay kinetics, suggesting that these photocarriers recombined non-radiatively. In contrast, the photocarriers probed at 5000 nm (the free/shallowly trapped electrons) exhibited much faster decay kinetics, which well explains the weak absorption <5000 cm−1 shown in Fig. S23. This result indicates that the free/shallowly trapped electrons were rapidly trapped by the mid-gap states in Mo:BiVO4, consistent with previous studies35,36. Regardless of such an unfavorable effect, the TA signals for this kind of photocarriers were still observed to vary significantly after loading Pd or CoOx on Mo:BiVO4 (Fig. 3b). Loading Pd on the {010} facet of Mo:BiVO4 accelerated the decay of the free/shallowly trapped electrons since Pd captured electrons. In contrast, loading CoOx on the {110} facet lead to an opposite effect because CoOx captured holes and thus increased the electron population in Mo:BiVO4. At 50 ps, for instance, loading CoOx increased the intensity of TA signal for free/shallowly trapped electrons by ~66%, while loading Pd only decrease the intensity by ~6% compared to that of bare sample. The effect of CoOx on the dynamics of the free/shallowly trapped electrons was more intense than that of Pd because electron transfer to Pd competed with trapping of electrons into deep trap states below the CB. Electron trapping to deep trap states was represented by the strong TA signal probed at 781 nm as shown in Fig. 3a.

a Transient profiles of photocarriers probed at 505 nm (trapped holes), 781 nm (deeply trapped electrons), and 5000 nm (free/shallowly trapped electrons) in Mo:BiVO4. b Transient profiles of photocarriers probed at 5000 nm (free/shallowly trapped electrons) for Mo:BiVO4, CoOx/Mo:BiVO4 and Mo:BiVO4/Pd. Pump wavelength: 470 nm (4 μJ pulse−1).
Considering the crucial impact of electron trapping on electron-transfer processes in the picosecond region, the electron dynamics was further examined in the microsecond-millisecond region where photoexcited electrons in Mo:BiVO4 are expected to have already relaxed in the trap states. As shown in Fig. 4a, Pd and CoOx accelerated and decelerated the decay of free/shallowly trapped electrons, respectively, similar to the effects in picosecond region (Fig. 3b). This result indicates that in this time region, Pd and CoOx captured electrons and holes, respectively, as expected. The population of free/shallowly trapped electrons that remained in Mo:BiVO4 after electrons are captured by Pd can be determined by estimating the ratio of ΔAbsorbance displayed in Fig. 4a with respect to bare Mo:BiVO4 and CoOx/Mo:BiVO4 for Mo:BiVO4/Pd and CoOx/Mo:BiVO4/Pd, respectively. At 200 μs, for instance, 59% and 42% of free/shallowly trapped electrons remained in Mo:BiVO4 for Mo:BiVO4/Pd and CoOx/Mo:BiVO4/Pd, respectively. This implies that 41 and 58% of free/shallowly trapped electrons transferred to Pd for Mo:BiVO4/Pd and CoOx/Mo:BiVO4/Pd, respectively. More electrons transferred to Pd on CoOx/Mo:BiVO4/Pd than on Mo:BiVO4/Pd because of more efficient charge separation (synergistic charge separation) in the former case. To further justify this effect, the impact of CoOx and Pd on the decay kinetics of trapped holes in Mo:BiVO4 was investigated. To further justify this effect, the impact of CoOx and Pd on the decay kinetics of trapped holes in Mo:BiVO4 was investigated. As depicted in Fig. 4b, CoOx and Pd accelerated and decelerated the decay of accumulated trapped holes, respectively. Surprisingly, in CoOx/Mo:BiVO4/Pd, the effect of CoOx on capturing photogenerated holes in Mo:BiVO4 was compensated by the effect of Pd on accumulating trapped holes by efficiently trapping electrons. Furthermore, CoOx/Mo:BiVO4/Pd accumulated more trapped holes in Mo:BiVO4, even higher than that in Mo:BiVO4/Pd. This finding reveals that electrons and holes were efficiently separated at different facets. Aside from accumulating photogenerated holes in Mo:BiVO4 and facilitating the transfer of free/shallowly trapped electrons to Pd, loading cocatalysts also activated the deeply trapped electrons in Mo:BiVO4 for photocatalysis. As shown in Fig. S24, the TA signal and decay kinetics of the deeply trapped electrons in CoOx/Mo:BiVO4/Pd were similar to that of Pd/Mo:BiVO4 and apparently different from that of Mo:BiVO4 and CoOx/Mo:BiVO4. This result suggests that the deeply trapped electrons transferred to Pd and became available for subsequent surface reactions in Mo:BiVO4/Pd and CoOx/Mo:BiVO4/Pd. The charge transfer involving deeply trapped electrons to Pd cocatalyst is possible via tunneling and trap-to-trap hopping. A similar diffusion of trapped electrons is often proposed to take place on long-lived persistent phosphor materials37, wherein the trap states are situated at ~0.5–1.0 eV below the conduction band minimum. Aside from this diffusion process, re-excitation of deeply trapped electrons to the CB and eventually transfer to the Pd will be possible. Taken together, the above findings demonstrate that selectively coloading Pd and CoOx on the expected facets of Mo:BiVO4 significantly enhanced the charge separation and suppressed rapid charge-carrier trapping and recombination. Such positive effects are supposed to be achieved by tuning the energetics between cocatalysts and respective Mo:BiVO4 facet (Fig. 4c and S25).

Transient profiles of photocarriers probed a at 2000 nm (free/shallowly trapped electrons) and b 505 nm (trapped holes) for Mo:BiVO4, CoOx/Mo:BiVO4, Mo:BiVO4/Pd, and CoOx/Mo:BiVO4/Pd. Samples were excited by 470 nm laser pulses (Surelite I, duration: 6 ns, fluence: 3 mJ pulse−1, repetition: 1 Hz). c Impacts of cocatalysts on the energetics of Mo:BiVO4 facets.
In conclusion, we developed an inorganic semiconductor-based system for efficient overall photocatalytic H2O2 generation. Faceted Mo:BiVO4 particles was used as a light absorber and its {110} and {010} facets were selectively loaded with CoOx and Pd as WOR and ORR cocatalysts, respectively. These cocatalysts in such a configuration greatly improved the kinetics and selectivity for surface reactions. Furthermore, the spatial separation of cocatalysts on different facets of Mo:BiVO4 significantly enhanced the charge separation and suppressed rapid charge-carrier trapping and recombination, a key challenge in improving the efficiency of inorganic photocatalysts. With these merits, CoOx/Mo:BiVO4/Pd generated H2O2 with an AQY of 1.2% at full spectrum and a STH of 0.29%, a new record for inorganic semiconductor-based systems.
Methods
Catalyst preparation
Single crystal Mo:BiVO4 was prepared by heating the mixture of K2CO3 (1.047 g), MoO3 (1.8 mg) and V2O5 (2.272 g) in a ceramic crucible at a heating rate of 1.5 °C/min to 450 °C and annealing for 5 h in a muffle furnace. The obtained Mo:K3V5O14 (2 g) was mixed with Bi(NO3)3•5H2O (0.326 g) and dispersed in 50 mL deionized water under ultrasonication for 30 min. The mixture was stirred and heated at 70 °C for 10 h under ultrasonication, separated by centrifugation, washed with deionized water, and dried at 70 °C for 8 h. As-prepared Mo:BiVO4 (0.2 g) was dispersed in 100 mL water, followed by addition of 0.1 mol NaIO3 and 0.27 mL Co(NO3)2 stock (1.5 g/L). The mixture was irradiated at λ > 420 nm for 3 h using a xenon lamp solar simulator (model 300 DUV; Perfect Light, Inc., light intensity = 0.1 W/cm2), filtered, washed with deionized water, and dried at 60 °C for 8 h. The as-prepared CoOx/Mo:BiVO4 (0.15 g) was dispersed in 100 mL pure water, followed by addition of 0.18 mL Na2PdCl4 stock (3.3 g/L). The mixture was irradiated at λ > 420 nm for 3 h. As-prepared CoOx/Mo:BiVO4/Pd was filtered, washed with deionized water, and dried at 60 °C for 8 h. Same photodeposition method was also applied in preparing Mo:BiVO4/Pd except for using Mo:BiVO4 as the starting material. Mo:BiVO4-CoOx-Pd was prepared following an impregnation procedure by air-purging the mixture of Mo:BiVO4 (0.2 g), Co(NO3)2 (0.4 mg) and Na2PdCl4 (0.8 mg) (from stock) until dry, followed by heating in a ceramic crucible at a heating rate of 5 °C/min to 200 °C and annealing for 0.5 h under reductive condition (10% H2 and 90% Ar) in a tube furnace. The elemental compositions were analyzed by EDS, XPS, and ICP-MS (Figs. S26 and 27 and Table S3).
Photocatalyst characterizations
XPS measurements were performed with a Thermo Scientific 250Xi system with monochromatic Al Kα as the excitation source. The XRD patterns were recorded with a Bruker D8 Advance X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å) operated at 40 kV and 40 mA. The BET tests were performed by an ASAP 2460 with N2 analysis adsorptive at 77.2 K. SEM images were taken using a Hitachi SU-8010 microscope equipped with EDS at 30 kV. TEM images were taken using a Hitachi 7650 microscope operated at 100 kV. UV-DRS spectra was taken with a Shimadzu UV-3600 with a resolution of 0.1 nm. The ICP-MS measurement was performed with a NexION 300X (detection limit 1 μg/L).
Photocatalytic activity tests
Photocatalyst (24 mg) was dispersed in 12 mL deionized water with 1 M phosphate buffer (pH 7.4) in a custom-made reactor containing a quartz window. The catalyst was dispersed by ultrasonication for 10 min and purged with O2 for 20 min. All the equipment needed is shown in Fig. S28. Photocatalytic production of H2O2 was assessed by irradiating photocatalyst suspension using a xenon lamp solar simulator (model 300 DUV; Perfect Light Inc.) under water bath (12 ± 0.5 °C). The light intensity was adjusted to 100 mW/cm2 (AM 1.5 G; irradiation area = 1.83 cm2). The Xenon lame and the standard AM1.5 G (ASTMG 173) spectrum is shown in Fig. S29. For the wavelength-dependent AQY analysis, the photolysis was performed using LED light irradiation (model slight; Perfect Light, Inc.). At designated time points, 50 μL suspension was taken for analysis of H2O2 productions and diluted with phosphate buffer (pH = 7.4) to a H2O2 concentration (2–20 μM) that is most suitable for accurate H2O2 quantification, followed by centrifugation. Then 50 μL supernatant was taken and mixed with 50 μL solutions containing phosphate buffer (50 mM, pH = 7.4), ampliflu red (100 µM) and horseradish peroxidase (0.05 U/mL). Ampliflu red selectively reacted with H2O2 in the presence of horseradish peroxidase and formed the product resorufin. Resorufin in the mixture solution was quantified using an Agilent high-performance liquid chromatography coupled to a photo-diode array detector (detection at 560 nm); 50 µL of each sample was injected. The calibration in Fig. S30 is used to quantitatively analyze the H2O2 concentration. Separation was carried out in a C18 column at 20 °C with an isocratic mobile phase of 55% sodium citrate buffer (with 10% methanol (v/v), pH 7.4) and 45% methanol (v/v) at a flow rate of 0.5 mL min−1. The AQY was determined using:
where [H2O2]1h is the H2O2 concentration, Itot_p is the total photo flux of simulated sunlight irradiation (4.4 × 10−3 mol/m2/s, calculation details were shown in section S3), V is the volume of suspension (12 mL), A is the irradiation area (1.91 cm2 in this study), and t is the reaction time (1 h).
The STH was determined using:
Where ΔG(H2O2) is the free energy for H2O2 formation (117 kJ mol–1), Itot_e is the total intensity of simulated sunlight irradiation (0.1 W/cm2), V is the volume of suspension (12 mL), A is the irradiation area (1.91 cm2 in this study), and t is the reaction time (1 h).
Photoelectrochemical characterizations
Particle-based electrodes was prepared by a particle transfer method38. Firstly, 10 mg prepared CoOx/Mo:BiVO4 particles were suspended in a 450 μl isopropanol, followed by sonicated for 5 min. Secondly, the uniform suspension solution was dropped casting on a 1 × 3 cm glass substrate and fully dried in air. Thirdly, a thin layer of Ti (2–5 nm) was sputtered on the CoOx/Mo:BiVO4 particles. At last, the transferred electrode was sonicated for 10 s in water to remove the excessive particles on the surface. The electrochemical properties were assessed on a Biologic SP150 electrochemical analyzer using a three-electrode cell with the as-prepared electrode as the working electrode, Ag/AgCl as the reference electrode, and glassy carbon as the counter electrode. Cyclic voltammetry curves were obtained in a N2- or O2-saturated phosphate buffer solution (0.5 M, pH = 6.5). All potentials versus Ag/AgCl were converted to values vs. RHE.
Photocarriers dynamics by TAS measurement
Microsecond-millisecond TAS characterizations were performed using Nd:YAG laser system (Continuum, Surelite I) equipped with custom-built spectrometers39,40. Briefly, the TA spectra was measured from 20,000 cm−1 (500 nm)–1600 cm−1 (6250 nm) after band-gap excitation using 470 nm laser pulses (duration: 6 ns, fluence: 3 mJ pulse−1). For the IR probing, the light emitted from the MoSi2 coil was focused on the sample and then the reflected light from the sample was introduced to a grating spectrometer. The photoexcited electrons was probed at 2000 cm−1 (2000 nm). The monochromated light was detected by a mercury cadmium telluride (MCT) detector (Kolmar). Meanwhile, the photogenerated trapped electrons and holes in the non-modified and (CoOx, Pd)-modified Mo:BiVO4 were probed at 12,800 cm−1 (781 nm) and 19,800 cm−1 (505 nm, 2.45 eV), respectively. The output electric signal was amplified with an AC-coupled amplifier (Stanford Research Systems, SR560, 1 MHz). The time resolution of the spectrometer was limited to 1 μs by the response of the MCT detector. The output electric signal was amplified using AC-coupled amplifier with a bandwidth of 1 MHz, which measures responses from one microsecond to milliseconds. Three thousand responses were accumulated to obtain the intensity trace at one particular wavenumber or probe energy. The experiments were carried out in vacuum and at room temperature.
Femtosecond time-resolved absorption measurements were performed by employing a pump-probe technique based on femtosecond Ti:Sapphire laser system (Spectra Physics, Solstice & TOPAS prime; duration = 90 fs; repetition rate = 1 kHz)40. The time resolution of this spectrometer was ~90 fs. Briefly, in this experiment, the photoexcited charge carriers in the photocatalysts were probed at 19,800 cm−1 (505 nm), 12,800 cm−1 (781 nm), and 2000 cm−1 (5000 nm). In the mid-IR absorption measurement, the probe light transmitted from the sample was detected by an MCT detector (Kolmar), while in the visible to near-infrared region, the diffuse-reflected probe light was detected by photomultiplier (Hamamatsu Photonics, H11903-20). The samples were excited by 470-nm pulses (duration: 90 fs, fluence: 4 μJ pulse−1). To obtain the absorbance change with a good signal-to-noise ratio, the pump pulses were chopped using an optical chopper at 500 Hz and the signal acquisition was carried out on a shot-by-shot basis at a rate of 1 kHz. The decay curves were obtained at 10 ps intervals and accumulated signals were averaged over 1000–4000 scans for one point. TAS measurements were performed in vacuum (base pressure ~ 10−5 Torr). For sample preparation, each Mo:BiVO4 and (Pd, CoOx)-loaded Mo:BiVO4 powders was prepared by dispersing the powder on isopropanol and then drop-casted on a circular CaF2 substrate and subsequently dried naturally in air to obtain a powder film with a density of ~ 1.25 mg cm−2.
Data availability
Source data are provided with this paper.
References
Ma, J. J. et al. Photochemical intermolecular dearomative cycloaddition of bicyclic azaarenes with alkenes. Science 371, 1338–1345 (2021).
Romero, E., Novoderezhkin, V. I. & van Grondelle, R. Quantum design of photosynthesis for bio-inspired solar-energy conversion. Nature 543, 355–365 (2017).
Chen, S. S., Qi, Y., Li, C., Domen, K. & Zhang, F. X. Surface strategies for particulate photocatalysts toward artificial photosynthesis. Joule 2, 2260–2288 (2018).
Shiraishi, Y. et al. Resorcinol-formaldehyde resins as metal-free semiconductor photocatalysts for solar-to-hydrogen peroxide energy conversion. Nat. Mater. 18, 985–993 (2019).
Xia, C., Xia, Y., Zhu, P., Fan, L. & Wang, H. T. Direct electrosynthesis of pure aqueous H2O2 solutions up to 20% by weight using a solid electrolyte. Science 366, 226–231 (2019).
Xue, Y., Wang, Y. T., Pan, Z. H. & Sayama, K. Electrochemical and photoelectrochemical water oxidation for hydrogen peroxide production. Angew. Chem. Int. Edit. 60, 10469–10480 (2021).
Mase, K., Yoneda, M., Yamada, Y. & Fukuzumi, S. Seawater usable for production and consumption of hydrogen peroxide as a solar fuel. Nat. Commun. 7, 11470 (2016).
Shi, X. J. et al. Understanding activity trends in electrochemical water oxidation to form hydrogen peroxide. Nat. Commun. 8, 701 (2017).
Fuku, K. et al. Photoelectrochemical hydrogen peroxide production from water on a WO3/BiVO4 photoanode and from O2 on an Au cathode without external bias. Chem. Asian J. 12, 1111–1119 (2017).
Tian, Z. L. et al. Efficient photocatalytic hydrogen peroxide generation coupled with selective benzylamine oxidation over defective ZrS3 nanobelts. Nat. Commun. 12, 2039 (2021).
Hisatomi, T. & Domen, K. Reaction systems for solar hydrogen production via water splitting with particulate semiconductor photocatalysts. Nat. Catal. 2, 387–399 (2019).
Qureshi, M. et al. Contribution of electrolyte in nanoscale electrolysis of pure and buffered water by particulate photocatalysis. Sustain. Energy Fuels 2, 2044–2052 (2018).
Teng, Z. et al. Atomically dispersed antimony on carbon nitride for the artificial photosynthesis of hydrogen peroxide. Nat. Catal. 4, 374–384 (2021).
Wu, Q. Y. et al. A metal-free photocatalyst for highly efficient hydrogen peroxide photoproduction in real seawater. Nat. Commun. 12, 483 (2021).
Wang, X. C. et al. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 8, 76–80 (2009).
Xiao, J. D. et al. Is C3N4 chemically stable toward reactive oxygen species in sunlight-driven water treatment? Environ. Sci. Technol. 51, 13380–13387 (2017).
Hirakawa, H. et al. Au nanoparticles supported on BiVO4: effective inorganic photocatalysts for H2O2 production from water and O2 under visible light. Acs Catal. 6, 4976–4982 (2016).
Zhang, K. et al. Near-complete suppression of oxygen evolution for photoelectrochemical H2O oxidative H2O2 synthesis. J. Am. Chem. Soc. 142, 8641–8648 (2020).
Wang, Q. et al. Scalable water splitting on particulate photocatalyst sheets with a solar-to-hydrogen energy conversion efficiency exceeding 1%. Nat. Mater. 15, 611–615 (2016).
Kudo, A., Omori, K. & Kato, H. A novel aqueous process for preparation of crystal form-controlled and highly crystalline BiVO4 powder from layered vanadates at room temperature and its photocatalytic and photophysical properties. J. Am. Chem. Soc. 121, 11459–11467 (1999).
Li, R. G. et al. Spatial separation of photogenerated electrons and holes among {010} and {110} crystal facets of BiVO4. Nat. Commun. 4, 1432 (2013).
Campos-Martin, J. M., Blanco-Brieva, G. & Fierro, J. L. G. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Edit. 45, 6962–6984 (2006).
Jin, Z. et al. Hydrophobic zeolite modification for in situ peroxide formation in methane oxidation to methanol. Science 367, 193–197 (2020).
Adams, J. S. et al. Solvent molecules form surface redox mediators in situ and cocatalyze O2 reduction on Pd. Science 371, 626–632 (2021).
Shiraishi, Y., Ueda, Y., Soramoto, A., Hinokuma, S. & Hirai, T. Photocatalytic hydrogen peroxide splitting on metal-free powders assisted by phosphoric acid as a stabilizer. Nat. Commun. 11, 3386 (2020).
Gamelin, D. R. Water splitting: Catalyst or spectator? Nat. Chem. 4, 965–967 (2012).
Klahr, B., Gimenez, S., Fabregat-Santiago, F., Bisquert, J. & Hamann, T. W. Photoelectrochemical and impedance spectroscopic investigation of water oxidation with “Co-Pi”-coated hematite electrodes. J. Am. Chem. Soc. 134, 16693–16700 (2012).
Maeda, K. & Domen, K. Photocatalytic water splitting: recent progress and future challenges. J. Phys. Chem. Lett. 1, 2655–2661 (2010).
Freakley, S. J. et al. Palladium-tin catalysts for the direct synthesis of H2O2 with high selectivity. Science 351, 965–968 (2016).
Schroder, M. et al. Hydrogen evolution reaction in a large-scale reactor using a carbon nitride photocatalyst under natural sunlight irradiation. Energy Technol-Ger. 3, 1014–1017 (2015).
Yang, J. H., Wang, D. G., Han, H. X. & Li, C. Roles of cocatalysts in photocatalysis and photoelectrocatalysis. Acc. Chem. Res. 46, 1900–1909 (2013).
Li, R. G., Han, H. X., Zhang, F. X., Wang, D. G. & Li, C. Highly efficient photocatalysts constructed by rational assembly of dual-cocatalysts separately on different facets of BiVO4. Energ Environ. Sci. 7, 1369–1376 (2014).
Qi, Y. et al. Redox based visible-light-driven Z-scheme overall water splitting with apparent quantum efficiency exceeding 10%. Joule 2, 2393–2402 (2018).
Ma, Y. M., Pendlebury, S. R., Reynal, A., Le Formal, F. & Durrant, J. R. Dynamics of photogenerated holes in undoped BiVO4 photoanodes for solar water oxidation. Chem. Sci. 5, 2964–2973 (2014).
Yamakata, A., Ranasinghe, C. S. K., Hayashi, N., Kato, K. & Vequizo, J. J. M. Identification of individual electron- and hole-transfer kinetics at CoOx/BiVO4/SnO2 double heterojunctions. Acs Appl. Energ. Mater. 3, 1207–1214 (2020).
Suzuki, Y. et al. Rational interpretation of correlated kinetics of mobile and trapped charge carriers: analysis of ultrafast carrier dynamics in BiVO4. J. Phys. Chem. C 121, 19044–19052 (2017).
Xu, J., Murata, D., Ueda, J., Viana, B. & Tanabe, S. Toward rechargeable persistent luminescence for the first and third biological windows via persistent energy transfer and electron trap redistribution. Inorg. Chem. 57, 5194–5203 (2018).
Zhong, M. et al. Surface modification of CoOx loaded BiVO4 photoanodes with ultrathin p-Type NiO layers for improved solar water oxidation. J. Am. Chem. Soc. 137, 5053–5060 (2015).
Yamakata, A., Vequizo, J. J. M. & Kawaguchi, M. Behavior and energy state of photogenerated charge carriers in single-crystalline and polycrystalline powder SrTiO3 studied by time-resolved absorption spectroscopy in the visible to mid-infrared region. J. Phys. Chem. C 119, 1880–1885 (2015).
Vequizo, J. J. M. et al. Trapping-induced enhancement of photocatalytic activity on brookite TiO2 powders: comparison with anatase and rutile TiO2 powders. Acs Catal. 7, 2644–2651 (2017).
Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC, No. 22006129), the Fundamental Research Funds for the Central Universities (No. 2020FZZX001-06) and JSPS KAKENHI Grant Number JP20K22556. We are grateful to Sudan Shen (State Key Laboratory of Chemical Engineering at Zhejiang University) and analysis center of agrobiology and environmental sciences for help in TEM and SEM measurements, respectively.
Author information
Authors and Affiliations
Contributions
C.C., Z.P., and K.D. designed research; T.L. and Z.P. synthesized the catalysts and conducted performance test; T.L. conducted SEM, TEM, and XPS measurements; J.J.M.V., K.K., A.Y. performed TAS measurements and analysis. T.L., C.C., Z.P., B.C., K.K., and K.D. analyzed data; T.L., C.C., Z.P., B.C., and K.D. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Jong Hyeok Park, Thierry Toupance and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, T., Pan, Z., Vequizo, J.J.M. et al. Overall photosynthesis of H2O2 by an inorganic semiconductor. Nat Commun 13, 1034 (2022). https://doi.org/10.1038/s41467-022-28686-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28686-x
This article is cited by
-
Keto-anthraquinone covalent organic framework for H2O2 photosynthesis with oxygen and alkaline water
Nature Communications (2024)
-
Linkage-engineered donor–acceptor covalent organic frameworks for optimal photosynthesis of hydrogen peroxide from water and air
Nature Catalysis (2024)
-
Efficient and stable visible-light-driven Z-scheme overall water splitting using an oxysulfide H2 evolution photocatalyst
Nature Communications (2024)
-
Synergistic interaction and chemically bonded association between ZIF-8 and C-doped g-C3N4 for enhancement of visible light photocatalytic H2O2 production
Journal of Sol-Gel Science and Technology (2024)
-
Exploring the Roles of Single Atom in Hydrogen Peroxide Photosynthesis
Nano-Micro Letters (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
