
Abstract
Previous genome-wide association studies revealed multiple common variants involved in eczema but the role of rare variants remains to be elucidated. Here, we investigate the role of rare variants in eczema susceptibility. We meta-analyze 21 study populations including 20,016 eczema cases and 380,433 controls. Rare variants are imputed with high accuracy using large population-based reference panels. We identify rare exonic variants in DUSP1, NOTCH4, and SLC9A4 to be associated with eczema. In DUSP1 and NOTCH4 missense variants are predicted to impact conserved functional domains. In addition, five novel common variants at SATB1-AS1/KCNH8, TRIB1/LINC00861, ZBTB1, TBX21/OSBPL7, and CSF2RB are discovered. While genes prioritized based on rare variants are significantly up-regulated in the skin, common variants point to immune cell function. Over 20% of the single nucleotide variant-based heritability is attributable to rare and low-frequency variants. The identified rare/low-frequency variants located in functional protein domains point to promising targets for novel therapeutic approaches to eczema.
Similar content being viewed by others


Introduction
Eczema is a chronic inflammatory skin disease that is often associated with immunoglobulin E (IgE)-mediated allergies. Like in other complex human diseases, there is a substantial heritable contribution to the disease risk. In recent years, several large genome-wide association studies (GWAS) identified a total of 32 susceptibility loci for eczema1,2,3,4,5,6,7,8,9. However, despite increasing sample sizes, the reported associations of single nucleotide polymorphisms (SNPs) have explained only a small fraction of the estimated heritability of eczema6,10.
It has been hypothesized that missing heritability may originate from rare risk variants that are not well captured on SNP arrays designed to study common variants with a minor allele frequency (MAF) > 0.0511. Thus, the majority of low-frequency SNPs (0.01 ≤ MAF < 0.05) and rare variants (MAF < 0.01) were out of reach in this approach. Furthermore, imputation of SNPs with low allele frequency at high quality was unattainable since until recently, a reference panel of sufficient size was not available.
There is increasing evidence that rare variants contribute to disease risk not only in Mendelian diseases but also in complex diseases. Recent studies suggested a role for low-frequency and rare variants in a number of complex disorders including inflammatory bowel disease, asthma, and cancer12,13,14. With the Haplotype Reference Consortium panel (HRC), a new resource has now become available including 32,488 sequenced individuals covering almost 65,000 haplotypes15. This allows for the first time the reliable imputation of rare variants.
In our meta-GWAS on rare variants in eczema, we uncover disease-associated exonic variants in the genes encoding dual specificity phosphatase 1 (DUSP1), neurogenic locus notch homolog protein 4 (NOTCH4), and solute carrier family 9 member A4 (SLC9A4). In DUSP1 and NOTCH4, the identified missense variants are located in functional domains. By extending the GWAS to common variants, another five new eczema-associated loci are detected. We estimate a contribution of over 20% for rare and low frequencies to eczema heritability.
Results
Study design and quality control
In total 21 study populations comprising 20,016 cases and 380,433 controls were included in our GWAS on rare variants in eczema (Supplementary Data 1). Of them, 19 study populations with available genotypes including 10,703 cases and 30,845 controls were analyzed with RVTESTS and combined in the rare variant set (RV set). The other two data sets were the publicly available FINNGEN study comprising 2,663 cases and 88,760 controls and UK Biobank (UKBB) contributing 6,650 cases and 260,828 controls (Fig. 1). Our study had 80% power to detect risk alleles with an allele frequency of 0.004 and a risk ratio of 1.6 using an allelic model with 10% prevalence, alpha < 1 × 10−8, and unselected controls (Supplementary Fig. 1)16. Taking into account that rare alleles usually have a higher impact on disease, our study was well-powered to detect rare variants associated with eczema. The genome-wide significance threshold was set at 1 × 10−8, as suggested previously for GWAS analyzing variants with a MAF of 0.00117.

Workflow of the meta-GWAS on rare variants in eczema is shown. Analyzing only rare/low-frequency variants, 11 loci were detected. In the complete data set, 38 loci were detected of which 6 were not reported to be associated with eczema or allergic disease previously. The significance threshold for the gene-level association analysis corresponds to 13,000 genes under study (carrying at least two variants with MAF < 0.05 and a CADD score ≥ 12.37). SNP single nucleotide polymorphism, MAF minor allele frequency.
A total of 9,397,031 variants passed quality control in all three data sets. There was no evidence of inflation of the test statistics (Supplementary Fig. 2).
The combined summary statistics of 4,269,731 rare and low-frequency variants were additionally used to perform the gene-level association test which combines the effects of multiple variants within a gene thus increasing power of revealing disease-relevant genetic variants in the presence of locus heterogeneity.
Loci associated with eczema at the single SNP level
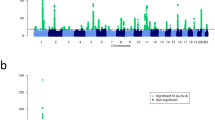
Performing serial conditional analyses, we identified 48 independent SNPs at 38 loci to be associated with eczema at genome-wide significance (Supplementary Data 2). Focusing only on 4,269,731 rare and low-frequency variants, a total of 273 SNPs at 11 loci were associated with eczema at genome-wide significance (Fig. 2, Supplementary Data 3). The best associated rare/low-frequency SNP at each locus is presented in Table 1. Of those, four loci were exclusively associated with rare/low-frequency variants; at profilaggrin (FLG), LOC101928093/DUSP1, arrestin domain containing 1 (ARRDC1), and C-C motif chemokine receptor 7 (CCR7)/SWI/SNF related, matrix associated, actin-dependent regulator of chromatin, subfamily E, member 1 (SMARCE1), rare/low-frequency variants were identified as independently associated lead SNPs (Table 1, Supplementary Data 2). LOC101928093/DUSP1 has not been reported to be associated with any allergic disease previously. Variants at ARRCD1 and CCR7/SMARCE1 were previously identified in a study on the broader phenotype “dermatitis or eczema” in UKBB18. Both SNPs revealed the same risk alleles and similar effect sizes in RV set, FINNGEN, and UKBB verifying association of the two loci with eczema. Notably, serial conditional analysis on chromosome 1 identified the three most frequent FLG null mutations at genome-wide significance with no residual association in the epidermal differentiation complex (EDC) at 1q21.3 (Supplementary Data 2).

Meta-analysis results for all variants with a minor allele frequency < 5% are shown. For each SNP (dots), the genomic location (x-axis) and the association P value (y-axis) are indicated. The red line corresponds to the genome-wide significance threshold of 1 × 10−8.
At seven loci, Src homology 2 domain containing E (SHE), T helper type 2 locus control region associated RNA (TH2LCRR), natural cytotoxicity triggering receptor 3 (NCR3)/allograft inflammatory factor 1 (AIF1), interleukin-15 receptor subunit alpha (IL15RA)/interleukin 2 receptor subunit alpha (IL2RA), proline rich 5 like (PRR5L), EMSY/leucine rich repeat containing 32 (LRRC32), and regulator of telomere elongation helicase 1 (RTEL1) common variants were more significantly associated with eczema than the respective rare/low-frequency variant although the effect sizes were smaller (Supplementary Data 4). Conditioning on the common variants abolished genome-wide significance of rare/low-frequency variants at all loci except for TH2LCRR and EMSY/LRRC32. Conditioning on rare/low-frequency variants often revealed a residual effect of the common variant, which may point to a larger or exclusive impact of the common variant. Alternatively, unidentified rare variants in linkage disequilibrium with a common SNP may underlie the association signal.
Genome-wide investigation of the complete data set identified 48 independent SNPs at 38 loci (Supplementary Data 2). Of those SATB1 antisense RNA 1 (SATB1-AS1)/potassium voltage-gated channel subfamily H member 8 (KCNH8), tribbles pseudokinase 1 (TRIB1)/LINC00861, zinc finger and BTB domain containing 1 (ZBTB1), T-box transcription factor 21 (TBX21)/oxysterol binding protein like 7 (OSBPL7), and colony stimulating factor 2 receptor subunit beta (CSF2RB) contain common eczema-associated SNPs which were previously not identified. All loci showed consistent effects in RV set, FINNGEN, and UKBB (Supplementary Data 2, Supplementary Fig. 24–28). Common variants in or near the genes encoding RUNX family transcription factor 3 (RUNX3), TNF superfamily member 4 (TNFSF4), D-2-hydroxyglutarate dehydrogenase (D2HGDH), DEAD-box helicase 6 (DDX6)/C-X-C motif chemokine receptor 5 (CXCR5), TNF receptor associated factor 3 (TRAF3), and mitogen-activated protein kinase kinase kinase 14 (MAP3K14)/Rho GTPase activating protein 27 (ARHGAP27) were previously associated in UKBB with a broad “dermatitis or eczema” phenotype18,19. We demonstrate association of these loci with the stricter definition of eczema (Supplementary Data 2).
Replication of eczema-susceptibility loci from previous GWAS
In order to verify the eczema-susceptibility loci identified in previous GWAS1,2,3,4,5,6,7,8,9, we looked-up in our study the best-associated SNP reported at each locus (Supplementary Data 5). Using a significance threshold of P < 0.0015 (=0.05/32 loci), we replicated 24 out of 32 loci. An independent locus near FLG on chromosome 1 (rs7512552)6 could not be confirmed, it was eliminated after adjusting for the three identified FLG variants (Supplementary Data 2, Supplementary Data 5). Six SNPs previously identified in GWAS on Asian populations2,3 did not replicate. However at three of these loci, on chromosomes 3, 5, and 20, SNPs in close proximity which were not in LD with the previously reported SNP passed the genome-wide significance threshold (Supplementary Data 5).
Gene-level association with eczema
Limited power is the main drawback when studying the association of individual rare variants in complex diseases. Gene-level association tests which combine the effects of multiple variants within a gene may overcome this problem in the presence of locus heterogeneity. In an explorative approach we used RV set to test different variant selection strategies and two different gene-level association tests, GRANVIL and SKAT which are implemented in the RVTESTS software package20 (Supplementary Data 6). Selecting variants based on the deleteriousness score (CADD) in combination with the variance-component test (SKAT) performed best for the different scenarios and identified FLG, DUSP1, and NOTCH4 to be significantly associated with eczema in RV set.
We then performed a gene-level association test for the meta-analysis of RV set, FINNGEN, and UKBB. For the analysis, we used the SNP-wise Mean model of MAGMA21 which is equivalent to the SKAT model with the advantage of allowing the usage of summary statistics. We included all variants with a MAF < 0.05 and a CADD score threshold of ≥ 12.37 for deleterious variants as suggested by Kircher et al (2014)22 capturing approximately the 5% most deleterious mutations. 13,000 genes with at least two deleterious variants were tested for association with eczema. Genes at five loci reached genome-wide significance at P < 3.8 × 10−6 (= 0.05/13,000; Supplementary Data 7). All variants contributing significantly to the gene-level association were also included in Table 1. All rare/low-frequency variants from Table 1 revealed consistent effects in the study populations of RV set, FINNGEN, and UKBB (Supplementary Fig. 3–23). As identified in RV set, MAGMA confirmed FLG, DUSP1, and NOTCH4 to be significantly associated with eczema in the complete data set (Supplementary Data 7). In addition, SLC9A4 and PRR5L passed the genome-wide significance threshold.
Since within the EDC on chromosome 1 and the MHC on chromosome 6, SNPs in multiple genes reached genome-wide significance (Supplementary Fig. 29), we conditioned the results on the three identified FLG variants and the two significantly associated NOTCH4 variants from MAGMA, rs8192591 and rs526945, and repeated the gene-level association test. All significantly associated genes within the EDC and within the MHC disappeared (Supplementary Fig. 29) suggesting that the best-associated genes FLG and NOTCH4 represented the main eczema-susceptibility genes in these regions.
Functional assessment of the rare/low-frequency eczema-associated variants
We evaluated the potential impact of the identified rare/low-frequency variants on protein function by analyzing their sequence context and reviewing their annotations in databases using the GTEx Portal23, MutationTaster24, and HaploReg v4.125. We included the lead SNPs from the single variant and gene-based analysis (Table 1). In addition, all SNPs in high LD with the associated variants (r2 > 0.8) were identified and functionally assessed (Supplementary Data 8).
New exonic variants were identified in DUSP1, NOTCH4, and SLC9A4. In the dual specificity phosphatase 1 gene (DUSP1) at 5q35.1, the best associated low-frequency variant rs114503346 was in high LD (D’ = 0.95; r2 = 0.70) with two low-frequency missense variants, rs34471628 and rs34013988 (Table 1). Both missense variants were in complete LD (D’ = 1, r2 = 1). Accordingly, their minor alleles, rs34471628_G and rs34013988_T, are located on the same haplotype which had a protective effect on eczema. Conditioning on rs114503346 did not yield any further association signal in that region (Supplementary Data 2). DUSP1 encodes a phosphatase which specifically dephosphorylates serine and threonine residues. DUSP1 is ubiquitously expressed with high mRNA levels in human skin (GTEx Portal on 12/04/20)23. The eczema-associated variants were located within regulatory regions in a wide range of tissues including skin and blood (Supplementary Data 8). Two functional motifs were previously identified to be important for DUSP1 function, the kinase interacting motif which binds the respective kinases and the phosphotyrosine-binding loop which is the active site of DUSP126,27. We used molecular modelling to investigate whether the variants identified in our study would affect these motifs and consequently impact protein function (Fig. 3).

Eczema-associated variants change the amino acids (in pink) at positions 56 (alanine, Ala, to threonine, Thr) and 187 (tyrosine, Tyr, to histidine, His). The kinase-interacting motif (blue), the phosphotyrosine-binding loop (green), and Asp227 (light blue) which belongs to the catalytic site are indicated. Acid and base groups in the functional sites are marked red and blue respectively.
Variant rs34013988_T leads to a substitution of alanine by threonine at residue 56 (p.Ala56Thr) and rs34471628_G changes tyrosine to histidine at residue 187 (p.Tyr187His). Both wild-type amino acids, Ala56 and Tyr187, are highly conserved among vertebrates (phastCons28 scores of 1). Ala56 is located within the kinase interacting motif of DUSP1 (Fig. 3a) which is found to be identical in other vertebrates and in human DUSP4 which is another member of this protein family26. Substitution of non-polar alanine by polar threonine may alter the kinase-binding capacity and thus affect the activity or specificity of DUSP1. Within the amino acid sequence, Tyr187 is located distant from the functional motifs. In the predicted 3D-structure of DUSP1, however, it is shifted close to the phosphotyrosine-binding loop (Fig. 3b) which is the catalytic site of DUSP1. Accordingly, replacement of Tyr187 with histidine could affect the catalytic activity of the protein. Histidine is frequently found in the active site of proteins because of its distinctive property to function as base or as acid in catalysis. Notably, His187 is located at a similar distance from the catalytic site as Asp227 which has been shown to contribute to the catalytic activity of DUSP1 as a general acid/base27. In addition, we found DUSP1 expression to be dysregulated in lesional skin of eczema patients further supporting a role in eczema (Supplementary Fig. 30).
While serial conditional analysis on chromosome 6 identified three independent common SNPs (rs2844594, rs6913664 and rs13208697) within the major histocompatibility complex (MHC) (Supplementary Data 2), the gene-level analysis on rare variants identified NOTCH4 as the best-associated gene in this region (Supplementary Data 7). Two variants with high CADD score contributed to the association with NOTCH4; rs8192591 and rs526945 (Table 1) which were in LD with the common variants rs6913664 and rs13208697 (Supplementary Data 4). Conditioning on the two NOTCH4 variants eliminated genome-wide significance of rs6913664 and rs13208697 suggesting that low-frequency, functional variants may underlie association of common variants at this locus. The association signal of the third common SNP, rs2844594, remained thus representing an independent signal.
The best-associated low-frequency variant from the single SNP analysis in the MHC region, the intergenic variant rs28732147, also pointed to the functional variant rs8192591 since both SNPs were in high LD (D’ = 0.92, r2 = 0.53) and revealed almost identical effect sizes (Table 1). rs8192591 is located in exon 9 of NOTCH4 (NM_004557.4). It is a missense variant causing a substitution of nonpolar glycine to polar serine at residue 534 within the highly conserved (phastCons score of 0.97) EGF-like domain 13 (Table 1, Supplementary Data 8) which may affect the domain’s calcium-binding capacity. The second variant rs526945 is located 4.5 kb upstream of NOTCH4 in a predicted promoter/enhancer region (CADD score of 15.2, Supplementary Data 8). Highest expression of NOTCH4 has been reported in lung, adipose, and breast tissue, as well as in monocytes23,29.
On chromosome 2, the common SNP rs3755274 in IL18R1 which revealed the strongest association signal (Supplementary Data 2) was in LD (D’ = 0.85, r2 = 0.09) with the low-frequency missense variant rs61731289 in SLC9A4 which was identified in the gene-level analysis (Supplementary Data 4, Supplementary Data 7). We propose the coding variant rs61731289 (CADD score of 15.2) changing arginine to lysine in position 640 of SLC9A4 as potential causal allele and effector transcript at this locus. SLC9A4 is mainly expressed in stomach, at lower levels in kidney and esophageal mucosa (GTEx Portal on 12/04/20)23.
Pathway enrichment analysis and tissue-specific gene expression
We performed a pathway enrichment analysis for GO terms and curated gene-sets30 and a tissue expression analysis for the 54 tissues included in GTEx23 with MAGMA21. In the tissue-specific gene expression analysis, we observed a significant correlation between the distribution of P values of SNPs and gene expression in small intestine, spleen, lung, whole blood, visceral omentum, and colon (Supplementary Fig. 31–33).
Using all GWAS SNPs, we observed a significant enrichment of immune-related pathways involved in cytokine signaling, T-cell differentiation and activation as well as enrichment of genes related to the cornified cell envelope in keratinocytes (Supplementary Data 9). Stratifying the results according to allele frequency revealed that rare/low-frequency SNPs were associated with the cornified cell envelope whereas common SNPs were associated with immune functions (Supplementary Data 10).
Moreover, we extended the analysis to candidate genes mapped from genome-wide significant variants, and evaluated their enrichment among differentially expressed gene (DEG) sets as defined for 54 different tissues. Prioritized genes from all genome-wide significant variants were enriched in sets of up-regulated DEGs in whole blood and skin (Supplementary Fig. 34a). Prioritized genes from only common variants were also enriched in sets of up-regulated DEGs in whole blood, as well as in small intestine (Supplementary Fig. 34b). In contrast, prioritized genes from rare/low-frequency variants were overrepresented in sets of up-regulated DEGs in skin and esophagus mucosa (Supplementary Fig. 34c). To exclude that the results for the rare/low-frequency variants were exclusively driven by FLG mutations, we repeated the analysis after conditioning on the three known FLG variants identified. This eliminated all prioritized genes from the epidermal differentiation complex and still identified a significant enrichment of the remaining genes in up-regulated DEGs from skin (Supplementary Fig. 34d).
Heritability explained by rare variants
In order to quantify the contribution of rare and low-frequency variants to the heritability of eczema, we used an in house data set comprising 7,092 unrelated individuals (20.1% cases) with HRC-imputed genotypes. GREML-LDMS was used to estimate heritability for 3 frequency bins including rare variants (MAF < 0.01), low-frequency variants (0.01 < MAF < 0.05), and common variants (MAF > 0.05) respectively. In total 5,575,243 high-quality SNPs (imputed at r2 > 0.9) were included in the analysis. The heritability explained by rare and low-frequency variants was 4.5% and 9.1% respectively (Supplementary Data 11 and 12). They accounted for 23.0% of the SNP-based heritability supporting a substantial role for rare and low-frequency in eczema susceptibility.
Discussion
We performed a meta-GWAS of 21 study populations including 20,016 cases and 380,433 controls to determine the role of rare variants in the development of eczema. Single SNP and gene-level analyses identified 11 loci carrying rare/low frequency variants at genome-wide significance. Exonic variants were detected in DUSP1, NOTCH4 and SLC9A4. In DUSP1 and NOTCH4 the identified missense variants were predicted to impact conserved functional domains. The analysis of common variants yielded another five eczema-susceptibility loci not reported previously. Over 20% of the SNP-based heritability was explained by rare and low-frequency variants, emphasizing the importance of studying rare variants in eczema.
In recent years, a number of studies investigated the role of rare variants in complex diseases. They discovered single novel loci with rare or low-frequency variants associated with ulcerative colitis12, age-related macular degeneration31 or schizophrenia32. For eczema, an exome-chip based analysis in a discovery set of 2,000 cases and 15,000 controls identified a single new eczema susceptibility locus containing low-frequency variants in the docking protein 2 gene (DOK2)33. Utilizing HRC-imputed data allowed us to significantly increase sample size and thus the power in our study on rare variants. Accordingly, we identified rare/low-frequency variants at 11 loci including three genes which contained amino acid-changing variants not reported previously33. However, it is clear that much larger data sets will be required to dissect the contribution of rare variants to complex diseases. Current studies on quantitative rather than binary phenotypes identified 22 and 512 rare or low-frequency variants underlying blood pressure34 and various blood cell traits, respectively35. They investigated up to 1.3 million study participants.
We discovered eczema-associated missense variants in DUSP1, NOTCH4, and SLC9A4. DUSP1 variants had a protective effect in the single variant and in the gene-level analysis. The two missense variants rs34471628 and rs34013988 were located near active sites. Both may alter the functional activity or specificity of DUSP1. Target proteins which are dephosphorylated by DUSP1 are extracellular signal-regulated kinases (ERK) 1 and 2, c-Jun N-terminal protein kinase (JNK) 1, and a kinase negative form of mitogen-activated protein kinase (MAPK) p38α36. Dysregulated DUSP1 expression was reported in cultured keratinocytes of eczema patients; association of a common candidate SNP near DUSP1 however was weak37. Interestingly, DUSP1 has been reported to mediate the anti-inflammatory effect of corticosteroids which are a mainstay of eczema therapy38. Dexamethasone-inducedDUSP1 expression repressed the ERK, JNK and p38α MAPK pathways and reduced the expression of inflammatory genes in human pulmonary epithelial cells39. Hence, novel therapies directed at modifying DUSP1 function may be beneficial in eczema treatment.
In the gene-level analysis focusing on rare variants with high functional impact, NOTCH4 was the best-associated gene in the MHC region. The identified NOTCH4 missense mutation rs8192591 was located in the EGF-like domain 13. While the number of EGF-like domains vary between the NOTCH1 and NOTCH4 receptors, the first 13 EGF-like domains are highly conserved40, with repeats 11–13 involved in ligand binding41. NOTCH4 belongs to a family of four highly conserved transmembrane receptors involved in proliferation, apoptosis, and cell fate decisions42. Expression of all NOTCH receptors and their ligands has been demonstrated by immunostaining in human skin sections43; NOTCH4 was expressed in the suprabasal layers of the epidermis and it was the major NOTCH receptor in the stratum corneum. NOTCH4 has recently been identified as a master molecular switch in a new pathway that decreases regulatory T (Treg) cell function in chronic inflammatory conditions44,45. Upregulation of NOTCH4 receptor Treg cells was necessary for allergen induced airway inflammation. NOTCH4 and its downstream effector pathways were upregulated on Treg cells of individuals with asthma and correlated with disease severity44.
Due to the high LD across the MHC region on chromosome 6, association signals are notoriously difficult to dissect. Conditioning on the two functional NOTCH4 SNPs eliminated genome-wide significance of two of the three independently associated common SNPs, suggesting that rare functional variants may underlie the common association signals at this locus. However, an independent association signal persists in the MHC (rs2844594), indicating that additional eczema risk variants remain to be discovered in that region. Functional studies will be required to confirm a role of NOTCH4 in eczema.
On chromosome 2, the identified missense variant rs61731289 pointed to SLC9A4 as the risk gene at this locus. However, conditioning on the low-frequency variant yielded strong residual association of the common variant rs3755274 demonstrating that other genetic risk factors exist at this locus. SLC9A4 belongs to the SLC9 family of sodium/hydrogen exchangers and is localizing to the plasma membrane in renal, intestinal, and other epithelia46. The eczema associated variant rs61731289 is located within the cytoplasmic tail of SLC9A4. While this region of SLC9A4 has not been functionally characterized, truncation studies of this domain in SLC9A3 have revealed its involvement in regulating ion transporter activity47. An SLC9A4 knockout mouse revealed major histological changes in the gastric mucosa consistent with impaired maturation and/or differentiation of gastric epithelial cells, as well as an inflammatory infiltrate dominated by eosinophils48. Thus SLC9A4 may play a role in epithelial integrity and eosinophilic inflammatory responses which are also characteristic of eczema49.
Of the 5 new eczema-susceptibility loci captured by common variants, SATB1-AS1/KCNH8, TRIB1/LINC00861, and TBX21/OSBPL7 were previously identified in a GWAS combining diverse inflammatory diseases excluding eczema50. Involvement of these loci in immune functions suggests that the same mechanisms also play a role in eczema. SATB1-AS is expressed in a wide range of tissues including the spleen, whole blood, and gut, whereas KCNH8 encodes a potassium channel that is primarily expressed in the nervous system23. SATB1-AS expression is modified by mutations in SATB151. SATB1 in turn is predominantly expressed in CD4 + CD8 + lymphocytes exerting both activating and repressive regulatory functions implicated in autoimmunity and cancer52. The pseudokinase TRIB1 is a key regulator of eosinophil differentiation that are important effector cells in allergic inflammation53. Moreover, it has been shown recently that TRIB1 restrains antiviral effector T cell function54. Little is known about LINC00861 function. Highest expression is found in the spleen and whole blood. Variants in TBX21 were associated with asthma in a candidate gene study55. TBX21 encodes the Th1-specific transcription factor T-bet which is involved in Th1/Th2 balance and suppressed atopic dermatitis-like skin inflammation in mice56.
ZBTB1 which was previously identified in a GWAS as a psoriasis susceptibility locus57 is a critical determinant of T cell development. ZBTB1 knockout mice completely lacked T cells58. The differentiation of common lymphoid progenitors of B and NK cell lineages was also affected59.
The CSF2RB locus was associated with asthma in a previous GWAS60. CSF2RB encodes the common beta subunit of the interleukin 5 (IL-5), interleukin 3 (IL-3), and granulocyte-macrophage colony-stimulating factor (GM-CSF) receptors. It plays a critical role in the regulation of Th2 immunity and allergic inflammation. In Csf2rb-deficient mice, allergen-induced expansion and accumulation of eosinophils in the lung was abolished and proliferation and activity of Th2 cells was reduced61.
Tissue-specific gene expression analysis and pathway enrichment analysis identified tissues and gene-sets related to immune functions and to skin development. Interestingly rare/low frequency variants and their target genes exerted their effects predominantly in skin which was still evident after adjusting for the strong genetic effect of the low-frequency variants in FLG. In contrast, common GWAS variants pointed to pathways and tissues of the immune system as supported by the reported functions of the new candidate genes from the common SNP analysis. Focusing on cutaneously expressed genes could therefore be a promising strategy for rare variant studies in eczema.
In order to avoid cost-intensive whole-exome or whole-genome sequencing we performed our study on HRC-imputed data. Before this dataset was published, imputation quality of rare variants was generally low and many rare variants were not even detected due to the low number of sequenced haplotypes of a population. HRC comprises the phased haplotypes of more than 32,000 sequenced individuals and allows imputation of rare variants with high confidence15. Accordingly, the majority of genome-wide significant low-frequency variants was imputed at high quality (r2 > 0.8) (Supplementary Data 13). We additionally determined the non-reference sensitivity (NRS) and non-reference discordancy (NRD)62 for the identified coding variants in an in house data set of 892 individuals whose HRC-imputed genotypes (after genotyping with Illumina’s HumanOmniExpressExome-8 SNP array) and whole exome sequences were available. Genotypes correlated very well for all coding variants (Supplementary Data 14). Even variants with an imputation quality r2 < 0.8 (Supplementary Data 13) confirmed association results at known eczema loci (FLG, ARRCD1, CCR7/SMARCE1). In addition, imputed loss-of-function mutations in FLG almost perfectly matched the previously reported allele frequencies and effect sizes63. Finally, effects of the identified exonic variants in DUSP1, NOTCH4 and SLC9A4 were homogeneous over all study populations, supporting the validity of the HRC-imputed approach.
Our analysis of the association between FLG and eczema has illustrated potential pitfalls of strategies for analyzing rare variants on the gene-level. In contrast to the single SNP analysis, gene-level tests have to take into account the genetic architecture of a locus including different effect sizes and effect directions of variants within a gene. Both, the SNP selection strategy and the gene-level association test had an impact on the results (Supplementary Data 6). SNP selection based on the deleteriousness score (CADD) in combination with the variance component test (SKAT) was most powerful for the different scenarios. Likewise a previous study on rare and low-frequency variants in eczema focused only on missense variants in the gene-level association test since they are more likely to have a deleterious effect33.
We restricted our analysis to variants with a MAF > 0.001. It has been suggested that rarer mutations could play a significant role in complex diseases. However, our power calculation indicated that larger populations are required to study those variants. In addition, sequence data would be essential for individual mutations which are unlikely to be included in reference data sets for imputation. Although the number of patients affected by a specific rare mutation may be low, the pinpointed genes and effects on the protein level could give important clues for unraveling pathways involved in eczema susceptibility. Our heritability estimation for rare and low-frequency variants yielded a contribution of 13.6% to the overall heritability on the observed scale (22.2% on the liability scale). For rare variants (MAF < 0.01) a heritability estimate of 4.5% on the observed scale (7.4% on the liability scale) was calculated. Mucha et al. reported a heritability estimate of 12.56% on the liability scale for rare variants included in the exome chip which is likely due to the large proportion (97%) of missense and other deleterious rare variants on the chip33. Interestingly, the heritability explained by the known loci was mainly attributable to common variants (Supplementary Data 11 and 12) indicating that the majority of eczema-associated rare variants remains to be discovered.
In summary, our study on rare variants in eczema identified novel exonic variants in 3 genes and intronic or intergenic variants at another 8 loci supporting the involvement of rare and low-frequency variants in the development of eczema. The localization of the missense variants and their potential functional impact on the newly-identified eczema-associated genes DUSP1 and NOTCH4 suggest promising targets for future therapies.
Methods
Study populations and phenotype definition
Characteristics of the study populations are summarized in Supplementary Data 1. The majority of them had participated in the previous genome-wide association study (GWAS) on eczema performed by the EArly Genetics and Lifecourse Epidemiology (EAGLE) Consortium6. Two additional studies from Sweden, CATTS, and SALTY, as well as two large population-based studies, FINNGEN and UKBB, were included. Eczema was defined based on a physician’s diagnosis according to standard diagnostic criteria64,65, on a doctor’s diagnosis reported in a questionnaire, or on self-reported disease. The phenotype characteristics of the individual study populations are indicated in detail in Supplementary Data 1 and Supplementary Note 1. All study populations were of European ancestry. All relevant ethical regulations were followed and informed consent was obtained from all participants or their legal guardians. All studies were approved by the local ethics committees (Supplementary Note 1).
Genotyping and imputation
In total, 19 study populations with individual genotype data were included in “RV set”. Genotyping, quality control (QC), genotype imputation, and association analysis were performed in each study population separately and are described in detail in the Supplementary Note 1. In brief, after genotyping and appropriate QC, phasing of the genotype data into haplotypes was carried out using SHAPEIT v266. Imputation of rare variants was performed using the Haplotype Reference Consortium Panel on imputation servers at the Michigan University or at the Wellcome Trust Sanger Institute15. In each data set, SNPs with poor imputation quality (r² or info score < 0.5), a MAF less than 0.001 or deviation from Hardy-Weinberg equilibrium in controls (P < 1 × 10−12) were excluded.
Details on genotyping, QC, genotype imputation, and association analysis in UKBB and FINNGEN are described in Supplementary Note 1.
Imputation quality of the identified exonic variants was confirmed by comparing imputed and sequenced genotypes in an in house data set of 892 individuals whose HRC-imputed genotypes (after genotyping with Illumina’s HumanOmniExpressExome-8 SNP array) and whole exome sequences were available. Non-reference sensitivity (NRS) and non-reference discordancy (NRD) were calculated (Supplementary Data 14)62.
Association analysis at the single SNP level
Individual genotype data available in the 19 study populations of RV set allowed us to analyze them with RVTESTS20, a tool which implements a broad set of rare variant association statistics and which was specifically developed to study rare variants at the genome-wide scale. GWA analysis of the imputed SNPs with eczema was carried out per study population using a score test as implemented in the RVTESTS software20. A covariance matrix describing the covariance between SNPs in the population was thereby generated. After association analysis, SNPs with a minor allele count < 1 in cases or controls were excluded in each data set. To be included in the single SNP meta-analysis, variants had to be present in at least 3 study populations. Single-SNP meta-analysis was performed using summary statistics of the individual study populations and a fixed-effects model as implemented in RareMETALS2 (https://genome.sph.umich.edu/wiki/RareMETALS2), an add-on R-package to RAREMETAL67 for the meta-analysis of rare variant association results from binary traits. Covariance matrices were included as covariates. Subsequently, summary data on eczema from UKBB and from FINNGEN were meta-analyzed with the summary data of RV set by using the sample size method of METAL68 which uses P values and effect directions, weighted according to sample size. All SNPs with data in RV set, UKBB, and FINNGEN were included. To identify all independent association signals at a locus, we performed serial conditional analyses with GCTA69. A ± 1 Mb-window around each lead SNP was defined and association signals within the window were conditioned on the lead SNP. This procedure was repeated until all genome-wide signals at a locus were eliminated (Supplementary Data 3).
If a rare and a common variant were associated with eczema at genome-wide significance at a specific locus, we mutually conditioned the results using GCTA69. P values before and after conditional analysis were included in Supplementary Data 4.
Association analysis at the gene level
In RV set different SNP selection strategies and gene-level association tests were explored. For aggregating SNPs into gene units, 3 different selection strategies were applied. First, SNPs predicted to severely impact protein levels (start lost, stop lost, stop gained, splice acceptor, splice donor) by the Ensembl Variant Effect Predictor70 were combined. Secondly, SNPs predicted to be missense SNPs were included on top of the first selection. Thirdly, to include potentially functional SNPs located in intronic or promotor regions of genes and to exclude coding SNPs with a predicted minor impact on protein function, a deleteriousness score for SNPs was used. For the third strategy, only SNPs with a PHRED score > 15 calculated by the Combined Annotation Dependent Depletion (CADD) tool22 were combined.
Two different tests implemented in RareMETALS were applied to perform association tests on the gene level by combining score statistics of rare variants per gene; the Gene- or Region-based ANalysis of Variants of Intermediate and Low-frequency (GRANVIL)71 and the SNP-set Kernel Association Test (SKAT)72. GRANVIL averages the score statistics of individual variants per gene and has most power if all variants have the same effect direction. SKAT is a variance component test and remains powerful if variants with opposite effect directions are combined. Finally, gene-level association analysis of the combined data set including FINNGEN and UKBB was conducted with MAGMA21 which is equivalent to SKAT with the advantage of allowing the usage of summary statistics.
Functional annotation
Linkage disequilibrium (LD) between SNPs was calculated using the single-nucleotide polymorphism annotator with LD information from the European population of the 1000 Genomes project phase 3v573. eQTLs for the lead SNPs were identified using the single-nucleotide polymorphism annotator73. eQTLs were only reported, if the queried SNP was the most significant eQTL SNP for a gene in a relevant tissue per LD group. Information about eQTL SNPs in relevant tissues was collected from the GTEx Consortium database version 6p23. SNP functional annotations were predicted using the Ensembl Variant Effect Predictor70 and HaploReg v4.125. Pathogenicity prediction was performed using the MutationTaster24.
Homology modeling
The 3D structures of both wild-type and mutant DUSP1 were modeled using a locally installed version of I-TASSER 5.1 using standard settings. Figures were prepared using PyMOL 2.4 (The PyMOL Molecular Graphics System, Version 2.4, Schrödinger, LLC).
Pathway enrichment analysis and tissue expression analysis
Pathway enrichment analysis and tissue expression analysis both were performed with MAGMA21 via the FUMA webpage (https://fuma.ctglab.nl)74. Pathway enrichment analysis included GO terms and curated gene-sets from the Molecular Signatures Database (MSigDB)30. Expression profiles were from 54 tissue types from GTEx23. To identify a potential difference between rare and common variants, we stratified our results according to the minor allele frequency. Candidate genes were prioritized from genome-wide significant SNPs based on a combination of three SNP-to-gene mapping strategies: positional mapping (physical position on the genome within 10 kb), eQTL mapping (eQTL associations from any tissue type) and chromatin interaction mapping (3D chromatin interactions based on any built-in chromatin interaction data available) using default parameters in FUMA74, except for maximum P value of lead SNPs which was set at 1 × 10−8. Candidate genes were prioritized from: all variants, common variants (MAF ≤ 0.05) and rare variants (MAF < 0.05%; before and after conditioning on the 3 identified FLGloss-of-function variants), respectively. We tested for tissue specificity of eczema prioritized genes using hypergeometric tests to evaluate its overrepresentation in sets of differentially expressed genes (DEG; sets of genes which are more or less expressed in a specific tissue compared to others) in 54 tissue types based on GTEx v8 RNA-seq data.
SNP-based heritability analysis
We built an in-house sample of 8,134 individuals with available genotypes and HRC-imputed data. Using GCTA69, we identified and removed related individuals (—grm-cutoff 0.05). SNP variants with MAF < 0.001, Hardy–Weinberg exact test < 1 × 10−12 and imputation quality r2 < 0.9 were excluded using PLINK75. The final dataset included 7,092 unrelated individuals (20.1% eczema cases) and 5,575,243 high-quality SNPs. Principal Components (PCs) were calculated in GCTA. LD score was calculated for all SNPs as the sum of LD r2 between a SNP and all the rest of SNPs within a region of 200 kb. All 5,575,242 SNPs were stratified in 4 bins based on the segment-based LD score of the SNPs and each bin further divided in 3 bins based on allele frequency (MAF < 0.01, 0.01 < MAF < 0.05, MAF > 0.05). Genomic Relationship Matrix (GRM; 12 random genetic effects) were constructed from SNPs annotated to the 12 different bins. To estimate SNP-based heritability (h2SNP) we implemented a GREML-LDMS model in GCTA that fit all 12 GRM together while adjusting by sex and the first 10 PCs76. This method is unbiased regardless of the properties of the underlying causal SNPs, therefore allowing us to robustly estimate h2SNP explained by bins of SNPs at different MAF while correcting for the region-specific LD heterogeneity across the genome76. Then, 2,663 significant variants (P < 1 × 10−8) from the Meta-Analysis as well as all SNPs within a window of ±500 kb were selected as significant loci and removed from the 12 bins annotated previously. 12 new GRM were constructed from these 12 bins of which a total of 82,621 SNPs were excluded. Estimations of h2SNP fitting components with and without the significant loci were compared to assess the h2SNP explained by significant loci identified in our meta-analysis. The estimates of variance explained on the observed scale were transformed to that on the underlying scale considering a population prevalence of 10%.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Summary statistics of the meta-analysis which were generated in this study have been deposited in the GWAS Catalog (https://www.ebi.ac.uk/gwas/home) under study accession GCST90044763.
References
Esparza-Gordillo, J. et al. A common variant on chromosome 11q13 is associated with atopic dermatitis. Nat. Genet. 41, 596–601 (2009).
Sun, L. D. et al. Genome-wide association study identifies two new susceptibility loci for atopic dermatitis in the Chinese Han population. Nat. Genet. 43, 690–694 (2011).
Hirota, T. et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat. Genet. 44, 1222–1226 (2012).
Ellinghaus, D. et al. High-density genotyping study identifies four new susceptibility loci for atopic dermatitis. Nat. Genet. 45, 808–812 (2013).
Schaarschmidt, H. et al. A genome-wide association study reveals 2 new susceptibility loci for atopic dermatitis. J. Allergy Clin. Immunol. 136, 802–806 (2015).
Paternoster, L. et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 47, 1449–1456 (2015).
Weidinger, S. et al. A genome-wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Hum. Mol. Genet. 22, 4841–4856 (2013).
Baurecht, H. et al. Genome-wide comparative analysis of atopic dermatitis and psoriasis gives insight into opposing genetic mechanisms. Am. J. Hum. Genet. 96, 104–120 (2015).
Paternoster, L. et al. Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nat. Genet. 44, 187–192 (2011).
Ferreira, M. A. et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat. Genet. 49, 1752–1757 (2017).
Visscher, P. M. et al. 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22 (2017).
Luo, Y. et al. Exploring the genetic architecture of inflammatory bowel disease by whole-genome sequencing identifies association at ADCY7. Nat. Genet. 49, 186–192 (2017).
Torgerson, D. G. et al. Resequencing candidate genes implicates rare variants in asthma susceptibility. Am. J. Hum. Genet. 90, 273–281 (2012).
Mancuso, N. et al. The contribution of rare variation to prostate cancer heritability. Nat. Genet. 48, 30–35 (2016).
McCarthy, S. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 48, 1279–1283 (2016).
Purcell, S., Cherny, S. S. & Sham, P. C. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149–150 (2003).
Fadista, J., Manning, A. K., Florez, J. C. & Groop, L. The (in)famous GWAS P-value threshold revisited and updated for low-frequency variants. Eur. J. Hum. Genet. 24, 1202–1205 (2016).
Kichaev, G. et al. Leveraging polygenic functional enrichment to improve GWAS Power. Am. J. Hum. Genet. 104, 65–75 (2019).
Johansson, A., Rask-Andersen, M., Karlsson, T. & Ek, W. E. Genome-wide association analysis of 350 000 Caucasians from the UK Biobank identifies novel loci for asthma, hay fever and eczema. Hum. Mol. Genet. 28, 4022–4041 (2019).
Zhan, X., Hu, Y., Li, B., Abecasis, G. R. & Liu, D. J. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics 32, 1423–1426 (2016).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315 (2014).
GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660 (2015).
Schwarz, J. M., Rodelsperger, C., Schuelke, M. & Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 7, 575–576 (2010).
Ward, L. D. & Kellis, M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 44, D877–D881 (2016).
Owens, D. M. & Keyse, S. M. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene 26, 3203–3213 (2007).
Gumpena, R. et al. Crystal structure of the human dual specificity phosphatase 1 catalytic domain. Protein Sci. 27, 561–567 (2018).
Siepel, A. & Haussler, D. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol. Biol. Evol. 21, 468–488 (2004).
Fraser, A. et al. Cohort Profile: the Avon Longitudinal Study of Parents and Children: ALSPAC mothers cohort. Int J. Epidemiol. 42, 97–110 (2013).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Fritsche, L. G. et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet 48, 134–143 (2016).
Singh, T. et al. Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat. Neurosci. 19, 571–577 (2016).
Mucha, S. et al. Protein-coding variants contribute to the risk of atopic dermatitis and skin-specific gene expression. J. Allergy Clin. Immunol. 145, 1208–1218 (2020).
Surendran, P. et al. Discovery of rare variants associated with blood pressure regulation through meta-analysis of 1.3 million individuals. Nat. Genet. 52, 1314–1332 (2020).
Vuckovic, D. et al. The polygenic and monogenic basis of blood traits and diseases. Cell 182, 1214–1231 e11 (2020).
Slack, D. N., Seternes, O. M., Gabrielsen, M. & Keyse, S. M. Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. J. Biol. Chem. 276, 16491–16500 (2001).
Rebane, A. et al. Mechanisms of IFN-gamma-induced apoptosis of human skin keratinocytes in patients with atopic dermatitis. J. Allergy Clin. Immunol. 129, 1297–1306 (2012).
Abraham, S. M. et al. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J. Exp. Med 203, 1883–1889 (2006).
Shah, S., King, E. M., Chandrasekhar, A. & Newton, R. Roles for the mitogen-activated protein kinase (MAPK) phosphatase, DUSP1, in feedback control of inflammatory gene expression and repression by dexamethasone. J. Biol. Chem. 289, 13667–13679 (2014).
Li, L. et al. Cloning, characterization, and the complete 56.8-kilobase DNA sequence of the human NOTCH4 gene. Genomics 51, 45–58 (1998).
Cordle, J. et al. A conserved face of the Jagged/Serrate DSL domain is involved in Notch trans-activation and cis-inhibition. Nat. Struct. Mol. Biol. 15, 849–857 (2008).
Bray, S. J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 17, 722–735 (2016).
Nickoloff, B. J. et al. Jagged-1 mediated activation of notch signaling induces complete maturation of human keratinocytes through NF-kappaB and PPARgamma. Cell Death Differ. 9, 842–855 (2002).
Harb, H. et al. A regulatory T cell Notch4-GDF15 axis licenses tissue inflammation in asthma. Nat. Immunol. 21, 1359 (2020).
Jin, K. et al. NOTCH-induced rerouting of endosomal trafficking disables regulatory T-cells in vasculitis. J Clin Invest (2020).
Donowitz, M., Ming Tse, C. & Fuster, D. SLC9/NHE gene family, a plasma membrane and organellar family of Na(+)/H(+) exchangers. Mol. Asp. Med. 34, 236–251 (2013).
Donowitz, M. et al. NHE3 regulatory complexes. J. Exp. Biol. 212, 1638–1646 (2009).
Gawenis, L. R. et al. Impaired gastric acid secretion in mice with a targeted disruption of the NHE4 Na+/H+ exchanger. J. Biol. Chem. 280, 12781–12789 (2005).
Leiferman, K. M. Eosinophils in atopic dermatitis. J. Allergy Clin. Immunol. 94, 1310–1317 (1994).
Ellinghaus, D. et al. Analysis of five chronic inflammatory diseases identifies 27 new associations and highlights disease-specific patterns at shared loci. Nat. Genet. 48, 510–518 (2016).
Shademan, M. et al. Expression profile analysis of two antisense lncRNAs to improve prognosis prediction of colorectal adenocarcinoma. Cancer Cell Int. 19, 278 (2019).
Zelenka, T. & Spilianakis, C. SATB1-mediated chromatin landscape in T cells. Nucleus 11, 117–131 (2020).
Mack, E. A. et al. Trib1 regulates eosinophil lineage commitment and identity by restraining the neutrophil program. Blood 133, 2413–2426 (2019).
Rome, K. S. et al. Trib1 regulates T cell differentiation during chronic infection by restraining the effector program. J. Exp. Med. 217(2020).
Suttner, K. et al. TBX21 gene variants increase childhood asthma risk in combination with HLX1 variants. J. Allergy Clin. Immunol. 123, 1062–1068 e1-8 (2009).
Makita, S. et al. T-bet and STAT6 coordinately suppress the development of IL-9-mediated atopic dermatitis-like skin inflammation in mice. J. Invest Dermatol 141, 1274–1285 e5 (2021).
Zuo, X. et al. Whole-exome SNP array identifies 15 new susceptibility loci for psoriasis. Nat. Commun. 6, 6793 (2015).
Siggs, O. M., Li, X., Xia, Y. & Beutler, B. ZBTB1 is a determinant of lymphoid development. J. Exp. Med. 209, 19–27 (2012).
Punwani, D. et al. Transcription factor zinc finger and BTB domain 1 is essential for lymphocyte development. J. Immunol. 189, 1253–1264 (2012).
Moffatt, M. F. et al. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 363, 1211–1221 (2010).
Asquith, K. L. et al. The IL-3/IL-5/GM-CSF common receptor plays a pivotal role in the regulation of Th2 immunity and allergic airway inflammation. J. Immunol. 180, 1199–1206 (2008).
Mitt, M. et al. Improved imputation accuracy of rare and low-frequency variants using population-specific high-coverage WGS-based imputation reference panel. Eur. J. Hum. Genet. 25, 869–876 (2017).
Sandilands, A. et al. Prevalent and rare mutations in the gene encoding filaggrin cause ichthyosis vulgaris and predispose individuals to atopic dermatitis. J. Investigative Dermatol. 126, 1770–1775 (2006).
Hanifin, J. M. & Rajka, G. Diagnostic features of atopic-dermatitis. Acta Derm.-Venereologica 60, 44–47 (1980).
Williams, H. C., Burney, P. G., Strachan, D. & Hay, R. J. The U.K. Working Party’s Diagnostic Criteria for Atopic Dermatitis. II. Observer variation of clinical diagnosis and signs of atopic dermatitis. Br. J. Dermatol. 131, 397–405 (1994).
Delaneau, O., Zagury, J. F. & Marchini, J. Improved whole-chromosome phasing for disease and population genetic studies. Nat. Methods 10, 5–6 (2013).
Feng, S., Liu, D., Zhan, X., Wing, M. K. & Abecasis, G. R. RAREMETAL: fast and powerful meta-analysis for rare variants. Bioinformatics 30, 2828–2829 (2014).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011).
McLaren, W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016).
Morris, A. P. & Zeggini, E. An evaluation of statistical approaches to rare variant analysis in genetic association studies. Genet Epidemiol. 34, 188–193 (2010).
Wu, M. C. et al. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 89, 82–93 (2011).
Arnold, M., Raffler, J., Pfeufer, A., Suhre, K. & Kastenmuller, G. SNiPA: an interactive, genetic variant-centered annotation browser. Bioinformatics 31, 1334–1336 (2015).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Yang, J. et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat. Genet. 47, 1114–1120 (2015).
Acknowledgements
Acknowledgments for each of the participating studies are provided in Supplementary Note 1.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Consortia
Contributions
Study coordination/study design: S.Gr., I.M., J.E.-G., Y.-A.L. Clinical data collection/sample contribution: C.A., A.A., H.Bi., K.B., M.B., A.C., S.C.D., A.E., A.F., S.Ge., C.G., H.H., P.G.H., G.H., M.-R.J., G.H.K., S.L., P.K.E.M., G.B.M., M.M.N., C.E.P., A.P., C.F.R., S.S., A.Si., M.S., D.S., K.S., A.T., P.J.T., J.M.V, S.W., Y.-A.L. Data analysis: S.Gr., I.M., J.E.-G., A.A.-S., E.P.-C., F.R., T.S.A., H.Ba., S.J.B., J.A.C., M.F., D.F.-O., M.A.R.F, N.H., P.G.Hy., R.K., M.L., M.M.-N., L.P., K.R., E.R., P.M.A.S., A.Sz., V.U., A.V., C.A.W., M.W., C.L.W., C.-J.X. Manuscript writing: S.Gr., I.M., Y.-A.L. All authors reviewed, revised, and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review informationNature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Grosche, S., Marenholz, I., Esparza-Gordillo, J. et al. Rare variant analysis in eczema identifies exonic variants in DUSP1, NOTCH4 and SLC9A4. Nat Commun 12, 6618 (2021). https://doi.org/10.1038/s41467-021-26783-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-26783-x
This article is cited by
-
Immunosuppression causes dynamic changes in expression QTLs in psoriatic skin
Nature Communications (2023)
-
Comprehensive analyses of fatty acid metabolism-related lncRNA for ovarian cancer patients
Scientific Reports (2023)
-
European and multi-ancestry genome-wide association meta-analysis of atopic dermatitis highlights importance of systemic immune regulation
Nature Communications (2023)
-
DNA methylation and aeroallergen sensitization: The chicken or the egg?
Clinical Epigenetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
