
Abstract
Photoactivatable drugs targeting ligand-gated ion channels open up new opportunities for light-guided therapeutic interventions. Photoactivable toxins targeting ion channels have the potential to control excitable cell activities with low invasiveness and high spatiotemporal precision. As proof-of-concept, we develop HwTxIV-Nvoc, a UV light-cleavable and photoactivatable peptide that targets voltage-gated sodium (NaV) channels and validate its activity in vitro in HEK293 cells, ex vivo in brain slices and in vivo on mice neuromuscular junctions. We find that HwTxIV-Nvoc enables precise spatiotemporal control of neuronal NaV channel function under all conditions tested. By creating multiple photoactivatable toxins, we demonstrate the broad applicability of this toxin-photoactivation technology.
Similar content being viewed by others


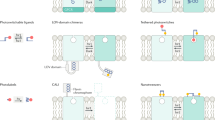
Introduction
Ion channels are pore-forming transmembrane proteins that allow the regulated flow of cations or anions across membranes. Due to their important biological roles in many cell types, ion channels constitute drug targets for the treatment of diseases, such as type-2 diabetes, hypertension, epilepsy, chronic pain, cardiac arrhythmia, and anxiety, and many are part of the classical drugs on the WHO’s list of essential medicines (https://list.essentialmeds.org). Ion channels are regarded as difficult targets for drugs due to the challenge to achieve subtype selectivity and because they represent complex protein structures embedded in the plasma membrane. Biological compounds, such as peptides found in animal venoms, demonstrate their usefulness in reaching high selectivity and affinity towards their targets owing to chemical surfaces larger than those of small organic compounds. As such, they represent exquisite high affinity and selective tools for the pharmacological control of ion channels and of cellular excitability1 and play a vital role in the deorphanization and classification of ion channels2. While animal peptide toxins originate from a wide variety of venomous species, they have in common a compact 3D structure, largely imposed by internal disulphide bridges that promote the formation, organization, and stabilization of secondary structures3. Mechanistically, peptide toxins targeting ion channels can inhibit the pore or modify the gating process to alter channel activation or inactivation properties and thereby act as inhibitors or activators4. Another interesting feature is the diversity in selectivity encountered so far. Some venom peptides such as ω-conotoxin-GVIA (N-type CaV2.2 channel5), BeKm1 (hERG channel6) or µ-conotoxin KIIIA (NaV1.27) are selective for a particular target. Others bind to virtually an entire class of ion channels (i.e. AaHII and NaV channels8). Hence, venom peptides appear as a promising class of compounds to control the activity of excitable cells with a user-defined selectivity.
When it comes to provide a light-mediated control of cell excitability, the technology developed the furthest is optogenetics as witnessed by the substantial amount of literature on the topic9,10. Undoubtedly, optogenetics have transformed many areas of biological research demonstrating the usefulness for a precise control of biological functions thanks to the unsurpassed spatial and temporal resolution of focussed light. However, this technology does suffer from several drawbacks that are challenging to overcome for therapeutic applications, except on rare occasions, such as recently shown for vision treatment11. Optogenetics is largely based on the use of genetically encoded material that needs to be supplied externally for invasive modification of the cell proteome. In contrast, photopharmacology does not require any modification of the cell proteome or genetic background and responds more favorably to the exigences of regulatory agencies for therapeutic development. Likewise to optogenetics, photoactivatable caged compounds are powerful tools for modulating the function of native proteins with high spatiotemporal resolution. This concept has shown promise in animal models for vision restoration11 and pain management12. Although uncaging of chemicals for ligand-gated channels enabled seminal optopharmacology studies informing on their function and subcellular location in complex biological environments13,14,15,16, caged ligands targeting voltage-gated ion channels more specifically remain rare. The example of saxitoxin demonstrates, however, that it is an interesting axis of development17. It is this challenge to deliver a greater number of interesting ligands amenable to light control that our project addresses. Indeed, with recent progresses in chemistry, it is now feasible to transform a venom peptide into a photosensitive tool. The most straightforward chemical strategy to achieve this aim is to add a photosensitive cage on the side chain of a residue that is key for activity18,19,20,21. Actually, a shared feature of most peptide toxins is that activity generally relies on a limited set of amino acids. Typically, a Lys residue (or a residue that can be substituted by Lys), is often critical for biological activity22. A similar approach has been used in the past on Cys residues to control region-selective disulfide bonding of oxytocin23, α-conotoxin IMI23, and human insulin23, but not to our knowledge to endow the pharmacological action of structurally complex toxins with a photosensitivity property. Based on our comprehension of peptide pharmacophores, we present herein a general caging strategy, involving covalent attachment of an orthogonal o-nitroveratryloxycarbonyl (Nvoc) protecting group on the lateral chain of a Lys residue that remains compatible with solid-phase peptide synthesis and that can be cleaved by photolysis under physiological conditions. We use both brain slices and in vivo experiments (mice neuromuscular junction and zebrafish larvaes) to validate the fact that illumination leads to selective inhibition of voltage-gated Na+ (NaV) channel with high spatial resolution. Finally, we illustrate that the technique can be generalized to peptide toxins possessing more or less ion channel selectivity, and is applicable to both inhibitors and activators.
Results
Design, chemical synthesis, and structure of HwTxIV-Nvoc
To demonstrate the applicability of modifying a venom peptide into a photosensitive tool in spite of the structural complexity and presence of multiple disulfide bridges, we synthetized a pure HwTxIV-Nvoc compound based on a highly effective analogue of native HwTxIV (nHwTxIV) (Fig. 1a). nHwTxIV was first identified as a NaV1.7 inhibitor but shown recently to target also NaV1.6 with a lower affinity24. We first aimed to develop a HwTxIV analogue with convergent blocking potencies for hNaV1.1, hNaV1.2, hNaV1.6, and hNaV1.7 channels. Based on earlier structure-function analyses using nHwTxIV single amino acid mutations25,26,27 and several 3D structures of HwTxIV-NaV1.7 complex (Supplementary Fig. 1a)28,29,30, several residues were targeted to produce a more general and potent NaV channel inhibitor. By synthesizing 20 analogues and evaluating their potencies by building dose-response curves thanks to an automated patch-clamp setup on hNaV1.1, hNaV1.2, hNaV1.6 and hNaV1.7 channels, we determined that the nHwTxIV analogue (HwTxIVG1G4K36 that we shall term HwTxIV afterwards for simplicity) was the most potent for a pan-NaV action (equivalent potencies on several NaV channels evaluated as IC50 values for inhibition of peak Na+ currents in the whole-cell configuration; hNaV1.1: 18.9 ± 1.2 nM, n = 3-10 cells per data point vs. 50.7 ± 1.2 nM for nHwTxIV, n = 3–6; hNaV1.2: 11.9 ± 1.1 nM, n = 4–9 vs. 8.3 ± 1.2 nM for nHwTxIV, n = 6–11; hNaV1.6: 19.5 ± 1.3 nM, n = 6–10 vs. 154.3 ± 1.2 nM, n = 3–7 for nHwTxIV; hNaV1.7: 15.2 ± 1.1 nM, n = 7–17 vs. 9.9 ± 0.9 nM, for nHwTxIV, Supplementary Fig. 1b and Supplementary Table 1). The improvement was most remarkable for hNav1.6, opening applications for this peptide. By mutating only the K32 into N32 (IC50 > 1 µM, n = 4–7 for nHwTxIV-N32 on hNaV1.7 vs. IC50 = 9.9 nM ± 0.9 nM, n = 5–6 for nHwTxIV on hNaV1.7), we confirmed that the K32 residue is an ideal amino acid residue for chemical modifications aimed at reducing HwTxIV potency30,31 (Supplementary Fig. 1c). Since N32 is sterically similar to K32, these results indicate the importance of the positive charge carried by K32 for activity. Before proceeding with the chemical synthesis of HwTxIV-Nvoc, we noted that K32 interacts with E811, D816, and E818 of the loop between S3 and S4 of NaV1.7-NavAb (pdb code 7K48)30. Based on those observations, we confirmed that, if the binding of HwTxIV-Nvoc onto the channel had to occur similarly to the non-caged HwTxIV, the presence of the Nvoc protecting group would introduce steric clashes that predictably should reduce peptide affinity (Fig. 1b). Similarly, the masking of the protonated lysine as an electrostatically neutral carbamate, also removes the positive charge that is suspected to contribute to toxin activity. HwTxIV-Nvoc was assembled stepwise using Fmoc-based SPPS on a 2-chlorotrityl-polystyrene resin, preloaded with a rink amide linker. The crude peptide was obtained in quantitative yield and of good quality to be directly oxidized. The presence of the Nvoc protecting group on K32 which is located next to the sixth Cys residue within the HwTxIV sequence can compromise the proper assembly of the three disulfide bridges within an ICK fold. The folding process was followed by analytical RP-HPLC and showed completion after 16 h with the disappearance of the unfolded peptide and formation of one main oxidation peak occurring with a slight left shift on the elution profile. Finally, the pH of the reaction mixture was adjusted to three and the oxidized peptide was purified to homogeneity by preparative RP-HPLC. Two successive purifications, first using a C18 (5 µm, 130 Å) Waters CSH column, second using a C18 (10 µm, 100 Å) Phenomenex Luna stationary phase (eluent system H2O/MeCN + 0.1% TFA), were needed to complete production of pure HwTxIV-Nvoc with a reasonably good global yield of 8.2% including SPPS, folding and purifications (Fig. 1c). The peptide was obtained with a measured exact mass of 722.1785 [M + 6H]6+ by LC-ESI QTOF (calculated mass of 4327.08 Da, which is consistent with the theoretical monoisotopic mass of 4327.00 Da) (Fig. 1d). The proper mass indicates that the Nvoc protecting group was not removed by the TFA cleavage treatment of the synthetic crude unfolded peptide or during oxidative folding. As attested by the dispersion of the resonances, noncaged HwTxIV is well structured (Fig. 1e black spectra, and Supplementary Data 1). As expected, the presence of the aromatic Nvoc group on K32 for HwTxIV-Nvoc disturbs the chemical environments of a series of residues on the side of the molecule where K32 is located without affecting the global fold of the peptide (Fig. 1e, f, orange spectra). The presence of the grafted Nvoc group generates a doubling of certain peaks, possibly indicating different orientations of the Nvoc protecting group. HwTxIV-Nvoc showed excellent dark stability in solution for over 1 week (Supplementary Fig. 2).

a Structure of HwTxIV-Nvoc with K32 and Nvoc group reported in blue and orange sticks respectively. HwTxIV structure has been isolated from PDB 7K48. The sequence chosen for caging is shown upon sequence alignment with nHwTxIV. b Illustration of steric van der Walls clashes (red cylinders) resulting from the addition of the Nvoc protecting group onto K32 upon binding of HwTxIV onto the chimeric Nav1.7-NavAb in the position as published30 (PDB code 7K48). c RP-HPLC elution profiles of crude, crude folded/oxidized, and purified folded/oxidized HwTxIV-Nvoc. RP-HPLC elution profile of purified folded/oxidized non-caged HwTxIV has been superimposed for comparison. d LC-ESI QTOF analysis of HwTxIV-Nvoc denotes an exact mass of 722.1785 [M + 6H]6+. e Superimposition of TOCSY spectra (sidechains area only for clarity) of the non-caged HwTxIV (in black), and of HwTxIV-Nvoc before illumination (in orange). f Residues showing major chemical shifts modifications in the presence of Nvoc are reported in orange on the surface of the molecule (180°views). K32 is highlighted in blue. The Nvoc group is reported in sticks. Source data are provided as a Source Data File.
HwTxIV-Nvoc drastically reduces the potency of the peptide for NaV channels
Next we validated functionally that the steric clashes introduced by grafting the Nvoc group on HwTxIV combined with the elimination of K32 positive charge is sufficient to reduce the NaV channel blocking potency of the peptide. Compared to non-caged HwTxIV, the caged toxin induces a 292- and 293-fold increase of the IC50 value on hNaV1.6 and hNaV1.7, respectively (Fig. 2a, d) and even greater shifts are observed on hNaV1.1 and hNaV1.2 (Fig. 2b, c). Concentrations above 1 µM are needed for HwTxIV-Nvoc to start exhibiting inhibition of all NaV channel subtypes tested. Remarkably, the toxin also slows the fast inactivation of hNaV1.6 (Fig. 2a, Supplementary Fig. 3a,b). We interpret this effect as due to the existence of a low-affinity binding locus on domain IV voltage sensor4 that is now revealed by the absence of a high-affinity block due to Nvoc presence. To test this hypothesis, we transplanted the S3-S4 motif from each of the four voltage-sensor domains (VSDI-IV) of hNaV1.6 into the homotetrameric rKV2.1 channel according to previously described boundaries32,33,34. The transferred region in all of the functional chimeras contains the crucial basic residues that contribute to gating charge movement in KV channels35,36,37. Examination of conductance-voltage (G-V) relationships for the hNaV1.6/rKV2.1 chimeras revealed that each of the four voltage-sensor motifs has a distinct effect on the gating properties of KV2.1. We next examined the effect of HwTxIV-Nvoc on the NaV1.6/KV2.1 VSD chimeras. 5 µM toxin did not alter the G-V relationship of the VSDI and VSDIII chimera, whereas VSDII and VSDIV were clearly inhibited (Supplementary Fig. 3c). HwTxIV-Nvoc binding to VSDII typically results in NaV channel inhibition, whereas VSDIV is involved in channel fast inactivation32,37 although a role in channel opening may also be possible38,39. Thus, we conclude that, above 1 μM, HwTxIV-Nvoc influences hNaV1.6 gating primarily by interacting with the S3-S4 motif in VSDII and VSDIV.

a i) Representative recording of hNaV1.6 currents elicited at 10 mV from a holding potential of -100 mV illustrating the extent of current block by different concentrations of noncaged HwTxIV (black) and HwTxIV-Nvoc (orange). ii) Average dose-response curves for hNaV1.6 current inhibition by non-caged HwTxIV (black) and HwTxIV-Nvoc (orange). The data were fitted according to a Hill equation (non-caged HwTxIV IC50 = 22.4 nM (n = 5) and HwTxIV-Nvoc IC50 = 6539 nM (n = 7) (292-fold reduction in IC50)). Scales: 2 ms, 1 nA. Note that on hNav1.6, high concentrations of HwTxIV-Nvoc produce a slowing of channel inactivation due to binding on a low-affinity site absent on hNav1.2 (Supplementary Figure 3). b i) Representative recording of hNaV1.1 current elicited at 10 mV from a holding potential of -100 mV illustrating the extent of current block by different concentrations of non-caged HwTxIV (black) and HwTxIV-Nvoc (orange). ii) Average dose-response curves for hNav1.1 current inhibition by noncaged HwTxIV (black) and HwTxIV-Nvoc (orange). The data were fitted according to a Hill equation (noncaged HwTxIV IC50 = 3.60 nM (n = 12); HwTxIV-Nvoc IC50 = 2794 nM (n = 8) (776-fold reduction in IC50). Scales: 2 ms, 1 nA. c i) Representative recording of hNaV1.2 current elicited at 10 mV from a holding potential of -100 mV illustrating the extent of current block by different concentrations of noncaged HwTxIV (black) and HwTxIV-Nvoc (orange). Scales: 2 ms, 1 nA. ii) Average dose-response curves for hNav1.2 current inhibitions by non-caged HwTxIV (black) and HwTxIV-Nvoc (orange). The data were fitted according to a Hill equation (noncaged HwTxIV IC50 = 3.23 nM (n = 11); HwTxIV-Nvoc IC50 = 5141 nM (n = 6) (1592-fold reduction in IC50)). d i) Representative recording of hNaV1.7 current elicited at 10 mV from a holding potential of -100 mV illustrating the extent of current block by different concentrations of non-caged HwTxIV (black) and HwTxIV-Nvoc (orange). Scales: 2 ms, 0.5 nA. ii) Average dose-response curves for hNav1.7 current inhibitions by non-caged HwTxIV (black) and HwTxIV-Nvoc (orange). The data were fitted according to a Hill equation (non-caged HwTxIV IC50 = 15.23 nM (n = 92 cells); HwTxIV-Nvoc IC50 = 4467 nM (n = 83 cells) (293-fold reduction in IC50)). All data are presented as mean ± SEM. Source data are provided as a Source Data File.
UV-dependent uncaging of HwTxIV-Nvoc restores a fully functional HwTxIV
UV illumination at 365 nm of HwTxIV-Nvoc fully produced uncaged HwTxIV with an uncaging half-time of 3.6 min (45 mW/cm2) and half-power of 11.8 mW/cm2 (Fig. 3a, b). HwTxIV released from uncaging of HwTxIV-Nvoc has the same elution time by RP-HPLC and molecular weight as synthetic noncaged HwTxIV (Fig. 3c, d). After illumination, the NMR spectra of uncaged HwTxIV and noncaged HwTxIV are perfectly superimposable indicating that illumination restores the predicted non-caged peptide (Fig. 3e). Also, photolysis of HwTxIV-Nvoc occurred from to λ = 365 nm up to 405 nm but was negligible at wavelengths in the 435–740 nm range (10 min, power> 18 mW/cm2) which offers the possibility to use this light-triggered uncaging technology in combination with fluorophores excited above 405 nm for additional monitoring techniques (Supplementary Fig. 4). Complete uncaging of HwTxIV-Nvoc leads to inhibitory properties on hNaV1.6 that are identical to noncaged toxin, confirming structure preservation upon uncaging, but also indicating that the addition of the Nvoc protecting group did not lead to an abnormal disulphide bridging during peptide synthesis (Fig. 3f, g). As expected from the various concentration-response curves measured so far, illumination of 100 nM HwTxIV-Nvoc that is by itself inactive at this concentration, leads to a significant inhibition of hNaV1.6 currents (Fig. 4a–c). Ending the illumination period does not restore hNav1.6 current levels, as Nvoc deprotection is irreversible (Fig. 4b). Similar light-induced block of Nav currents were observed with hNaV1.2 (Fig. 4d, e). Also, as expected from a lipophilic toxin, HwTxIV blocked hNaV1.6 in a poorly reversible manner after partial uncaging of 100 nM HwTxIV-Nvoc. Indeed, 11 min peptide washing was required to observe 16% of current recovery (n = 6; Supplementary Fig. 5). We also demonstrated that the extent of hNaV1.6 inhibition increases as a function of illumination time and power which is expected by the progressively larger extent of uncaging occurring with illumination (Fig. 4f, g). As negative control, we show that illumination alone (365 nm, 250 s, 45 mW/cm2) has no impact on Na+ current amplitude (Supplementary Fig. 6a). Moreover, the Nvoc photolysis by-product resulting from uncaging of 100 nM BeKm1-Nvoc toxin, used as a negative control since BeKm-1 does not act on NaV channels, is inert on hNaV1.6 (Supplementary Fig. 6b).

a Top: Analytical RP-HPLC profiles of HwTxIV-Nvoc after different durations of illuminations (365 nm, 45 mW/cm2) demonstrating time-dependent control of uncaging. Bottom: Uncaged/Caged ratio of HPLC chromatogram peaks area versus irradiation time at 365 nm (45 mW/cm2). b Top: Analytical RP-HPLC profiles of HwTxIV-Nvoc after different intensities of illuminations (365 nm, 5 min) demonstrating intensity-dependent control of uncaging. Bottom: Uncaged/Caged ratio of HPLC chromatogram peaks area versus irradiation power at 365 nm (3 min). c Analytical RP-HPLC profiles of non-caged HwTxIV (top), 50:50 ratio of noncaged HwTxIV after partial uncaging of HwTxIV-Nvoc (middle) and of HwTxIV-Nvoc (bottom). d Mass analyses of noncaged HwTxIV and uncaged HwTxIV by LC-ESI QTOF ([M + 6H]6+ values). e Superimposition of TOCSY spectra (same area as Fig. 1e) of the noncaged HwTxIV (in black), and of the uncaged HwTxIV after illumination of HwTxIV-Nvoc (in purple). f Representative recording of hNaV1.6 currents elicited at 10 mV from a holding potential of -100 mV illustrating the extent of current block by different concentrations of non-caged HwTxIV (black) and uncaged HwTxIV (purple). Scales: 2 ms, 1 nA. g Average dose-response curves for hNav1.6 current inhibitions by non-caged HwTxIV (black) and uncaged HwTxIV (purple). The data were fitted according to a Hill equation (noncaged HwTxIV IC50 = 22.4 nM (n = 5) and uncaged HwTxIV IC50 = 15.20 nM (n = 6)). Data are presented as mean ± SEM. Source data are provided as a Source Data File.

a–c Light-induced inhibition of hNaV1.6 current by photolysis of 100 nM HwTxIV-Nvoc. Note that not all of the HwTxIV-Nvoc is uncaged by photolysis during this exposure time. a Representative recordings of hNaV1.6 current with caged HwTxIV (orange) and after various illumination times (purple) and (b) average normalized time courses of hNaV1.6 current inhibition during and following light application (n = 11, mean ± SEM). Scale: 2 ms, 1 nA. c Average normalized current at steady-state in control (dark), caged (orange) and uncaged (purple) conditions (n = 11, ***p < 0.001, two-sided repeated-measures 1-way ANOVA followed by Bonferroni’s post-test.). d Representative recordings of hNaV1.2 current with caged HwTxIV (orange) and after illumination (purple). Scale: 2 ms, 1 nA. e Average normalized current at steady-state in control (dark), caged (orange), and uncaged (purple) conditions (n = 12, mean ± SEM ***p < 0.001, two-sided repeated-measures 1-way ANOVA followed by Bonferroni’s post-test). f–g Controllable uncaging of HwTxIV-Nvoc via UV irradiation. f Left: Superimposition of normalized hNaV1.6 currents at steady-state recorded after varying durations of illumination. Middle: Average normalized time courses of hNaV1.6 current inhibition (n = 40 cells, mean ± SEM). Arrow indicates steady-state. Right: Plot of current amplitude at the end of 800 s recording versus duration of illumination at 365 nm. Scale: 2 ms, 20% of amplitude. g Left: Superimposition of normalized hNav1.6 currents at steady-state recorded after varying power of illumination (4 min). Middle: Average normalized time courses of hNav1.6 current inhibition (n = 43 cells, mean ± SEM). Arrow indicates steady-state. Right: Plot of current amplitude at the end of 800 s recordings versus duration of illumination at 365 nm. Scale: 2 ms, 20% of amplitude. h Representative examples of photocontrol (365 nm, 45 mW/cm2) of 1 nM AaHII-R62K-Nvoc activity on hNaV1.2 current (left, AaHII PDB code 6NT48), 100 nM BeKm1-Nvoc activity on hERG current (middle, BeKm-1 PDB code 1J5J40) and 100 nM charybdotoxin-Nvoc activity on KV1.2 current (right, ChTx PDB code 4JTA41). AaHII-R62K toxin induces slowing of inactivation of hNaV1.2, while BeKm1 and charybdotoxin induce block of hERG and KV1.2 channels, respectively. For AaHII-R62K-Nvoc and BeKm1-Nvoc, scale is 2 ms and 1 nA. For charybdotoxin-Nvoc, scale is 400 ms and 200 pA. Source data are provided as a Source Data File.
Generalisation of caging approach for toxins modulating ion channels
Similar inhibitory toxins displaying an important amine function on a key residue for pharmacology such as BeKm1 (hERG blocker)40 and charybdotoxin (KV1.2 blocker)41 were also caged with a Nvoc protecting group (on K18 for BeKm1 and on K27 for charybdotoxin based on previous SAR studies40,41). In addition, a Nvoc-grafted analogue of AaHII (a NaV channel activator) was also produced wherein Arg62, another essential residue for function8, was replaced by Lys62 with a Nvoc protecting group to enlarge the applicability of the technique. Those toxins present similar uncaging efficacies, indicating that cage removal is largely toxin amino acid sequence- and conformation-independent (Supplementary Fig. 7). These results suggest that this peptide toxin caging/uncaging approach is widely applicable regardless of amino acid sequences. As expected for an appropriate positioning of Nvoc on the toxin sequence, uncaging by illumination at 365 nm of AaHII-R62K-Nvoc, BeKm1-Nvoc, or charybdotoxin-Nvoc lead to UV-dependent effects on hNaV1.2 (activation), hERG (block), and KV1.2 (block) currents (Fig. 4h), respectively. This result indicates that this approach can be generalized to toxin activators and pore blockers for the control of a wide diversity of ion channels.
Spatiotemporal control of HwTxIV activity in brain slices
To expand our approach to biological systems, we assessed the use of HwTxIV-Nvoc in mouse brain slices during optical measurements of Na+ influx. We first determined that action potentials (APs) recorded in neocortical layer-5 (L5) pyramidal neurons were inhibited by local application of 500 nM noncaged HwTxIV from the surface of the brain slice near the cell body (Fig. 5a). Similar experiments conducted with HwTxIV-Nvoc show that AP shape is unaltered (Fig. 5b, c). Unsurprisingly, photolysis of HwTxIV-Nvoc leads to a significant decrease of the maximal membrane potential (Vm) 1 min after illumination (Fig. 5b, c). As expected from the time-course of reversal in the uncaging experiments performed on the hNaV1.6-expressing cell line (Supplementary Fig. 5), the effect of uncaged HwTxIV was poorly reversible in brain slices. It took 30 min to get a reasonable washout of the peptide toxin (Supplementary Fig. 8). Next, spatial selectivity was first examined using a ~100 μm diameter 365 nm UV LED spot. In this configuration, the somatic AP was not affected by illuminating a spot centered ~100 μm away from the soma, whereas direct illumination of the soma inhibited the somatic AP (Fig. 5d, e). We next used a more precise ~40 μm diameter illumination spot to define the spatial conditions for toxin uncaging/AP blocking by positioning the spot at varying distances from the soma (Fig. 5f, g). The somatic AP was not affected when the cell was >80 µm from the spot center, whereas it was partially inhibited at distances between 60 and 20 µm from the spot center (Fig. 5h). These results provide information about the spatial resolution that can be attained by uncaging of HwTxIV-Nvoc inside the area of photolysis. We then recorded Na+ influx via NaV channels, associated with an AP, to unambiguously assess the effect of the uncaged toxin on the channels expressed in the axon initial segment (AIS) (Fig. 5i) using an ultrafast Na+ imaging approach42. Illumination of HwTxIV-Nvoc in the soma (500 ms, 2 mW) fully prevented Na+ influx in the AIS, even at the same depolarized Vm of the AP (Fig. 5j, k, Supplementary Movie 1). This result demonstrates that the photo-release of HwTxIV blocks NaV channels and AP propagation even at positive Vm. Altogether, these results provide proof-of-principle that controlled photolysis of HwTxIV-Nvoc can be used to precisely target AP initiation and Na+ influx in the AIS in brain slices.

a Inhibition of AP by 500 nM non-caged HwTxIV (n = 6). b AP in control condition, in presence of 2.5 µM HwTxIV-Nvoc and 1 min after uncaging. c Left: mean ± SEM (n = 8 cells) of normalized AP peak after addition of HwTxIV-Nvoc and 1 min after photolysis (*p = 0.0078, two-sided paired t test). Right: mean ± SEM (n = 5 cells) of normalized AP width after addition of HwTxIV-Nvoc. d APs 1 min after HwTxIV-Nvoc uncaging over a spot centered ~100 µm from the cell body or over the soma. e Mean ± SEM (n = 4 cells) of the normalized AP peak after uncaging 100 µm away from the soma or directly on the soma. (*p = 0.0005, two-sided paired t test). f Image of the ~40 µm diameter UV spot. Purple curve represents the light intensity profile on cell schematic. g Top: images of a L5 pyramidal neuron relative to UV (405 nm) illumination spot position. Bottom: AP in the presence of HwTxIV-Nvoc (orange traces) and 1 min after uncaging (purple traces) with cell positioned at 100, 80, 60, 40, 20, and 0 µm distance from spot. h Mean ± SEM (n = 5 cells) of normalized AP peak 1 min after uncaging with the cell positioned at decreasing distances from spot center (100 µm; 80 µm, p = 0.2302; 60 µm, *p = 0.0434; 40 µm, *p = 0.0420; 20 µm, *p = 0.0133; 0 µm, ***p < 0.0001). Two-sided paired t-test. i Left, L5 pyramidal neuron filled with 500 µM of ING-2. AIS area of Δ[Na+] measurement in red cylinder. Right, images before and during UV uncaging pulse illustrating photolysis area. j Left, somatic AP (top) and associated Δ[Na+] signal in the presence of HwTxIV-Nvoc. Right, after toxin uncaging, the cell was depolarized to +20 mV to correspond to control AP peak and measure Δ[Na+] signal in the same condition. k mean ± SEM (n = 4 cells) of the Δ[Na+] signal maximum (peak) before and after uncaging the toxin. (*p < 0.0174, two-sided paired t-test). Source data are provided as a Source Data File.
In vivo spatiotemporal control of caged toxins
In order to evaluate the functional in vivo effects of our approach, we first validated that intraperitoneal injection of HwTxIV-Nvoc has negligible effects on mice activity contrary to a 10-fold lower amount of noncaged HwTxIV which drastically reduces motor behavior (Fig. 6a–c). Using in situ EDL muscle contraction monitoring in anesthetized mice, we validated that muscle illumination at 365 nm has no effects on EDL twitches by itself in control mice but demonstrated that a similar illumination paradigm of mice injected with HwTxIV-Nvoc (365 nm, 50 mW/cm²) significantly reduces contractile force thanks to uncaging of the peptide at the neuromuscular junctions where Nav1.6 is expressed (Fig. 6d–e). This result demonstrates in vivo activation of caged peptide toxins using UV light in superficial tissues. The data also indicate that the caged peptide reaches concentrations at the neuromuscular junctions high enough for light activation in spite of a simple intraperitoneal injection. By using a zebrafish larvae model, we confirmed the efficacy of this approach using AaHII-R62K-Nvoc peptide. Indeed, zebrafish illumination induces a significant reduction of swimming movements and a paralysis in all (6/6) larvaes previously injected with 50 µM AaHII-R62K-Nvoc (Supplementary Fig. 9 and Supplementary Movies 2 and 3). Illumination of control larvaes in the same conditions induce a nonsignificant reduction of movements largely due to habituation to the new environment in which the larvae were positioned. Those in vivo approaches demonstrate the potential of caged peptide toxins to be ineffective prior to illumination but fully active in spatially restricted regions after illumination in order to control ion channels in an integrated model.

a Representative trajectory plots of mice injected with vehicle (left), non-caged HwTxIV (center) or HwTxIV-Nvoc (right). b–c Quantification of total movements (b) and rearing (c) in mice injected with vehicle (white circles), HwTxIV (black circles) or HwTxIV-Nvoc (orange circles). (n = 5 in each group, mean ± SEM *p < 0.05; **p < 0.01; ***p < 0.001 versus vehicle, two-sided repeated-measures 2-way ANOVA test followed by Tukey’s post test). d Representative twitches obtained from mice injected with vehicle or HwTxIV-Nvoc at t = 0; t = 5; t = 10; t = 15; t = 20 min after injection. e EDL twitch force normalized to muscle mass (g/mg of EDL) in vehicle or HwTxIV-Nvoc mice before and after illumination (365 nm, 50 mW/cm2). (n = 5 in each group, mean ± SEM **p = 0.0041; ***p = 0.0002 versus t0, two-sided repeated-measures Friedman test followed by Dunn’s post-test). Source data are provided as a Source Data File.
Discussion
In summary, we report the development and application of a robust, generalizable, and in vivo compatible strategy for producing photoactivatable peptide toxins modulating voltage-gated ion channels and cell excitability. By using HwTxIV-Nvoc, we demonstrate that caged compounds can be activated by wavelengths <435 nm and is therefore compatible with dyes used for optical imaging or fluorescent compounds to monitor voltage-dependent structural changes in ion channels43. It is worth noting that this approach allows a spatial and temporal control of voltage-gated ion channel function. Our findings provide opportunities for gaining insights into the functional role of ion channels in physiological or pathological processes such as sensory perception disorders, muscle and brain channelopathies44 or cardiac muscle diseases45. The utility of classical pharmacology in vivo is limited, because local drug delivery is temporally ill-defined and imprecise. Often also, the far-from-perfect selectivity profile of peptide toxins leads to unwanted side effects in vivo. In contrast, photopharmacology is able to mimic the timing, amplitude, and spread of naturally occurring modulatory signals. By offering a real-time control of voltage-gated ion channels complexes and their functionalities in precisely defined regions that are poorly accessible to electrophysiological or pharmacological manipulations, photoactivatable peptide toxins open a window for the understanding of the physiology of ion channels. With the methodology described herein, the need for selective compounds is also attenuated because a channel subtype of interest can be targeted selectively thanks to the precision of spatial uncaging. In the case of HwTxIV, acting like a pan-NaV inhibitor for NaV1.1, NaV1.2, NaV1.6, and NaV1.7, the selectivity of action on NaV1.6 is provided by uncaging the compound at the neuromuscular junction, within a tissue, the skeletal muscle, where NaV1.6 predominates but lacking the three other channel subtypes. Similar approaches have been previously reported for ligand-gated ion channels46 and an elegant approach using coumarin-derived protected group grafted on saxitoxin targeting sodium channels has been recently published17. Despite very fast activation, this approach has been used on a nonpeptidic toxin and proof of in vivo efficacy has not been demonstrated yet. In our study, we demonstrate the ability of the caged peptide to be used, in vivo, as light-sensitive modulators of ion channels both in mice and zebrafish larvae. Given the number of peptides being isolated from venoms that target membrane proteins, the use of these photosensitive groups will first enable the development of a large number of additional photoactivable toxins to better understand the functional heterogeneity of ion channels and next, by establishing causal relationships between a protein activity and a cellular or physiological output, photoactivatable peptide toxins hold strong potential for identifying new therapeutic targets in many ion channel-related diseases. Peptide toxins of venom origin seem to hold the greatest promises as they have been tailored by nature for proper pharmacological action in vivo (adequate distribution and stability) and display a variety of properties in terms of selectivity and reversibility. In our case, low reversibility of HwTxIV is a desired property for prolonged muscle paralysis or chronic pain treatment for instance. In brain tissues, where experimentalists like to repeat the observation from multiple cells, more reversible peptide toxins than HwTxIV would be preferred for rapid clearance of the effect. The main limitation that can be invoked is the fact that this technology is based on peptides, poorly delivered by oral ingestion, more expensive to be produced than small chemicals and with more limited stability in vivo, although considerable progress is witnessed lately in using peptides as therapeutics.
Methods
Ethical Statement
All animal care and experimental procedures were performed in the animal facilities that have been accredited by the French Ministry of Agriculture. The experimental procedures were approved by the regional ethic committees (CEEA-006 Pays de la Loire; CEEA-012 Grenoble; CEEA-LR Languedoc Roussillon, France) according to the Directive 2010/63/EU of the European Union.
Molecular modelling
To understand if modifications in toxin activity are connected to alterations in peptide/channel interaction, molecular simulations were performed. The structures of NaVAb/NaV1.7-VS2A chimera with m3-Huwentoxin-IV (pdb code 7K48)30 was used as a template for i) illustrating the docking of HwTxIV on NaV1.7, ii) deciding that K32 was a good candidate for caging, and iii) highlighting the clashes induced by the Nvoc presence on K32. Discovery Studio (Dassault System) was used to build HwTxIV-Nvoc structure. After applying a fast, Dreiding-like forcefield in order to clean geometry of designed peptide, the van-der Waals (vdW) repulsion forces between HwTxIV-Nvoc and NaV1.7 were calculated using the “Show bumps” plugin, implemented in PyMOL (http://www.pymolwiki.org/index.php/Show_bumps).
Chemical syntheses of HwTxIV and peptide analogues
All peptides were assembled stepwise using Fmoc-based Solid Phase Peptide Synthesis (SPPS) on a PTI Symphony synthesizer at a 0.05 mmol or 0.1 mmol scale on 2-chlorotrityl chloride polystyrene resin (substitution approximately 1.6 mmol/g). For caged peptides, Fmoc-L-Lysine residues at position 32 (HwTxIV), 18 (BeKm1), 27 (charybdotoxin), and 62 (AaHII-R62K) were replaced by Fmoc-L-Lys(Nvoc)-OH (Iris Biotech, Marktredwitz, Germany) during assembly. The Fmoc protecting group was removed using 20% piperidine in DMF and free amine was coupled using tenfold excess of Fmoc amino acids and HCTU/DIEA activation in NMP/DMF (3×15 min). Linear peptides were de-protected and cleaved from the resin with TFA/H2O/1,3-dimethoxybenzene(DMB)/TIS/2,2′-(Ethylenedioxy)diethanethiol(DODT) 85.1/5/2.5/3.7/3.7 (vol.), then precipitated out in cold diethyl ether. The resulting white solids were washed twice with diethyl ether, re-suspended in H2O/acetonitrile, and freeze dried to afford crude linear peptide. Oxidative folding of the crude linear HwTxIV-Nvoc was successfully conducted at RT in the conditions optimized for the HwTxIV using a peptide concentration of 0.1 mg/mL in a 0.1 M Tris buffer at pH 8.0 containing 10% of DMSO. Similar conditions were used for the syntheses of AaHII-R62K-Nvoc, BeKm1-Nvoc, and charybdotoxin-Nvoc. Purification of the peptides were performed on Chromolith®HighResolution RP-18e column (100 ×4.6 mm) using a 5–65% buffer B run in 14 min at 2 mL/min. Solvent system: A: H2O 0.1%TFA and B: MeCN 0.1%TFA.
LC-ESI-QTOF MS analyses
LC-ESI-MS data were acquired with an Accurate mass QTOF 6530 (Agilent) coupled to an Agilent 1290 UPLC system. If required, separation of the peptide sample (5 µL, approx. 10 µg/mL) was done on an Ascentis express 90 Å C18 column (2.0 μm, 2.1 mm ID × 100 mm L, Supelco) heated at 70 °C at a flow rate of 0.5 mL/min and with a 2-70% buffer B/A gradient over 11 min (buffer A: H2O/formic acid, 99.9/0.1 (v/v); buffer B: ACN/formic acid, 99.9/0.1 (v/v)). If not required, the sample was directly injected at 0.6 mL/min flow rate in H2O/MeCN +0.1% formic acid. The acquisition was done in the positive mode with a Dual ESI source, within a m/z 100-1700 mass range and analyzed via the Agilent MassHunter software version 10.0. Source parameters were set as follows: capillary voltage, 4 kV; fragmentor, 100 V; gas temperature, 350 °C; drying gas flow, 12 L/min; nebulizer 50 psig.
The observed monoisotopic masses of the peptides were as follow: AaHII-R62K: 7210.21 Da (theoretical: 7210.14); AaHII-R62K-Nvoc: 7449.23 (theoretical: 7449.18); BeKm-1: 4088.79 Da (theoretical 4088.78); BeKm-1-Nvoc: 4327.85 Da (theoretical: 4327.81); Charybdotoxin: 4292.84 Da (theoretical: 4292.87); Charybdotoxin-Nvoc: 4531.94 Da (theoretical: 4531.91); HwTxIV: 4088.04 Da (theoretical: 4087.96); HwTxIV-Nvoc: 4327.08 Da (theoretical: 4327.00). Source data are provided as a Source Data File.
Monitoring of Nvoc deprotection by analytical RP-HPLC
Analytical RP-HPLC was performed using an SPD M20-A system (Shimadzu) with a Luna OmegaPS C18 column (4.6 ×250 mm, 5 μm, 100 Å). Two µL (corresponding to 7 µg of material) was loaded and a 5-60% acetonitrile gradient (0.1% TFA v/v) was applied over 35 min at room temperature to detect analytes by UV absorbance at 214 nm. Illumination of samples was performed at 365 nm for different times (between 1 sec and 30 min illumination time) at 41.8 mW/cm2 or less (as specified in the Result section) for 10 min using a CoolLED pE4000 light source (CoolLED, UK). Flash energy has been measured using a High Sensitive Thermal Power Head (S401C, ThorLabs). Coelution of uncaged HwTxIV and non-caged HwTxIV was performed using a 50:50 ratio for both compounds.
NMR spectrometry
HwTxIV-Nvoc was illuminated for 30 min at 100% power (45 mW/cm2) using a CoolLED pE4000 light source (CoolLED, UK). Three 200 μL solutions were used in 3 mm NMR tubes for i) HwTxIV-Nvoc before illumination (500 μM); ii) HwTxIV-Nvoc after illumination (500 µM); and iii) non-caged HwTxIV (200 μM). For each sample, two-dimensional homonuclear (80 ms TOCSY and 160 ms NOESY) and heteronuclear 13C-HSQC (natural abundancy) spectra were acquired on a BRUKER 700 MHz NMR spectrometer equipped with a 5 mm TCI cryoprobe, at 298 K. Processing and analyses were performed with Bruker’s TopSpin3.2 and CcpNMR programs. Spectra are drawn with CcpNMR program. 3D structure is drawn with the PyMOL software (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
Cell cultures
HEK293 cells stably expressing the hNaV1.1 (XM_011511604), hNaV1.2 (XM_017004655) or hNaV1.6 (XM_011538651) channels, CHO cells transiently expressing hKV1.2 (NG_027997.2) and CHO cells stably expressing hNaV1.7 (XM_011511618) or the hERG (NM_000238) channels were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum, 1 mM pyruvic acid, 4.5 g/L glucose, 4 mM glutamine, 800 µg/mL G418, 10 U/mL penicillin and 10 μg/mL streptomycin (Gibco, Grand Island, NY). All cell lines were incubated at 37 °C in a 5% CO2 atmosphere. For electrophysiological recordings, cells were detached with trypsin and floating single cells were diluted (~300,000 cells/mL) in medium contained (in mM): 140 mM, 4 KCl, 2 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES (pH 7.4, osmolarity 298 mOsm).
Automated patch-clamp recordings and pharmacological studies
Whole-cell recordings were used to investigate the effects of HwTxIV peptides on HEK293 or CHO cells expressing hNaV1.1, hNaV1.2, hNaV1.6, or hNaV1.7 channels but also the effects of AaHII-R62K-Nvoc, BeKm1-Nvoc, and charybdotoxin-Nvoc on hNaV1.2, hERG, and hKV1.2, respectively. Automated patch-clamp recordings were performed using the SyncroPatch 384PE from Nanion (München, Germany). Chips with single-hole medium resistance of 4.52 ± 0.08 MΩ (n = 384) were used for recordings. Pulse generation and data collection were performed with the PatchControl384 v1.5.2 software (Nanion) and the Biomek v1.0 interface (Beckman Coulter). Whole-cell recordings were conducted according to the recommended procedures of Nanion. Cells were stored in a cell hotel reservoir at 10 °C with shaking speed at 60 RPM. After initiating the experiment, cell catching, sealing, whole-cell formation, liquid application, recording, and data acquisition were all performed sequentially and automatically. For sodium channel recordings, intracellular solution was (in mM): 10 CsCl, 110 CsF, 10 NaCl, 10 EGTA, and 10 HEPES (pH 7.2, osmolarity 280 mOsm), while being 10 KCl, 110 KF, 10 NaCl, 10 EGTA, and 10 HEPES (pH 7.2, osmolarity 280 mOsm) for potassium channel recordings. Extracellular solution was (in mM): 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 5 glucose, and 10 HEPES (pH 7.4, osmolarity 298 mOsm). For sodium channel recordings at room temperature (18–22 °C), holding potential was set at −100 mV and the sampling rate set at 20 kHz. Peptides were diluted in the extracellular solution containing 0.3% bovine serum albumin (BSA). Compounds were tested at 0 mV with 50 ms test potentials repeated every 5 s. Current inhibition percentages were measured at equilibrium after 14 min peptide application time. For hERG current, whole-cell experiments were performed at a holding potential of −80 mV and BeKm1-Nvoc was tested at a 200 ms test potential of +60 mV following a first activation step of 1000 ms at +60 mV and a 10 ms step at −120 to recover from inactivation with a pulse every 8 s. For KV1.2 current, whole-cell experiments were performed at a holding potential of −90 mV and charybdotoxin-Nvoc was tested at a 2000 ms test potential of +60 mV with a pulse every 12 s.
Photoactivation of HwTxIV-Nvoc on HEK293 cells
The pharmacology of HwTxIV-Nvoc, as well as the efficiency of the released product to block hNaV channels was studied using combined automated patch-clamp and UV illumination. After 2 min of control, HwTxIV-Nvoc was added to the external buffer and effects were recorded for 100 s prior to a 250 s-duration illumination for photocleavage induction of the caged compound. Different wavelengths, illumination powers’ and durations were used as specified in figure legends. For AaHII-R62K-Nvoc, BeKm1-Nvoc, and charybdotoxin-Nvoc, a 250 s-duration illumination at 45 mW/cm² was used to uncage compounds.
Construction of hNav1.6/rKv2.1 chimeras
Channel chimeras were generated using sequential PCR with KV2.1Δ747,48 (Genscript, USA) and hNaV1.6 (NM_014191, Origene Technologies, USA) as templates. cRNA was synthesized using T7 polymerase (mMessage mMachine kit, Thermo Fisher, USA) after linearizing cDNA with appropriate restriction enzymes. This chimeric approach was previously shown to robustly indicate the binding locus of toxins32. Boundaries of the chimeras were chosen as reported previously in Fig. S2 of Bosmans et al. 201133 and include the transfer of hNaV1.6 regions (MMAYITEFVNLGNVSALRTFRVLR (VSDI), GFIVSLSLMELSLADVEGLSVLRSFRLLR (VSDII), SLVSLIANALGYSELGAIKSLRTLRALR (VSDIII), SIVGMFLADIIEKYFVSPTLFRVIRLARIGRILR (VSDIV) into KV2.1.
Two-electrode voltage-clamp recordings
Two-electrode voltage-clamp recording techniques on Xenopus laevis oocytes (OC-725C, Warner Instruments, USA; 150 μL recording chamber) were used to measure channel currents 1 day after cRNA injection and incubation at 17 °C in ND96 that contained (in mM): 96 NaCl, 2 KCl, 5 HEPES, 1 MgCl2, and 1.8 CaCl2, 50 μg/mL gentamycin, pH 7.6. Data were filtered at 4 kHz and digitized at 20 kHz using pClamp software (Molecular Devices, USA). Microelectrode resistances were 0.5–1 MΩ when filled with 3 M KCl. For KV channel experiments, the external recording solution contained (in mM): 50 KCl, 50 NaCl, 5 HEPES, 1 MgCl2 and 0.3 CaCl2, pH 7.6 with NaOH. All experiments were performed at RT (~21 °C) and toxin samples were diluted in recording solution with 0.1% BSA. Voltage-activation relationships were obtained by measuring tail currents for KV channels. After the addition of toxin to the recording chamber, equilibration between toxin and channel was monitored using weak depolarizations elicited at 5–10 s intervals. For all channels, voltage-activation relationships were recorded in the absence and presence of the toxin. Off-line data analysis was performed using Clampfit 11 (Molecular Devices, USA) and Origin 8 (Originlab, USA).
Mice
For coronal slices, procedures were reviewed by the ethics committee affiliated to the animal facility of the university (D3842110001) and performed in accordance with European Directives 2010/63/UE on the care, welfare, and treatment of animals. Specifically, C57BL/6 J mice were housed with their mother with ad libitum access to food and water. Animals (21–35 postnatal days old) were anesthetised by isoflurane inhalation and the entire brain was removed after decapitation. For neuromuscular experiments, experiments were carried out on 25 male mice (C57BL/6 J mice) aged between 6 and 8 weeks. Mice were housed five per cage and maintained on a 12/12 h light/dark schedule in a temperature-controlled facility (22 ± 1 °C) with free access to food and water. Animals were kept undisturbed for 7 days before experiments. Animals were divided into five groups. For contractile in situ experiments, two groups of 5 mice received or a single dose of HwTxIV-Nvoc (5 mg/kg in 100 µL) or a similar intraperitoneally injection of 0.9% NaCl solution. For behavior actimeter experiments, three groups of five mice received a single intraperitoneally injection of HwTxIV (0.5 mg/kg in 100 µL) or HwTxIV-Nvoc (5 mg/kg in 100 µL) or similar intraperitoneally injection of 0.9% NaCl solution. All procedures were conducted in conformity with European rules for animal experimentation (French Ethical Committee APAFIS#8186-2016121315485337, January 10, 2017, for EDL contractile in situ experiments, APAFIS#1765-2016121315485337, January 10, 2017, for motor behavior experiments with native toxin).
Electrophysiological recordings from neocortical brain slices
Neocortical coronal slices (350 µm thick) were prepared and maintained using previously described procedures adapted from other preparations49,50,51,52. Briefly, slices were incubated in extracellular solution at 37 °C for 45 min and then maintained at room temperature before use. The extracellular solution contained (in mM): 125 NaCl, 26 NaHCO3, 1 MgSO4, 3 KCl, 1 NaH2PO4, 2 CaCl2, and 20 glucose, bubbled with 95% O2 and 5% CO2. The intracellular solution contained (in mM): 125 KMeSO4, 5 KCl, 8 MgSO4, 5 Na2-ATP, 0.3 Tris-GTP, 12 Tris-Phosphocreatine, 20 HEPES, adjusted to pH 7.35 with KOH. Layer-5 (L5) pyramidal neurons from the somatosensory cortex were selected and patched in a whole-cell configuration. Somatic action potentials (APs) were elicited by injecting current pulses of 3–5 ms duration and of 1–2 nA amplitude. In Na+ imaging experiments, after the photo-release of the toxin, the current intensity was increased to 5–10 nA in order to depolarize the cell to the same Vm corresponding to the AP peak in the control condition. The measured Vm was corrected for the junction and the bridge potentials. The caged toxin was dissolved in the extracellular solution at 2.5 μM concentration and locally delivered either using a SmartSquirt micro-perfusion system (WPI, Hitchin, UK) with a tip of 250 µm diameter, or by simple pressure ejection with a tip of ~10 µm. Photolysis of the caged compound was performed either on a spot of ~100 μm diameter and using the light of a 365 nm LED (~2 mW of power) controlled by an OptoLED (Cairn Research, Faversham, UK), or on a spot of ~40 µm using a 300 mW / 405 nm diode laser (Cairn Research). The protocol at 365 nm consisted of 1–3 pulses of 100–300 ms duration and 1 s interval, or by a single pulse of 500 ms duration. The protocol at 405 nm consisted of 2 pulses of 500 ms duration and 5 s interval. In the case of simultaneous fluorescence recordings and UV stimulation, uncaging was optically combined to imaging by coupling the two excitation wavelengths similarly to what was done for an equivalent application53.
Na+ imaging
Optical measurements of [Na+] from the AIS were obtained as previously described42. Briefly, neurons were loaded with 500 µM of the Na+ indicator ING-2 (IonBiosciences, San Marcos, TX, USA) for 20–30 min after establishing the whole-cell configuration. Fluorescence was excited using a 520-mW line of a LaserBank (Cairn Research) band-pass filtered at 517 ± 10 nm, directed to the preparation using a 538 nm long-pass dichroic mirror. Emitted fluorescence was band-pass filtered at 559 ± 17 nm before being recorded with a DaVinci 2 K CMOS camera (SciMeasure, Decatur, GA) at 10 kHz with a pixel resolution of 30×128. Optical data, obtained by averaging 4 trials under the same condition, were corrected for bleaching using a trial without current injection. The fractional change of Na+ fluorescence was expressed in terms of intracellular Na+ concentration change (Δ[Na+]) using the previously published calibration for which 1% corresponds to 0.175 mM42.
Actimeter
Mice were randomly assigned to 3 groups according to peptide toxin treatment as described above. The motor behavior was examined with an open field actimeter54. For this analysis, mice were individually placed in an automated photocell activity chamber (Letica model LE 8811, Bioseb, France) which consists of a plexiglass chamber (20 cm×24 cm×14 cm) surrounded by two rows of infrared photobeams. The first row of sensors was raised at a height of 2 cm for measuring the horizontal activity and the second row placed above the animal for vertical activity. Just after treatment administration via intraperitoneally injection, the spontaneous motor activity was measured for 10 min using a movement analysis system (Bioseb, France), which dissociates total movements and rearing (numbers). All the experiments were realized in a dark room.
Contractile properties of fast-EDL muscle using sciatic nerve stimulation
EDL muscle force properties were analyzed by using in situ muscle contraction measurements55,56. Briefly, mice were anesthetized by intraperitoneally injection of xylazine/ ketamine (10/100 mg/kg), and the adequacy of the anesthesia was monitored throughout the experiment. The skin was then carefully removed from the left leg, the sciatic nerve was carefully isolated and EDL muscle was dissected free with its blood supply intact. The foot and the tibia of the mice were fixed by two clamps, and the distal tendon of the EDL muscle was attached to a force transducer and positioned parallel to the tibia. Throughout the experimental procedure, the mice were kept on a heating pad to maintain normal body temperature, and the muscles were continuously perfused with Ringer solution. Stimulation electrodes were positioned at the level of the sciatic nerve, and connected to a pulse generator with stimulation characteristics of 0.2 ms duration, 6 V stimulation amplitude and 1 Hz frequency. The muscle was stretched before stimulation voltage was applied to produce the most powerful twitch contractions. Stimulation was maintained during all the experiments. Twitch parameters were measured prior to UV illumination, after 5 and 10 min of UV illumination (365 nm, 50 mW/cm²) and 5 and 10 min after UV illumination. At the end of the experiments, EDL muscles were rapidly dissected and weighted. Twitch forces were normalized in grams per milligram of fresh EDL muscles. All force data are expressed as percentage of the initial force, i.e. before illumination. All the experiments were realized in a dark room. Data were collected and stored for analyses with Chart v4.2.3 (PowerLab 4/25 ADInstrument, PHYMEP France).
Zebrafish larvaes movement tracking
Zebrafish larvaes (Danio rerio, wild type AB strain) were maintained under standardized conditions and experiments were conducted in accordance with local approval (APAFIS#4054-2016021116464098 v5) and the European Communities council directive 2010/63/EU. 120 h post fertilization (hpf) larvae were anaesthetized using tricaine methanesulfonate (MS222, Sigma-Aldrich). Larvae were injected (microinjector-World Precision Instruments) in the pericardium with 1 nL of either vehicle (Texas Red) or a 50 μM solution of AaHII-R62K-Nvoc. Locomotor activity was recorded in a dark environment inside a DanioVision observation chamber coupled with Ethovision video tracking v. 14 (Ethovision XT, Noldus Information Technology, Netherlands). After 10 min of tracking, larvae were exposed to 365 nm light (Prismatix) for 5 min. After a period of 30 min locomotor activity was recorded as before.
Statistics and data analyses
Values are represented as mean ± SEM. The significance of illumination tests on automated patch-clamp was tested by performing paired 1-way ANOVA and Bonferroni’s multiple and significance of in vivo experiments was tested by performing Friedman test with Dunn’s multiple comparisons test. A p-value lower than 0.05 was considered significant. Significance of pharmacological tests on electrophysiological and optical recordings in brain slices was tested by performing the paired t-test with 0.01 as probability threshold value.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data regarding chemical syntheses and electrophysiology generated in this study are provided in the Source Data File. HwTxIV BMRB and PDB codes used for NMR experiments are respectively 5527 and 1MB6. Source data are provided with this paper.
Code availability
R codes used for electrophysiological analyses and Matlab code related to sodium imaging in brain slices are available on GitHub (https://github.com/jerome-montnach/Caged-HwTxIV.git).
References
Kalia, J. et al. From foe to friend: using animal toxins to investigate ion channel function. J. Mol. Biol. 427, 158–175 (2015).
Gonoi, T., Sherman, S. J. & Catterall, W. A. Voltage clamp analysis of tetrodotoxin-sensitive and -insensitive sodium channels in rat muscle cells developing in vitro. J. Neurosci. 5, 2559–2564 (1985).
Mouhat, S., Jouirou, B., Mosbah, A., De Waard, M. & Sabatier, J. M. Diversity of folds in animal toxins acting on ion channels. Biochem. J. 378, 717–726 (2004).
Ahern, C. A., Payandeh, J., Bosmans, F. & Chanda, B. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 147, 1–24 (2016).
McCleskey, E. W. et al. Omega-conotoxin: direct and persistent blockade of specific types of calcium channels in neurons but not muscle. Proc. Natl. Acad. Sci. USA 84, 4327–4331 (1987).
De Waard, S. et al. Functional Impact of BeKm-1, a High-Affinity hERG Blocker, on Cardiomyocytes Derived from Human-Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 21, 7167 (2020).
Pan, X. et al. Molecular basis for pore blockade of human Na(+) channel Nav1.2 by the mu-conotoxin KIIIA. Science 363, 1309–1313 (2019).
Clairfeuille, T. et al. Structural basis of alpha-scorpion toxin action on Nav channels. Science 363, aav8573 (2019).
Joshi, J., Rubart, M. & Zhu, W. Optogenetics: Background, Methodological Advances and Potential Applications for Cardiovascular Research and Medicine. Front. Bioeng. Biotechnol. 7, 466 (2020).
Entcheva, E. & Kay, M. W. Cardiac optogenetics: a decade of enlightenment. Nat. Rev. Cardiol. 18, 349–367 (2020).
Tochitsky, I., Kienzler, M. A., Isacoff, E. & Kramer, R. H. Restoring Vision to the Blind with Chemical Photoswitches. Chemical Rev. 118, 10748–10773 (2018).
Mourot, A. et al. Rapid optical control of nociception with an ion-channel photoswitch. Nat. Methods 9, 396–402 (2012).
Tazerart, S., Mitchell, D. E., Miranda-Rottmann, S. & Araya, R. A spike-timing-dependent plasticity rule for dendritic spines. Nat. Commun. 11, 4276 (2020).
Matsuzaki, M., Hayama, T., Kasai, H. & Ellis-Davies, G. C. R. Two-photon uncaging of gamma-aminobutyric acid in intact brain tissue. Nat Chem. Biol. 6, 255–257 (2010).
Ellis-Davies, G. C. Caged compounds: photorelease technology for control of cellular chemistry and physiology. Nat. Methods 4, 619–628 (2007).
Dembitskaya, Y., Wu, Y. W. & Semyanov, A. Tonic GABAA Conductance Favors Spike-Timing-Dependent over Theta-Burst-Induced Long-Term Potentiation in the Hippocampus. J. Neurosc. 40, 4266–4276 (2020).
Elleman, A. V. et al. Precise spatiotemporal control of voltage-gated sodium channels by photocaged saxitoxin. Nat. Commun.12, 4171 (2021).
Bourgault, S., Letourneau, M. & Fournier, A. Development of photolabile caged analogs of endothelin-1. Peptides 28, 1074–1082 (2007).
Wirkner, M. et al. Photoactivatable caged cyclic RGD peptide for triggering integrin binding and cell adhesion to surfaces. Chembiochem 12, 2623–2629 (2011).
Santos, F. C., Soares, A. M. S., Goncalves, M. S. T. & Costa, S. P. G. Phototriggered release of tetrapeptide AAPV from coumarinyl and pyrenyl cages. Amino Acids 49, 1077–1088 (2017).
Nguyen, A., Rothman, D. M., Stehn, J., Imperiali, B. & Yaffe, M. B. Caged phosphopeptides reveal a temporal role for 14-3-3 in G1 arrest and S-phase checkpoint function. Nat. Biotechnol. 22, 993–1000 (2004).
Dauplais, M. et al. On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 272, 4302–4309 (1997).
Karas, J. A. et al. 2-nitroveratryl as a photocleavable thiol-protecting group for directed disulfide bond formation in the chemical synthesis of insulin. Chemistry 20, 9549–9552 (2014).
Goncalves, T. C. et al. Direct evidence for high affinity blockade of NaV1.6 channel subtype by huwentoxin-IV spider peptide, using multiscale functional approaches. Neuropharmacology 133, 404–414 (2018).
Deng, M. et al. Synthesis and biological characterization of synthetic analogs of Huwentoxin-IV (Mu-theraphotoxin-Hh2a), a neuronal tetrodotoxin-sensitive sodium channel inhibitor. Toxicon 71, 57–65 (2013).
Agwa, A. J. et al. Spider peptide toxin HwTx-IV engineered to bind to lipid membranes has an increased inhibitory potency at human voltage-gated sodium channel hNaV1.7. Biochem. Biophys. Acta. Biomembr. 1859, 835–844 (2017).
Agwa, A. J. et al. Manipulation of a spider peptide toxin alters its affinity for lipid bilayers and potency and selectivity for voltage-gated sodium channel subtype 1.7. J. Biol. Chem. 295, 5067–5080 (2020).
Shen, H., Liu, D., Wu, K., Lei, J. & Yan, N. Structures of human Nav1.7 channel in complex with auxiliary subunits and animal toxins. Science 363, 1303–1308 (2019).
Peng, K., Shu, Q., Liu, Z. & Liang, S. Function and solution structure of huwentoxin-IV, a potent neuronal tetrodotoxin (TTX)-sensitive sodium channel antagonist from Chinese bird spider Selenocosmia huwena. J. Biol Chem. 277, 47564–47571 (2002).
Wisedchaisri, G. et al. Structural Basis for High-Affinity Trapping of the NaV1.7 Channel in Its Resting State by Tarantula Toxin. Mol Cell 81, 38–48 (2021). e34.
Gao, S. et al. Investigation of binding modes of spider toxin-human voltage-gated sodium channel subtybe 1.7. J. Biomol. Struct. Dyn, 1–9 (2020).
Bosmans, F., Martin-Eauclaire, M. F. & Swartz, K. J. Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456, 202–208 (2008).
Bosmans, F., Puopolo, M., Martin-Eauclaire, M. F., Bean, B. P. & Swartz, K. J. Functional properties and toxin pharmacology of a dorsal root ganglion sodium channel viewed through its voltage sensors. J. Gen. Physiol. 138, 59–72 (2011).
Osteen, J. D. et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature 534, 494–499 (2016).
Aggarwal, S. K. & MacKinnon, R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron 16, 1169–1177 (1996).
Seoh, S. A., Sigg, D., Papazian, D. M. & Bezanilla, F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron 16, 1159–1167 (1996).
Ahern, C. A., Payandeh, J., Bosmans, F. & Chanda, B. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 147, 1–24 (2016).
Gilchrist, J. & Bosmans, F. Using voltage-sensor toxins and their molecular targets to investigate Nav1.8 gating. J. Physiol. 596, 1863–1872 (2018).
Horn, R., Ding, S. & Gruber, H. J. Immobilizing the moving parts of voltage-gated ion channels. J. Gen. Physiol. 116, 461–476 (2000).
Korolkova, Y. V. et al. New binding site on common molecular scaffold provides HERG channel specificity of scorpion toxin BeKm-1. J. Biol. Chem. 277, 43104–43109 (2002).
Banerjee, A., Lee, A., Campbell, E. & Mackinnon, R. Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K(+) channel. Elife 2, e00594 (2013).
Filipis, L. & Canepari, M. Optical measurement of physiological sodium currents in the axon initial segment. J Physiol, https://doi.org/10.1113/JP280554 (2020).
Fletcher-Taylor, S. et al. Distinguishing Potassium Channel Resting State Conformations in Live Cells with Environment-Sensitive Fluorescence. ACS Chem. Neurosci 11, 2316–2326 (2020).
Matthews, E., Balestrini, S., Sisodiya, S. M. & Hanna, M. G. Muscle and brain sodium channelopathies: genetic causes, clinical phenotypes, and management approaches. Lancet Child Adolesc Health 4, 536–547 (2020).
Li, N. et al. Impaired neuronal sodium channels cause intranodal conduction failure and reentrant arrhythmias in human sinoatrial node. Nat. Commun. 11, 512 (2020).
Noguchi, J. et al. In vivo two-photon uncaging of glutamate revealing the structure-function relationships of dendritic spines in the neocortex of adult mice. J. Physiol. 589, 2447–2457 (2011).
Frech, G. C., VanDongen, A. M., Schuster, G., Brown, A. M. & Joho, R. H. A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature 340, 642–645 (1989).
Swartz, K. J. & MacKinnon, R. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron 18, 665–673 (1997).
Jaafari, N. & Canepari, M. Functional coupling of diverse voltage-gated Ca(2+) channels underlies high fidelity of fast dendritic Ca(2+) signals during burst firing. J. Physiol. 594, 967–983 (2016).
Jaafari, N., De Waard, M. & Canepari, M. Imaging fast calcium currents beyond the limitations of electrode techniques. Biophys. J. 107, 1280–1288 (2014).
Ait Ouares, K. & Canepari, M. The Origin of Physiological Local mGluR1 Supralinear Ca(2+) Signals in Cerebellar Purkinje Neurons. J. Neurosci. 40, 1795–1809 (2020).
Ait Ouares, K., Filipis, L., Tzilivaki, A., Poirazi, P. & Canepari, M. Two Distinct Sets of Ca(2+) and K(+) Channels Are Activated at Different Membrane Potentials by the Climbing Fiber Synaptic Potential in Purkinje Neuron Dendrites. J. Neurosci. 39, 1969–1981 (2019).
Vogt, K. E., Gerharz, S., Graham, J. & Canepari, M. Combining membrane potential imaging with L-glutamate or GABA photorelease. PLoS One 6, e24911 (2011).
Larcher, T. et al. Characterization of dystrophin deficient rats: a new model for Duchenne muscular dystrophy. PLoS One 9, e110371 (2014).
Carre-Pierrat, M. et al. Pre-clinical study of 21 approved drugs in the mdx mouse. Neuromuscul. Disord. 21, 313–327 (2011).
Auda-Boucher, G. et al. Fetal muscle-derived cells can repair dystrophic muscles in mdx mice. Exp. Cell Res. 313, 997–1007 (2007).
Acknowledgements
M. De Waard and M. Canepari thank the Agence Nationale de la Recherche (ANR) for financial support to the laboratory of excellence “Ion Channels, Science and Therapeutics” (grant N° ANR-11-LABX-0015). Both researchers also thank the ANR for a grant entitled OptChemCom (grant N° ANR-18-CE19-0024-02). This work was supported by the Fondation Leducq in the frame of its program of ERPT equipment support (purchase of an automated patch-clamp system), by a grant “New Team” of the Région Pays de la Loire to M. De Waard, the National Institutes of Health (R01NS091352 to F.B.), by an AFM grant for LED equipment (AFM22401) and by a European FEDER grant in support of the automated patch-clamp system of Nanion. The salary of S. Nicolas was supported by the Fondation Leducq. The fellowships of J. Montnach and L. Filipis are provided by the ANR OptChemCom. The scholarship of L.A. Blömer is provided by the laboratory of excellence “Ion Channels, Science and Therapeutics”.
Author information
Authors and Affiliations
Contributions
JM, LL, and SN performed cell culture, automated patch-clamp, RP-HPLC experiments and analyses. FB and DC performed chimera experiments. CC and RB performed the chemical syntheses peptides. CJ performed zebrafish experiments. CH, JM, and AL performed in vivo experiments. HM and CL performed NMR experiments. LAB and LF performed experiments and analyses in brain slices. JM, MC and MDW made the figures. MDW conceived and managed the project. JM, MDW, and MC wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Matthew Banghart, Rajesh Khanna and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Montnach, J., Blömer, L.A., Lopez, L. et al. In vivo spatiotemporal control of voltage-gated ion channels by using photoactivatable peptidic toxins. Nat Commun 13, 417 (2022). https://doi.org/10.1038/s41467-022-27974-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-27974-w
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
