
Abstract
Ultrafast infrared nano-imaging has demonstrated access to ultrafast carrier dynamics on the nanoscale in semiconductor, correlated-electron, or polaritonic materials. However, mostly limited to short-lived transient states, the contrast obtained has remained insufficient to probe important long-lived excitations, which arise from many-body interactions induced by strong perturbation among carriers, lattice phonons, or molecular vibrations. Here, we demonstrate ultrafast infrared nano-imaging based on excitation modulation and sideband detection to characterize electron and vibration dynamics with nano- to micro-second lifetimes. As an exemplary application to quantum materials, in phase-resolved ultrafast nano-imaging of the photoinduced insulator-to-metal transition in vanadium dioxide, a distinct transient nano-domain behavior is quantified. In another application to lead halide perovskites, transient vibrational nano-FTIR spatially resolves the excited-state polaron-cation coupling underlying the photovoltaic response. These examples show how heterodyne pump-probe nano-spectroscopy with low-repetition excitation extends ultrafast infrared nano-imaging to probe elementary processes in quantum and molecular materials in space and time.
Similar content being viewed by others


Introduction
Functional materials offer intriguing applications based on their unique optical and electronic properties, such as quantum phase transition based Mott transistors1, polaronic carrier transport in lead halide perovskite photovoltaics2, coherent phonons and vibrations driving singlet fission3, or electronic energy transfer in light-harvesting complexes4. These properties emerge from the interplay of the elementary electronic, vibrational, and phononic quantum states. By exciting one of these degrees of freedom out of equilibrium and probing their dynamic response, ultrafast spectroscopy disentangles mode-coupling and competing interactions that are otherwise convoluted in static spectroscopy5. In addition, when the system is perturbed far-from-equilibrium, it can be driven into new photoinduced quantum states, enabling ultrafast optical control of metallic, superconductive, and polaronic functionalities6. Further, strong and ultra-short laser fields lead to extreme nonlinear optical phenomena with applications from high harmonic generation7,8 to light-field petahertz electronics9.
However, these ultrafast processes and material functions often exhibit spatial heterogeneities associated with, e.g., lattice defects, strain, grain boundaries, and nonuniform doping from atomic to device scales10,11,12. To address the associated spatiotemporal dynamics, a variety of ultrafast nanoimaging techniques have been developed, including ultrafast transmission electron microscopy (TEM)13, photoemission electron microscopy (PEEM)14, or X-ray microscopy15. Yet, these techniques often cannot readily resolve the low-energy electronic and lattice coupling and dynamics that critically control the material properties.
In contrast to these established techniques, scattering scanning near-field optical microscopy (s-SNOM) has recently been implemented with ultrafast time resolution16,17,18,19,20,21,22,23,24,25,26,27,28. In particular, ultrafast infrared nanoimaging based on nano-FTIR spectroscopy19 and electro-optic sampling (EOS)24,29 has enabled access to nanoscale carrier dynamics in semiconductors19,21,24, correlated electron17, and polaritonic materials20,23 with spatiotemporal-spectral resolutions. Ultrafast infrared nanoimaging to date has probed strong and short-lived carrier and collective polaritonic excitations based primarily on high-repetition-rate (>10 MHz) laser pump sources, which offer a high duty cycle to enhance the signal-to-noise ratio.
Important long-lived transients in materials arise from many-body interactions induced by a strong perturbation, such as photoinduced phases in correlated electron systems30,31,32,33 or polaron dynamics in organic–inorganic hybrid photovoltaics34,35. In these systems, cooperative dynamics among the many degrees of freedom36,37 result in the formation of nonequilibrium states with nano- to microsecond lifetimes. However, high-repetition excitation precludes quantitative probing of the ultrafast dynamics of those states. Full spatiotemporal-spectral resolution with low-repetition excitation has only been achieved recently21,23, while the reduced duty cycle limits signal intensity and contrast. This calls for a generalized approach with enhanced excited-state contrast at low-repetition-rate excitation.
Here, we demonstrate non-degenerate heterodyne pump-probe infrared scattering scanning near-field optical microscopy (HPP IR s-SNOM) with low-repetition-rate modulated excitation, which provides simultaneous space, time, frequency, and phase resolutions with high sensitivity. A modulated optical pump excites the system into an excited state, followed by infrared heterodyne probing of the transient low-energy electronic and vibrational response. The induced third-order nano-localized polarization is isolated by sideband lock-in detection and directly detected interferometrically in the time domain, enabling ultrafast nanoimaging with high contrast even with the low-repetition excitation rate of 1 MHz. The excitation-modulated HPP IR s-SNOM thus provides the analog of ground-state nano-FTIR spectroscopy, resolving the transient and nano-localized excited-state response. The spectrally resolved near-field pump-probe signal is modeled to quantitatively extract the spatiotemporal evolution of the transient dielectric function of the material based on a combination of finite dipole and four-layer reflection model.
As a representative application to quantum materials, we perform nanoimaging of the electron dynamics associated with the ultrafast photoinduced insulator-to-metal transition (IMT) in vanadium dioxide (VO2), resolving transient domain dynamics that is distinct from the established strain-induced heterogeneity in the thermally induced transition10,16,17,38. In another application to soft molecular materials, based on transient nano-spectroscopy with the spectral resolution, we directly resolve heterogeneity in polaron–cation coupling that controls the photovoltaic response by probing the vibrational dynamics in a triple cation perovskite34,35.
The low-repetition-rate excitation with highly sensitive detection leads to the study of photoinduced phase transitions as well as soft photovoltaic materials with long-lived carrier and vibrational responses. The approach thus holds promise to establish the missing links between the elementary processes at the nanoscale and the associated macroscopic optical, photophysical, catalytic, or electronic properties of a wide range of functional materials.
Results
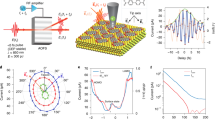
Figure 1a–c shows the schematics of HPP IR s-SNOM with femtosecond pump (Yb:KGW amplified laser with ~185 fs full-width-at-half-maximum (FWHM) pulse duration, 1030 nm center wavelength, ~1 MHz repetition rate, Pharos, Light Conversion), and broadband infrared nano-probe spectroscopy (tunable at 5–10 μm, ~170 fs FWHM pulse duration, Orpheus OPA/DFG, Light Conversion), with overall time resolution of ~200 fs (see Supplementary Fig. S1), and ~40 nm spatial resolution as given by the apex radius of the metallic scanning probe tip (Supplementary Fig. S4). IR s-SNOM is implemented with an asymmetric Michelson interferometer, consisting of sample and reference arms as established39,40. An atomic force microscope (Innova AFM, Bruker) in the sample arm is operated in tapping mode with a tip-tapping frequency ωt (for details, see Supplementary Note 1). Pump and probe pulses, separated by time delay T, are collinearly directed onto the AFM tip (ARROW-NCPt, NanoAndMore USA) with an off-axis parabolic mirror (NA = 0.45). The fundamental or frequency-doubled pump source is modulated at frequency ΩM by an acousto-optic modulator (AOM), or a mechanical chopper. The pump-induced excited-state population and corresponding ground-state bleach (Fig. 1b) are detected by pump-probe s-SNOM spectroscopy. Here, the tip-scattered near-field probe signal ENF is then optically heterodyned with the local oscillator field ELO from the reference arm, with variable delay t, and detected by a HgCdTe (MCT) detector. As in conventional IR s-SNOM, lock-in demodulation (HF2LI, Zurich Instruments) at nωt (n = 1, 2, 3. . . ), combined with the interferometric heterodyne detection, isolates \({I}_{{{{{{{{\rm{NF,Het}}}}}}}}} \sim {E}_{{{{{{{{\rm{NF}}}}}}}}}{E}_{{{{{{{{\rm{LO}}}}}}}}}^{* }\) that provides background-free nano-localized imaging contrast39,40.

a HPP IR s-SNOM, with the ~ 185 fs optical 1.2/2.4 eV (1030/515 nm) pump excitation, ~170 fs tunable mid-IR 0.12–0.25 eV (5–10 μm) probe, and interferometric heterodyne near-field detection. AOM acousto-optic modulator, DFG difference-frequency generation, OAP off-axis parabolic mirror, OPA optical parametric amplifier, Osc .+RA oscillator and regenerative amplifier laser system. b Far-from-equilibrium excitation followed by mid-infrared probe of the transient low-energy vibrational and electronic response coupled to the excited state. c The tip-localized time domain signal of \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\), from which the pump-induced change in the nano-localized complex dielectric function \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) is retrieved with spatiotemporal-spectral resolution. d Sideband-demodulated pump-probe interferogram ΔINF(t, T) for a Ge reference sample. e T-dependent pump-probe transients from two different ELO phase values ΔINF(ϕ = 0, T) and ΔINF(ϕ = π, T), the derived frequency-averaged heterodyne pump-probe amplitude relaxation RHPP(T), and the self-homodyne pump-probe signal relaxation ΔISHPP(T). Inset: tip-enhanced pump excitation in nano-localized probe volume, leading to faster relaxation from higher excited carrier density.
In contrast to previous pump-probe IR s-SNOM implementations with un-modulated pump excitation19,24, with excitation modulation at ΩM we perform sideband lock-in demodulation at nωt ± ΩM to directly detect the photoinduced change in the near-field signal ΔINF(T), similar to a recent implementation in ultrafast THz-EOS s-SNOM29. This detection scheme enhances the signal-to-noise ratio by a factor of >4 in comparison to the conventional scheme, corresponding to a reduction in the data acquisition time by more than one order of magnitude (Supplementary Fig. S9). By scanning ELO in time t, we then acquire the heterodyne pump-probe interferogram ΔIHPP(t, T) in the time domain.
ΔIHPP(t, T) is then Fourier transformed with respect to t and deconvolved with \({E}_{{{{{{{{\rm{LO}}}}}}}}}(\overline{\nu })\) to yield the real and imaginary parts of \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu },T)\) (Fig. 1c), and with much higher signal-to-noise ratio compared to the pump un-modulated case (see Supplementary Fig. S9 for examples). \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu },T)\) quantitatively relates to the transient photoinduced change in the complex dielectric function, which can be retrieved through the model fitting. HPP IR s-SNOM thus provides the full 4D characterization of the transient material response with spatial (X, Y), temporal (T), and spectral (\(\overline{\nu }\)) resolutions, with high excited-state contrast enabled by the selective detection of the pump-induced near-field response.
As an example, Fig. 1d shows a representative pump-probe interferogram ΔIHPP(t, T) of a germanium reference sample, acquired by continuously scanning the ELO delay t at selected pump-probe time delays T. The t-dependent profiles of ΔIHPP(t, T) is determined and limited by the spectral profile of the infrared-probe pulse due to the nearly instantaneous and spectrally broad transient free carrier Drude response of germanium41. To determine the spectrally averaged amplitude relaxation RHPP(T), ΔINF is recorded as T is scanned for sets of two distinct ELO phase values, ϕ = 0 and π (see Fig. 1d) for constructive and destructive interference near zero-path difference (ZPD), to yield the amplitude decay of the HPP signal as RHPP(T) = ΔINF(ϕ = 0, T) − ΔINF(ϕ = π, T) (Fig. 1e). This approach of extracting the spectrally averaged amplitude based on the two-phase measurement is applicable due to the narrower laser bandwidth of ~100 cm−1 FWHM compared to the much broader Drude response in germanium (see Supplementary Note 1).
The HPP signal relaxation RHPP(T) (red) probing the recombination of photoinduced carriers is notably distinct from the self-homodyne pump-probe (SHPP) decay ΔISHPP(T) (blue), recorded without interference with ELO. ΔISHPP(T) shows a small initial rise followed by a slow decay41, while RHPP(T) only shows a decay yet faster than ΔISHPP(T). We attribute this difference to the convolution of the time-dependent far-field background in ΔISHPP(T)22. The tip-enhanced pump field results in higher local carrier density compared to the far-field pumped background (Fig. 1e, inset). With its pure local probe character, RHPP(T) thus exhibits a faster relaxation, reflecting the enhanced recombination and scattering induced by the higher excited carrier density. This observation critically highlights the necessity of interferometric heterodyne detection to quantify the nano-localized pump-probe dynamics, particularly in the high fluence regime where the observed dynamics are highly sensitive to the local pump intensity.
Theory of HPP IR s-SNOM
To evaluate excited-state absorption resonances at the nanoscale18,21,23,24, which contain critical information associated with many-body interactions, we extend the dipole model in combination with multilayer reflection, established for both ground-state42,43,44 and ultrafast s-SNOM18,24, to quantitatively relate the experimentally observed sideband-demodulated pump-probe interferogram ΔIHPP(t) to the underlying transient dielectric function \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\). Importantly, we find that \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) as the direct observable in transient vibrational nano-spectroscopy is generally the convolution of the excited-state absorption and Fano-type interference. This underscores the importance of the theoretical framework to retrieve \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) that purely encodes the excited-state absorption to distinguish these two contributions.
We illustrate the application for the case of a transient molecular vibrational response coupled to photoinduced carriers35,45, with a 600-nm thick sample film coated on a substrate (Fig. 2a). We assume ground-state and excited-state dielectric functions, \({\tilde{\epsilon }}^{(0)}(\overline{\nu })\) and \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\), with vibrational resonances centered at \({\overline{\nu }}_{{{{{{{{\rm{gs}}}}}}}}}\) and \({\overline{\nu }}_{{{{{{{{\rm{ex}}}}}}}}}\), respectively (Fig. 2b). The pump excitation modifies the dielectric function of the sample film to \({\tilde{\epsilon }}^{{{{{{{{\rm{(e)}}}}}}}}}={\tilde{\epsilon }}^{(0)}+{{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}\), down to a depth d1 = 100 nm determined by the absorption coefficient. The remaining depth of d2 = 500 nm is left unperturbed at \({\tilde{\epsilon }}^{(0)}\). The near-field scatter with and without pump excitation is calculated based on a combination of finite dipole42 and four-layer models for a sinusoidally modulated tip-sample distance and is demodulated with the second harmonic tip-tapping frequency. While the finite dipole model is known to quantitatively retrieve vibrational resonances42, the four-layer reflection model46 accounts for partial excitation of a material layer. For details, see Supplementary Note 2.

a Theoretical framework to quantitatively relate the measured pump-induced near-field signal change ΔENF to the transient complex dielectric function \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}\). b Example ground-state complex dielectric function \({\tilde{\epsilon }}^{(0)}(\overline{\nu })\) and its pump-induced change \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) for a model vibrational response, with the ground-state resonance at \({\overline{\nu }}_{{{{{{{{\rm{gs}}}}}}}}}\) and the frequency-shifted excited-state resonance at \({\overline{\nu }}_{{{{{{{{\rm{ex}}}}}}}}}\). c Simulated heterodyne IR s-SNOM interferograms for the ground-state INF(t) and its pump-induced change ΔIHPP(t). d \({E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) and \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) in frequency-domain, obtained from Fourier transform and \({E}_{{{{{{{{\rm{LO}}}}}}}}}(\overline{\nu })\) deconvolution of the time-domain interferograms in panel c.
Figure 2c shows the calculated ground-state s-SNOM interferogram INF(t) (black). The pump-induced response \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\), which consists of a broadband carrier response and a vibrational excited-state absorption resonance, then gives rise to the pump-probe interferogram ΔIHPP(t) (red). As expected, the non-resonant term in \({\tilde{\epsilon }}^{(0)}(\overline{\nu })\) leads to a nearly instantaneous center burst of INF(t) limited by the duration of the probe pulse followed by a vibrational free-induction decay (FID)40. ΔIHPP(t) is phase-shifted due to the complex broadband response of photoinduced carriers, with the FID from the frequency-shifted photoinduced vibrational resonance.
The Fourier transforms of INF(t) and ΔIHPP(t), after deconvolution with \({E}_{{{{{{{{\rm{LO}}}}}}}}}(\overline{\nu })\), yield the spectral profiles of the ground-state \({E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) (black) and its photoinduced change \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) (red) as shown in Fig. 2d. As can be seen, \({E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) approximates the ground-state complex dielectric function \({\tilde{\epsilon }}^{(0)}(\overline{\nu })\) as established40,42. In contrast, the spectral profile of \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) is a complex convolution of ground- and excited-state dielectric responses. While \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) exhibits a resonance corresponding to the excited-state absorption at \({\overline{\nu }}_{{{{{{{{\rm{ex}}}}}}}}}\), it is also compounded by Fano-type interference of the vibrational response with the pump-induced broadband carrier response. In the analysis of the experimental data presented below, with the application of the finite dipole model42 based on Drude-carrier and Lorentzian-vibrational material responses, we retrieve the transient complex dielectric function \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) quantitatively from \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\). \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) purely describes the excited-state absorption and thus provides the fundamental electronic and vibrational coupling and their dynamics (see Supplementary Note 2).
We note that Fano-type interference is generally expected to give rise to a nontrivial lineshape in ΔENF whenever spectrally distinct and narrow resonances interfere with pump-induced broadband response, as is generally the case for visible-to-UV pump and infrared-to-THz probe nano-spectroscopy of a wide range of molecular and soft materials. The model is generalizable to other types of resonant excitation and is universally applicable to retrieve the underlying ground- and excited-state dielectric functions in HPP s-SNOM, enabling the quantitative separation of excited-state absorption and Fano-type interference.
Phase-controlled imaging of quantum IMT in vanadium dioxide nanobeam
We first apply HPP IR s-SNOM to nanoimaging heterogeneity in the photoinduced quantum phase transition and associated electron dynamics in a nanobeam of the correlated electron material vanadium dioxide (VO2). VO2 exhibits a thermal insulator-to-metal transition (IMT) at ~340 K, which involves a bandgap collapse with a change from an insulating monoclinic to a metallic rutile phase10,38,47,48. The IMT can also be induced optically10,16,17,30,32,33 with promising applications for, e.g., ultrafast photoswitches48.
The phase transition is believed to be caused by a complex interplay between electron–electron correlation and electron–phonon coupling, with details regarding the exact mechanism still remaining elusive49,50. As a manifestation of its intricacy, both the thermally induced and photoinduced transition of VO2 exhibit spatial heterogeneity and are susceptible to local strain and chemical heterogeneity10,16,17,38,47. Recent optical pump/infrared-probe s-SNOM of the photoinduced IMT of VO2 revealed a spatial profile distinct from that of thermal strain-induced heterogeneity and was attributed to possible stoichiometric zoning16. However, that measurement was performed in SHPP, and the influence of the far-field background on the measured dynamics could not be ruled out22.
Here, we apply HPP IR s-SNOM to VO2 nanobeams51 to quantify the heterogeneous electron dynamics associated with the IMT in a background-free and phase-resolved manner (Fig. 3a). The micro-Raman spectrum of the nanobeams on a silicon substrate shows the VO2 initially in the M2 phase at room temperature52 (see Supplementary Fig. S4). Following the NIR pump excitation above the bandgap at 1.2 eV (1030 nm, ~2 mJ/cm2 fluence), we measure the HPP interferogram ΔIHPP(t) with a 0.2 eV (6 μm, 1670 cm−1, ~50 μJ/cm2 fluence) mid-infrared probe at T = 0 ps (Fig. 3b). We adjust the phase of ELO relative to that of the photoinduced response ΔIHPP(t) as indicated by ϕ = 0 and π in the inset. Figure 3c (top) then shows the time-resolved HPP amplitude relaxation RHPP(T), acquired at three selected nanobeam locations based on this two-phase measurement.

a Schematic of NIR pump (1.2 eV, ~2 mJ/cm2) infrared-probe (0.2 eV) ultrafast nanoimaging of VO2 (left) probing the complex dielectric functions of the insulator and metallic states (right, adapted from a complex refractive index measurement by Wan et al.53). b Time-domain HPP near-field interferogram acquired on a VO2 nanobeam at T = 0 ps, with the shifted phase between the ground-state INF(t) and the photoinduced state ΔIHPP(t) (inset). c The HPP signal amplitude relaxation RHPP(T) measured by the two-phase T-dependent (TD) scans at selected locations (top) and the equivalent acquired based on space-domain (SD) scans (bottom). d Ultrafast HPP nanoimaging of the VO2 nanobeam with the AFM topography. e The HPP amplitude mapping at T = 0 ps, derived from mappings at the two ELO phases, ϕ = 0 and π. f Time-dependent evolution of the HPP signal along L1 and L2 in panel e with the corresponding topography of the nanobeam.
While at position 2 and 3 the photoinduced infrared response fully relaxes within ~2 ps and signifies excited carrier relaxation, at position 1 the pump-induced signal plateaus beyond 10 ps, characteristic for the formation of the metastable metallic state10,16,30,33. By adjusting the pump fluence, we can locally switch the behavior between ps-transient carrier relaxation in the photo-doped insulating state and the photoinduced IMT with its ultrafast nucleation of the metallic state followed by a slower transient domain growth (Supplementary Fig. S4).
Figure 3d then shows the corresponding ultrafast HPP nanoimaging, derived from imaging the sideband-demodulated intensity ΔINF for the two ELO phase values ϕ = 0 and π (example shown in Fig. 3e for T ps) as RHPP({X, Y}) = ΔINF(ϕ = 0, {X, Y}) − ΔINF(ϕ = π, {X, Y}). In addition to the non-uniformity of the decay at individual representative locations as shown in Fig. 3c (bottom), the line profiles across and along the cR axis of the VO2 crystal both demonstrate dynamically evolving spatial disorder in the HPP signal amplitude (Fig. 3f). While the heterogeneity along the cR axis is particularly pronounced in this example, in other cases a heterogeneity perpendicular to the cR axis stands out (see Supplementary Fig. S5). By measuring multiple nanobeams with different orientations, we verify that the nonuniform spatial profile of the pump beam does not account for the observed transient heterogeneity, being due to intrinsic heterogeneities in each nanobeam (Supplementary Fig. S6).
As is apparent from Fig. 3b (inset) comparing INF(t) and ΔIHPP(t) in time, the transient near-field signal ΔENF is phase-shifted from the ground-state response ENF, due to the different dielectric response between the insulating and metallic phases as is known for the thermally induced IMT of a VO2 film (see Fig. 3a, right)53. By fitting both the amplitude (∣ΔENF∣/∣ENF∣ ~ 0.2) and the optical phase shift (Δϕ = ϕ(ΔENF) − ϕ(ENF) ~ 57∘), we retrieve the actual photoinduced change in the dielectric constant of \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}} \sim 0.25\,+\,1.0i\) at the probe energy of \({\overline{\nu }}_{{{{{{{{\rm{probe}}}}}}}}}=1670\) cm−1. In comparison with \({\tilde{\epsilon }}_{{{{{{{{\rm{metal}}}}}}}}}^{{{{{{{{\rm{therm.}}}}}}}}}\approx 3\,+\,84i\) for the thermally induced metallic phase in an extended film sputtered on a silicon substrate53, our finding of \(| {{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}| \, < < \, | {\tilde{\epsilon }}_{{{{{{{{\rm{metal}}}}}}}}}|\) implies that the photoinduced excited state is only partially metallic at the pump fluence of ~2 mJ/cm2. \({{{{{{{\rm{Im}}}}}}}}[{{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}] \, > \,{{{{{{{\rm{Re}}}}}}}}[{{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}]\) is in agreement with the case of a thermally induced metallic phase, with the extracted nano-localized transient dielectric phase \(\arg ({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}})=76\pm {2}^{\circ }\) slightly smaller than that of a thermally induced metallic phase \({\tilde{\epsilon }}_{{{{{{{{\rm{metal}}}}}}}}}^{{{{{{{{\rm{therm.}}}}}}}}}=9{5}^{\circ }\pm 1{6}^{\circ }\), which is derived from ellipsometry literature values for VO2 films. With the samples prepared under different conditions in the literature47,53,54,55,56, the deviation might arise from different morphology, strain, or doping, but also a possibly distinct quantum nature of the photoinduced phase in comparison to a thermally induced metallic phase.
A pronounced heterogeneity along the cR axis (Fig. 3d, f) has been established for thermally induced metallic VO2 nanobeams and is attributed to nonuniform local strain10,17,38. In contrast, the transient heterogeneity in the photoinduced IMT is predominantly perpendicular with respect to the cR axis and is consistent with previous work that implied a distinct origin such as intrinsic stoichiometric zoning16. The co-existence of the two types of the dynamic heterogeneities both along and across the cR axis highlights the complexity of the photoinduced IMT, the exact mechanism of which remains unsolved to date. HPP IR s-SNOM, with its space and time resolutions, is thus applicable to address the intracrystalline heterogeneity of a photoinduced IMT in VO2 to guide the development of device applications of VO2 nanostructures with ultrafast control. With quantitative phase resolution, it also lays the groundwork to address the unsolved question regarding the distinct nature of quantum states underlying the photoinduced vs. thermally induced metallic phases30,32,49.
Transient vibrational nano-spectroscopy of the polaron–cation coupling in lead halide perovskites
In the extension of HPP IR s-SNOM to soft and molecular materials, we demonstrate ultrafast vibrational nano-spectroscopy of lead halide perovskites (Fig. 4a) to resolve electron-vibration coupling and its spatial heterogeneity. Lead halide perovskites exhibit an extraordinary optoelectronic response, characterized by the spontaneous formation of long-lived free carriers and long diffusion lengths57. Their unusual photovoltaic performance is believed to arise from polaron formation58, where the charge–phonon coupling extends across multiple unit cells and results in high defect tolerance and coherent carrier transport2.

a Chemical composition and structure of FAMACs perovskite (top), polaron–cation coupling and associated blue-shift of molecular vibration (bottom). b Far-field transmission visible-pump (2.4 eV) IR-probe spectrum at T = 0.5 ps (top), the ground-state (GS) vibrational absorbance Avib and transient absorbance ΔAvib (bottom).
As another unique aspect, lead halide perovskites exhibit heterogeneity over multiple length scales in their optoelectronic responses in, e.g., photoluminescence intensity, carrier lifetime, or open circuit voltage11. While several works have addressed the nanoscale heterogeneity in the lattice strain and elasticity in the electronic ground-state59,60, the direct relationship between the nonuniform optoelectronic response and the underlying polaronic heterogeneity has not yet been established. Ultrafast infrared vibrational spectroscopy has previously elucidated the coupling between a molecular cation and a photoinduced polaron in perovskites35,61 (Fig. 4a), yet was unable to address the underlying spatial heterogeneity due to the diffraction-limited resolution.
Using HPP IR s-SNOM, we aim to resolve the polaron–cation coupling on the nanoscale. To establish the ground and excited-state vibrational responses and their expected coupling to the polaron, we first perform conventional far-field visible (2.4 eV)-pump/infrared (0.2 eV)-probe transmission spectroscopy as shown in Fig. 4b for a thin film of the triple cation perovskite FAMACs, with chemical composition \([{({{{{{{{{\rm{FA}}}}}}}}}_{0.83}{{{{{{{{\rm{MA}}}}}}}}}_{0.17})}_{0.95}{{{{{{{{\rm{Cs}}}}}}}}}_{0.05}]{{{{{{{\rm{Pb}}}}}}}}{({{{{{{{{\rm{I}}}}}}}}}_{0.83}{{{{{{{{\rm{Br}}}}}}}}}_{0.17})}_{3}\). We estimate an injected carrier density of ~1019 cm−3 (for experimental details, see Supplementary Note 4). The pump-induced change in transmission (−ΔT/T) at the pump-probe delay of T = 0.5 ps exhibits the transient vibrational signature arising from the CN anti-symmetric stretch mode of the formamidinium (FA) cation61 in addition to the spectrally broad background from polaron absorption34. The vibrational excited-state absorption (ΔAvib) is compared to the ground-state absorption (Avib) in Fig. 4b (bottom), exhibiting a blue-shift as well as an absorptive lineshape, suggesting an enhancement in the transition dipole moment. These two observations are consistent with previous observations on similar perovskites35,61. As established35, these two features signify the coupling of the molecular vibration to polaron absorption with a large transition dipole moment and lower resonance energy. This gives rise to the blue-shift and enhanced transition dipole moment of the hybridized polaron-coupled vibration (Fig. 4a). Based on the reported resonance frequency of polaron absorption at 1100–1200 cm−134,35, the observed vibrational blue-shift of ~5 cm−1 corresponds to a spatially averaged polaron–cation coupling strength of ~50 cm−1 (see Supplementary Fig. S8 for details).
In ultrafast HPP IR s-SNOM, we then explore the associated nanoscale heterogeneity in polaron–cation coupling (Fig. 5a). Figure 5b shows ΔIHPP(t) for a pump-probe delay of T = 2 ps with the center burst arising from the instantaneous polaron absorption34 and the long-lived coherence associated with the transient vibrational response. Figure 5c then shows the Fourier transformed spectral profile of the pump-induced \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) and ground-state \({E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\). The transient vibrational resonant peak at \({\overline{\nu }}_{{{{{{{{\rm{ex}}}}}}}}}\) in \({{{{{{{\rm{Im}}}}}}}}[{{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })]\) is blue-shifted from the ground-state peak position \({\overline{\nu }}_{{{{{{{{\rm{gs}}}}}}}}}\) in \({{{{{{{\rm{Im}}}}}}}}[{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })]\) by ~5 cm−1, in agreement with the polaron–cation coupling observed in the far-field measurement.

a Ground-state IR s-SNOM \({{{{{{{\rm{Re}}}}}}}}[{E}_{{{{{{{{\rm{NF}}}}}}}}}]\) imaging (top) and AFM topography (bottom). b HPP IR s-SNOM interferogram ΔINF(t) acquired at T = 2 ps with instantaneous polaron absorption and long-lived transient vibrational coherence. c Phase- and frequency-resolved nano-localized pump-probe \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) and ground-state response \({E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\). d Decomposition of \({{{{{{{\rm{Im}}}}}}}}[{{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })]\) into the transient vibrational (\({{{{{{{\rm{Im}}}}}}}}[{{\Delta }}{E}_{{{{{{{{\rm{NF,vib}}}}}}}}}(\overline{\nu })]\)) and carrier (\({{{{{{{\rm{Im}}}}}}}}[{{\Delta }}{E}_{{{{{{{{\rm{NF,car}}}}}}}}}(\overline{\nu })]\)) contributions with a minor feature from Fano-type interference. e The nano-localized transient vibrational signal \({{{{{{{\rm{Im}}}}}}}}[{{\Delta }}{E}_{{{{{{{{\rm{NF,vib}}}}}}}}}(\overline{\nu })]\) at different sample locations shows nanoscale spatial heterogeneity in the polaron–cation coupling.
We then apply and fit the data to the model described above (Fig. 2) to retrieve the transient complex dielectric function \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\). The resonant spectral profile in the retrieved \({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\) (Supplementary Fig. S7) is essentially identical to \({{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })\), with the background carrier response (\({{\Delta }}{\tilde{\epsilon }}_{{{{{{{{\rm{NF}}}}}}}}} \sim -0.1\,+\,0.3i\)) qualitatively consistent with polaron absorption. The contribution from Fano-type interference to an apparent shift is negligible in this case compared to the transient vibrational response, due to the relatively small carrier background (Fig. 5d). We thus subtract the carrier background from \({{{{{{{\rm{Im}}}}}}}}[{{\Delta }}{E}_{{{{{{{{\rm{NF}}}}}}}}}(\overline{\nu })]\) to extract the nano-localized excited-state absorption \({{{{{{{\rm{Im}}}}}}}}[{{\Delta }}{E}_{{{{{{{{\rm{NF,vib}}}}}}}}}(\overline{\nu })]\) (see Supplementary Note 4).
Figure 5e then shows the nano-localized excited-state absorptions \({{{{{{{\rm{Im}}}}}}}}[{{\Delta }}{E}_{{{{{{{{\rm{NF,vib}}}}}}}}}(\overline{\nu })]\) at three different sample locations. The varying degree of vibrational peak shifts of 5–8 cm−1 suggests a spatial heterogeneity in polaron–cation coupling of 50–70 cm−1 (Fig. 5e, inset), obscured in the spatially averaged far-field spectroscopy above. Such disordered polaron–cation coupling is likely associated with a non-uniformity in the dynamic lattice elasticity. This interpretation is supported by other recent experimental and theoretical investigations59,60,62,63, which have identified a heterogeneity in chemical composition and resulting local lattice disorder and strain in perovskite films. With HPP IR s-SNOM probing the excited-state vibrational absorption, we resolve spatial heterogeneities in polaron–cation coupling arising from lattice disorder, which directly impacts polaron formation, lifetime, transport and, as such, photovoltaic device performance.
Discussion
Recent ultrafast infrared nanoimaging demonstrated the nanoscale probing of a range of low-energy phenomena in semiconductor, 2D, and other quantum materials16,17,18,19,20,22,24,26, yet mostly with high-repetition excitation that provides sufficient contrast against the simultaneously detected unpumped ground-state response. This conventional approach has therefore been limited primarily to the detection of short-lived nonequilibrium states. This has hampered the application of the technique to access long-lived transient states that often arise from cooperative dynamics associated with many-body interactions36,37 represented by, e.g., photoinduced phase transitions in correlated electron materials or polaron formation in molecular materials.
In the adaptation of modulated excitation with sideband detection for HPP IR s-SNOM, we facilitate the isolation of the excited-state response from the unperturbed ground-state response, establishing nano-FTIR spectroscopy of the purely transient and nano-localized response with low-repetition excitation. HPP IR s-SNOM thus universally endows ultrafast infrared nanoimaging with the ability to quantitatively resolve ultrafast dynamics associated with long-lived perturbations. We note that HPP s-SNOM, which is compatible with the full range of probe frequencies from visible to far-infrared together with its relatively facile implementation, is complementary to ultrafast EOS s-SNOM probing nanoscale dynamics in the THz regime29.
As demonstrated in VO2, HPP IR s-SNOM isolates the near-field pump-probe from the ground-state response to achieve ultrafast nanoimaging of the IMT dynamics with high contrast (Fig. 3d). Eliminating the convoluted far-field background contribution by heterodyning ΔENF with a phase-controlled ELO22, HPP IR s-SNOM also accurately determines the timescale of the purely nano-localized carrier dynamics (Fig. 1e). Further, by simultaneous and phase-locked recording of INF and ΔINF interferograms, HPP IR s-SNOM quantifies the transient complex dielectric response on the nanoscale (Fig. 3a, b), thus essentially performing ultrafast nano-ellipsometry based on interferometric heterodyne detection. The observed transient spatial heterogeneity suggests intricate co-existence of competing mechanisms such as nonuniform strain and stoichiometric zoning.
In extensions to transient vibrational nano-spectroscopy, we resolve the generally weak excited-state vibrational coherence of a lead halide perovskite in the time domain (Fig. 5b) with sensitive and selective detection of ΔINF(t) enabled by pump modulation. The resulting phase and spectrally resolved excited-state vibrational response quantifies the spatially varying polaron–cation coupling (Fig. 5e), which is central to the photovoltaic response of perovskites. Thus the combination of pump modulation and heterodyne detection in HPP IR s-SNOM provides pure transient and nano-localized contrast, extending the applicability of ultrafast infrared nanoimaging from the weakly perturbed to the far-from-equilibrium regime.
More generally, ultrafast nanoimaging based on an electronic excitation and a low-energy probe has a unique advantage in studying the spatial heterogeneity of electron–phonon coupling, a central topic in, e.g., two-dimensional materials64, hybrid photovoltaics2, or nanoscale thermal transport65. Ultrafast heterodyne infrared nanoimaging, with its direct access to vibational, phononic, and polaronic modes, can thus play an important role not only in mapping the inherent disorder in electron–phonon coupling11, but also the optical control of such coupling in combination with nanoscale quantum architectures23.
HPP IR s-SNOM also lays the foundation for adapting other state-of-the-art ultrafast spectroscopy to nanoimaging. For example, by implementing two pump pulses with controlled time delay, ultrafast non-degenerate two-dimensional nano-spectroscopy4,66 can be realized, probing coherence and population transfer among different modes on the nanoscale. Our implementation with low-repetition excitation is particularly beneficial to potentially implement nonlinear spectroscopy with infrared67 and THz68 excitations at the nanoscale, which would require a strong pump fluence that is only attainable in amplifier laser sources. Further, in combination with interferometric heterodyne detection, adiabatic plasmonic nano-focused electronic four-wave mixing27,28 would provide two-dimensional electronic nano-spectroscopy to characterize local electron or exciton dynamics in, e.g., two-dimensional materials69.
HPP s-SNOM can thus resolve the full spatiotemporal-spectral evolution of key elementary excitations which define the properties of a wide range of functional materials. With the pump modulation and heterodyne detection, HPP s-SNOM bridges the prolific success and plethora of modalities of far-field ultrafast spectroscopy to ultrafast nano-spectroscopy and -imaging to probe coupling and dynamics at the nanoscale.
Data availability
The data generated in this study have been deposited in the Open Science Framework (OSF) at https://osf.io/nkyta/.
Code availability
The MATLAB codes developed in this study have been deposited in the Open Science Framework (OSF) at https://osf.io/nkyta/.
References
Nakano, M. et al. Collective bulk carrier delocalization driven by electrostatic surface charge accumulation. Nature 487, 459–462 (2012).
Miyata, K., Atallah, T. L. & Zhu, X.-Y. Lead halide perovskites: crystal-liquid duality, phonon glass electron crystals, and large polaron formation. Sci. Adv. 3, e1701469 (2017).
Duan, H. G. et al. Intermolecular vibrations mediate ultrafast singlet fission. Sci. Adv. 6, 52–70 (2020).
Scholes, G. D. et al. Using coherence to enhance function in chemical and biophysical systems. Nature 543, 647–656 (2017).
Von Hoegen, A., Mankowsky, R., Fechner, M., Först, M. & Cavalleri, A. Probing the interatomic potential of solids with strong-field nonlinear phononics. Nature 555, 79–82 (2018).
Mitrano, M. et al. Possible light-induced superconductivity in K3C60 at high temperature. Nature 530, 461–464 (2016).
Yoshikawa, N., Tamaya, T. & Tanaka, K. High-harmonic generation in graphene enhanced by elliptically polarized light excitation. Science 356, 736–738 (2017).
Hafez, H. A. et al. Extremely efficient terahertz high-harmonic generation in graphene by hot Dirac fermions. Nature 561, 507–511 (2018).
Schlaepfer, F. et al. Attosecond optical-field-enhanced carrier injection into the gaas conduction band. Nat. Phys. 14, 560–564 (2018).
O’Callahan, B. T. et al. Inhomogeneity of the ultrafast insulator-to-metal transition dynamics of VO2. Nat. Commun. 6, 1–8 (2015).
Tennyson, E. M., Doherty, T. A. & Stranks, S. D. Heterogeneity at multiple length scales in halide perovskite semiconductors. Nat. Rev. Mater. 4, 573–587 (2019).
Jones, A. C., Kearns, N. M., Ho, J. J., Flach, J. T. & Zanni, M. T. Impact of non-equilibrium molecular packings on singlet fission in microcrystals observed using 2D white-light microscopy. Nat. Chem. 12, 40–47 (2020).
Feist, A. et al. Ultrafast transmission electron microscopy using a laser-driven field emitter: femtosecond resolution with a high coherence electron beam. Ultramicroscopy 176, 63–73 (2017).
Da̧browski, M., Dai, Y. & Petek, H. Ultrafast photoemission electron microscopy: imaging plasmons in space and time. Chem. Rev. 120, 6247–6287 (2020).
Wen, H., Cherukara, M. J. & Holt, M. V. Time-resolved X-ray microscopy for materials science. Annu. Rev. Mater. Res. 49, 389–415 (2019).
Dönges, S. A. et al. Ultrafast nanoimaging of the photoinduced phase transition dynamics in VO2. Nano Lett. 16, 3029–3035 (2016).
Huber, M. A. et al. Ultrafast mid-infrared nanoscopy of strained vanadium dioxide nanobeams. Nano Lett. 16, 1421–1427 (2016).
Huber, M. A. et al. Femtosecond photo-switching of interface polaritons in black phosphorus heterostructures. Nat. Nanotechnol. 12, 207–211 (2017).
Wagner, M. et al. Ultrafast dynamics of surface plasmons in InAs by time-resolved infrared nanospectroscopy. Nano Lett. 14, 4529–4534 (2014).
Ni, G. X. et al. Ultrafast optical switching of infrared plasmon polaritons in high-mobility graphene. Nat. Photonics 10, 244–247 (2016).
Charnukha, A. et al. Ultrafast nonlocal collective dynamics of Kane plasmon-polaritons in a narrow-gap semiconductor. Sci. Adv. 5, eaau9956 (2019).
Sternbach, A. J. et al. Artifact free time resolved near-field spectroscopy. Opt. Express 25, 28589 (2017).
Sternbach, A. J. et al. Programmable hyperbolic polaritons in van der Waals semiconductors. Science 371, 617–620 (2021).
Eisele, M. et al. Ultrafast multi-terahertz nano-spectroscopy with sub-cycle temporal resolution. Nat. Photonics 8, 841–845 (2014).
Cocker, T. L., Peller, D., Yu, P., Repp, J. & Huber, R. Tracking the ultrafast motion of a single molecule by femtosecond orbital imaging. Nature 539, 263–267 (2016).
Yao, Z. et al. Nanoimaging and nanospectroscopy of polaritons with time resolved s-SNOM. Adv. Opt. Mater. 8, 1–18 (2020).
Jiang, T., Kravtsov, V., Tokman, M., Belyanin, A. & Raschke, M. B. Ultrafast coherent nonlinear nanooptics and nanoimaging of graphene. Nat. Nanotechnol. 14, 838–843 (2019).
Kravtsov, V., Ulbricht, R., Atkin, J. M. & Raschke, M. B. Plasmonic nanofocused four-wave mixing for femtosecond near-field imaging. Nat. Nanotechnol. 11, 459–464 (2016).
Plankl, M. et al. Subcycle contact-free nanoscopy of ultrafast interlayer transport in atomically thin heterostructures. Nat. Photonics 15, 594–600 (2021).
Cavalleri, A. et al. Femtosecond structural dynamics in VO2 during an ultrafast solid-solid phase transition. Phys. Rev. Lett. 87, 237401–1–237401–4 (2001).
Wegkamp, D. et al. Instantaneous band gap collapse in photoexcited monoclinic VO2 due to photocarrier doping. Phys. Rev. Lett. 113, 216401 (2014).
Wegkamp, D. & Stähler, J. Ultrafast dynamics during the photoinduced phase transition in VO2. Prog. Surf. Sci. 90, 464–502 (2015).
Cocker, T. L. et al. Phase diagram of the ultrafast photoinduced insulator-metal transition in vanadium dioxide. Phys. Rev. B - Condens. Matter Mater. Phys. 85, 155120 (2012).
Munson, K. T., Kennehan, E. R., Doucette, G. S. & Asbury, J. B. Dynamic disorder dominates delocalization, transport, and recombination in halide perovskites. Chem 4, 2826–2843 (2018).
Munson, K. T., Doucette, G. S., Kennehan, E. R., Swartzfager, J. R. & Asbury, J. B. Vibrational probe of the structural origins of slow recombination in halide perovskites. J. Phys. Chem. C 123, 7061–7073 (2019).
Basov, D. N., Averitt, R. D., Van Der Marel, D., Dressel, M. & Haule, K. Electrodynamics of correlated electron materials. Rev. Mod. Phys. 83, 471–541 (2011).
Sundaram, S. K. & Mazur, E. Inducing and probing non-thermal transitions in semiconductors using femtosecond laser pulses. Nat. Mater. 1, 217–224 (2002).
Jones, A. C., Berweger, S., Wei, J., Cobden, D. & Raschke, M. B. Nano-optical investigations of the metal-insulator phase behavior of individual VO2 microcrystals. Nano Lett. 10, 1574–1581 (2010).
Huth, F. et al. Nano-FTIR absorption spectroscopy of molecular fingerprints at 20 nm spatial resolution. Nano Lett. 12, 3973–3978 (2012).
Muller, E. A., Pollard, B. & Raschke, M. B. Infrared chemical nano-imaging: accessing structure, coupling, and dynamics on molecular length scales. J. Phys. Chem. Lett. 6, 1275–1284 (2015).
Yeh, T. T. et al. Ultrafast carrier dynamics in Ge by ultra-broadband mid-infrared probe spectroscopy. Sci. Rep. 7, 1–10 (2017).
Govyadinov, A. A., Amenabar, I., Huth, F., Carney, P. S. & Hillenbrand, R. Quantitative measurement of local infrared absorption and dielectric function with tip-enhanced near-field microscopy. J. Phys. Chem. Lett. 4, 1526–1531 (2013).
Mooshammer, F. et al. Nanoscale near-field tomography of surface states on (Bi0.5Sb0.5)2Te3. Nano Lett. 18, 7515–7523 (2018).
Wirth, K. G. et al. Tunable s-SNOM for nanoscale infrared optical measurement of electronic properties of bilayer graphene. ACS Photonics 8, 418–423 (2021).
Glaser, T. et al. Infrared spectroscopic study of vibrational modes in methylammonium lead halide perovskites. J. Phys. Chem. Lett. 6, 2913–2918 (2015).
Katsidis, C. C. & Siapkas, D. I. General transfer-matrix method for optical multilayer systems with coherent, partially coherent, and incoherent interference. Appl. Opt. 41, 3978 (2002).
Qazilbash, M. M. et al. Mott transition in VO2 revealed by infrared spectroscopy and nano-imaging. Science 318, 1750–1753 (2007).
Liu, K., Lee, S., Yang, S., Delaire, O. & Wu, J. Recent progresses on physics and applications of vanadium dioxide. Mater. Today 21, 875–896 (2018).
Wall, S. et al. Ultrafast disordering of vanadium dimers in photoexcited VO2. Science 362, 572–576 (2018).
Vidas, L. et al. Does VO2 host a transient monoclinic metallic phase? Phys. Rev. X 10, 031047 (2020).
Wu, J. et al. Strain-induced self organization of metal-insulator domains in single-crystalline VO2 nanobeams. Nano Lett. 6, 2313–2317 (2006).
Atkin, J. M. et al. Strain and temperature dependence of the insulating phases of VO2 near the metal-insulator transition. Phys. Rev. B 85, 020101 (2012).
Wan, C. et al. On the optical properties of thin-film vanadium dioxide from the visible to the far infrared. Ann. Phys. 531, 1900188 (2019).
Ramirez-Rincon, J. A. et al. Thermal hysteresis measurement of the VO2 dielectric function for its metal-insulator transition by visible-IR ellipsometry. J. Appl. Phys. 124, 195102 (2018).
Choi, H., Ahn, J., Jung, J., Noh, T. & Kim, D. Mid-infrared properties of a film near the metal-insulator transition. Phys. Rev. B - Condens. Matter Mater. Phys. 54, 4621–4628 (1996).
Verleur, H. W., Barker, A. S. & Berglund, C. N. Optical properties of VO2 between 0.25 and 5 eV. Phys. Rev. 172, 788–798 (1968).
Stranks, S. D. et al. Electron-hole diffusion lengths exceeding 1 micrometer in an organometal trihalide perovskite absorber. Science 342, 341–344 (2013).
Zhu, X.-Y. & Podzorov, V. Charge carriers in hybrid organic-inorganic lead halide perovskites might be protected as large polarons. J. Phys. Chem. Lett. 6, 4758–4761 (2015).
Jones, T. W. et al. Lattice strain causes non-radiative losses in halide perovskites. Energy Environ. Sci. 12, 596–606 (2019).
Nishida, J., Alfaifi, A. H., Gray, T. P., Shaheen, S. E. & Raschke, M. B. Heterogeneous cation-lattice interaction and dynamics in triple-cation perovskites revealed by infrared vibrational nanoscopy. ACS Energy Lett. 5, 1636–1643 (2020).
Taylor, V. C. A. et al. Investigating the role of the organic cation in formamidinium lead iodide perovskite using ultrafast spectroscopy. J. Phys. Chem. Lett. 9, 895–901 (2018).
Ghosh, D., Smith, A. R., Walker, A. B. & Islam, M. S. Mixed A-cation perovskites for solar cells: atomic-scale insights into structural distortion, hydrogen bonding, and electronic properties. Chem. Mater. 30, 5194–5204 (2018).
Kubicki, D. J. et al. Phase segregation in Cs-, Rb- and K-doped mixed-cation (MA)x(FA)1−xPbI3 hybrid perovskites from solid-state NMR. J. Am. Chem. Soc. 139, 14173–14180 (2017).
Jin, C. et al. Interlayer electron-phonon coupling in WSe2/hBN heterostructures. Nat. Phys. 13, 127–131 (2017).
Wang, W. & Cahill, D. G. Limits to thermal transport in nanoscale metal bilayers due to weak electron-phonon coupling in Au and Cu. Phys. Rev. Lett. 109, 175503 (2012).
Hamm, P. & Zanni, M. T. Concepts and Methods of 2D Infrared Spectroscopy (Cambridge University Press, 2011).
Farrell, K. M. et al. Shot-to-shot 2D IR spectroscopy at 100 kHz using a Yb laser and custom-designed electronics. Opt. Express 28, 33584 (2020).
Reimann, K., Woerner, M. & Elsaesser, T. Two-dimensional terahertz spectroscopy of condensed-phase molecular systems. J. Chem. Phys. 154, 120901 (2021).
Moody, G. et al. Intrinsic homogeneous linewidth and broadening mechanisms of excitons in monolayer transition metal dichalcogenides. Nat. Commun. 6, 1–6 (2015).
Acknowledgements
We thank Lei Jin and Junqiao Wu (University of California, Berkeley) for providing VO2 nanobeams, and Jiselle Ye, Prachi Sharma, and Sean E. Shaheen (University of Colorado Boulder) for supplying perovskite samples. J.N., S.C.J., P.T.S.C., D.M.W., and M.B.R. acknowledge support from the NSF Science and Technology Center on Real-Time Functional Imaging (STROBE) under DMR-1548924 for instrument development and the perovskite work. S.A.D., O.K. and M.B.R. further acknowledge funding from the U.S. Department of Energy, Office of Basic Sciences, Division of Material Sciences and Engineering, under Award No. DE-SC0008807 for the work on VO2. J.N. acknowledges support from the Japan Society for the Promotion of Science for a JSPS Overseas Research Fellowship.
Author information
Authors and Affiliations
Contributions
J.N., O.K., S.A.D. and M.B.R. conceived the research. J.N., P.T.S.C., and D.M.W. performed the experiments. J.N., S.C.J., P.T.S.C., D.M.W. and M.B.R. analyzed the data. J.N., S.C.J. and M.B.R. discussed and wrote the manuscript. All the authors read and provided comments on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nishida, J., Johnson, S.C., Chang, P.T.S. et al. Ultrafast infrared nano-imaging of far-from-equilibrium carrier and vibrational dynamics. Nat Commun 13, 1083 (2022). https://doi.org/10.1038/s41467-022-28224-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-28224-9
This article is cited by
-
Near-field terahertz nonlinear optics with blue light
Light: Science & Applications (2023)
-
Energy transfer in N-component nanosystems enhanced by pulse-driven vibronic many-body entanglement
Scientific Reports (2023)
-
Nanoscale heterogeneity of ultrafast many-body carrier dynamics in triple cation perovskites
Nature Communications (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
