
Abstract
Barrier tissues are populated by functionally plastic CD4+ resident memory T (TRM) cells. Whether the barrier epithelium regulates CD4+ TRM cell locations, plasticity and activities remains unclear. Here we report that lung epithelial cells, including distinct surfactant protein C (SPC)lowMHChigh epithelial cells, function as anatomically-segregated and temporally-dynamic antigen presenting cells. In vivo ablation of lung epithelial MHC-II results in altered localization of CD4+ TRM cells. Recurrent encounters with cognate antigen in the absence of epithelial MHC-II leads CD4+ TRM cells to co-express several classically antagonistic lineage-defining transcription factors, changes their cytokine profiles, and results in dysregulated barrier immunity. In addition, lung epithelial MHC-II is needed for surface expression of PD-L1, which engages its ligand PD-1 to constrain lung CD4+ TRM cell phenotypes. Thus, we establish epithelial antigen presentation as a critical regulator of CD4+ TRM cell function and identify epithelial-CD4+ TRM cell immune interactions as core elements of barrier immunity.
Similar content being viewed by others


Introduction
Tissue-resident memory T (TRM) cells operate as front-line adaptive immune sentinels for barrier tissues, such as the skin, lungs, vagina, and intestine1,2. Barrier tissues experience unavoidable, transient, repeated exposures to environmental antigens. TRM cell formation and responses depend on diverse natures of antigenic stimuli, types of antigen-presenting cells (APC), and costimulatory signals and cytokines within the challenged tissue3,4. These signals diversify TRM cell responses beyond their initial phenotype specification, yielding T cell plasticity that enhances anti-microbial immunity and tissue barrier integrity3,5,6. Because TRM cells and T cell plasticity have been linked to barrier tissue pathologies like asthma, psoriasis, and inflammatory bowel disease (IBD)7,8,9,10, tissue-resident regulators of TRM cell plasticity have become an important knowledge gap.
As direct interfaces between the environment and the body, epithelial cells are poised to be critical regulators of barrier immunity4,11. Barrier epithelial cells recruit and maintain CD8+ TRM cells near the sites of antigen encounter, and reactivate them in the tissues via local antigen presentation12,13. In contrast to CD8+ TRM cell effects, epithelial roles governing CD4+ TRM locations, plasticity, and activities remain speculative. Distinct barrier epithelial cells, such as alveolar type 2 (AT2) cells of the lungs14, Lgr5+ stem cells of the small intestinal crypts15, enterocytes of the villi16, and keratinocytes of the skin all express MHC-II17. Although the only known function of MHC-II is antigen presentation to CD4+ T cells, whether barrier epithelial cells direct CD4+ TRM cell activities during exposures to relevant cognate antigens remains speculative.
Streptococcus pneumoniae (Spn) is a leading cause of pneumonia and infectious deaths worldwide18. Serotype replacement and antibiotic resistance have contributed to the classification of Spn as a priority pathogen by WHO in need of better antibiotics and vaccines. The protection offered by Spn-specific CD4+ TRM cells is TH17 polarized and serotype-independent19, representing a potential avenue for vaccination against all Spn serotypes. Especially considering that up to 65% of children and ~10% of adults are exposed to this pathobiont at any given time18, understanding the biology of Spn-specific CD4+ TRM cells is imperative to improving the outomes of recurrent Spn exposures.
In this work, we postulate that lung epithelial cells (LECs) mediate antigen presentation to pulmonary CD4+ TRM cells in order to regulate their localization and activities after Spn infection. We define molecular factors involved in this communication axis and show a physiological role of LEC MHC-II related to CD4+ TRM cell plasticity and immunity.
Results
Identification of SPClowMHChigh LECs
We sought to identify LECs with antigen-presenting abilities during homeostasis. Because MHC-II has been localized to AT2 cells14 and AT2 cells have no known cell-specific surface markers useful for flow cytometry, we used mice expressing GFP under control of human surfactant protein C (SPC) promoter (SPC-GFP mice)20 in order to discriminate the SPC-GFPhigh AT2 cells. At baseline, LECs from elastase digested lungs of (Fig. 1a) separated into two clusters based on MHC-II profiles. The MHC-IIlow cluster was largely comprised of airway LECs (i.e., club and multiciliated cells) with minimal contribution of alveolar type 1 (AT1) cells (Fig. 1a and Supplementary Fig. 1a). The MHC-IIhigh cluster was comprised of SPC-GFPhigh AT2 cells and a comparable-sized fraction of SPC-GFPlow cells (Fig. 1a and Supplementary Fig. 1a). Interestingly, the SPC-GFPlow cells expressed more MHC-II than AT2 cells, almost comparable to levels observed on CD45+CD24+MHC-II+ professional APCs (Fig. 1b and Supplementary Fig. 1b). These SPC-GFPlow cells exhibited smaller forward scatter, comparable to club cells (Fig. 1c), and higher β4 integrin (Fig. 1d) and stem cell antigen Sca-1 (Fig. 1e) when compared to AT2 cells. We sought to define the SPC-GFPlow cells using mRNA for lineage-specifying markers. The SPC-GFPlow cells coexpressed markers characteristic of AT2 cells (Spc) and club cells (Scgb1a1), but not markers for multiciliated cells (Foxj1) or AT1 cells (Aqp5) (Fig. 1f). To further characterize this LEC type, we used bronchioalveolar stem cell (BASC) reporter mice that express YFP and mCherry inserted into Spc and Scgb1a1 loci, respectively21 (Fig. 1g). These mice confirmed a population of SPClow cells with high levels of MHC-II, and revealed that they were distinguishable from club cells (SCGB1A1high) that were MHC-IIlow, bronchioalveolar stem cells22 (BASC; SCGB1A1medium SPCmedium) that were MHC-IImedium, and AT2 cells (SPChigh) that were MHC-IIhigh (Fig. 1g). The SPC-YFP+ cells were consistently found within alveoli and not in conducting airways, suggesting an alveolar location for SPClow LECs (Fig. 1h). Transmission electron microscopy (TEM) of sorted SPClow LECs revealed these cells contain both the AT2 cell-defining lamellar bodies, as well as electron-dense secretory vesicles characteristic of club cells (Fig. 1i). Lamellar bodies were exclusively observed in alveoli and never in airways upon inspection of >100 anatomically distinct areas of C57BL/6J mouse lungs (Supplementary Fig. 1c), leading us to conclude that SPClowMHC-IIhigh cells localize to alveoli rather than conducting airways. The SPClow cells also possessed a transcript profile, including Sox2, MHC-I molecule H2-K1, long non-coding RNA Aw112010, and cell cycle regulator Cdkn1a (Supplementary Fig. 1d), reminiscent of distal lung stem cells23. Thus, these data suggest a discrete set of SPClow LECs that express high amounts of MHC-I and MHC-II, exhibit transcriptional features of lung progenitor cells, and surface marker, transcriptional, and structural characteristics distinct from conventional LEC types, hereafter referred to as SPClowMHChigh LECs.

a Genetic construct of SPC-GFP mice and gating strategy to identify major LECs including club (yellow), multiciliated (red), alveolar type 1 (AT1, purple), SPClow (blue), alveolar type (AT2, green) lung epithelial cells in SPC-GFP mice. b Contour plot and quantification of LEC MHC-II. CD45+CD24+MHC-II+ APCs depicted in dark blue. One-Way ANOVA with Holm-Sidak’ multiple comparison test. c Histogram and quantification of LEC cell sizes, One-Way ANOVA with Holm-Sidak’s multiple comparison test. Histogram and quantification for d airway epithelial marker β4 integrin and e stem cell antigen, Sca-1 by SPC-GFPlow and AT2 cells, two-tailed Mann–Whitney test. f mRNA levels of select lineage-defining transcripts in sorted LECs, two-tailed Mann–Whitney test. Sorting strategy is the same as gating strategy in Fig. 1a. g Contour plot for identification of distinct LECs in bronchioalveolar stem cell (BASC) reporter mice and quantification of LEC MHC-II, One-Way ANOVA with Tukey’s multiple comparison test. Major LECs including club (yellow), BASC (magenta), SPClow (blue), alveolar type (AT2, green), and other cell (gray) are indicated. h Representative immunofluorescent micrograph showing anatomical location of SPC-YFP+ cells (green) and SCGB1A1-mCherry+ (red) with DAPI (blue) used as counterstain. Data is representative of n = 2 mice, 2 experiments. i Representative transmission electron micrographs for LECs sorted and pooled from n = 3 SPC-GFP mice. Experiment was repeated twice. Yellow asterisks indicate electron-dense secretory vesicles and green asterisks depict lamellar bodies. All data have n ≥ 5 mice, two independent experiments. All data are presented as mean ± SEM.
LECs exhibit anatomically segregated and temporally dynamic APC activities
To establish if antigen presentation is a feature of LECs, we examined their antigen uptake and processing abilities by intratracheal instillation of DQ-Ovalbumin (DQ-OVA). Consistent with antigen uptake and proteolytic degradation of DQ-OVA to release the quenched fluorophore, we observed fluorescence in all LECs at baseline and after infection with Spn serotype 19F (Fig. 2a and Supplementary Fig. 1a). Because antigen processing abilities were observed in cells with very low MHC-II at baseline, we tested whether MHC-II levels were altered in infection settings where antigen presentation becomes necessary. Indeed, Sp19F infection increased MHC-II on MHC-IIlow LECs while further enhancing its expression on distal LECs, which remained the highest MHC-II expressers at all times (Fig. 2b). Further supporting APC functions, all LECs sorted from infected lungs displayed cell-specific patterns of MHC-II related accessory genes (Supplementary Fig. 2a) and activated antigen-specific CD4+ T cells in MHC-II dependent fashions in ex vivo co-cultures (Supplementary Fig. 2b). Finally, to explicitly establish LECs as APCs, we generated mice lacking MHC-II in LECs by crossing H2-Abfl/fl mice24 to Nkx2.1Cre-ERT2 mice25 (henceforth MHC-IIΔEpi) (Fig. 2c). Administration of tamoxifen efficiently deleted all baseline or induced MHC-II on LECs (Supplementary Fig. 2c, d) while leaving MHC-II intact on other cell types (Supplementary Fig. 2e). Stimulation of antigen-specific CD4+ T cells by antigen-fed LECs was compromised by MHC-II gene targeting (Fig. 2c). Thus, our results reveal that all LECs respond to infection by increasing MHC-II on their surface, and LEC MHC-II mediates antigen presentation to CD4+ T cells.

a Histogram and quantification of in vivo DQ-ovalbumin (DQ-OVA) uptake and processing abilities of LECs from naïve and Sp19F-infected mice 48hours postinfection (hpi), two-tailed Mann–Whitney test. b Histogram and quantification for LEC MHC-II from naïve and Sp19F-infected mice 48hpi, two-tailed Mann–Whitney test. c Genetic construct and experimental timeline of MHC-IIΔEpi mice. Quantification of OT-II CD4+ T cell activation by MHC-IIfl/fl and MHC-IIΔEpi LECs sorted from Sp19F-infected mice 48 h postinfection (hpi), one-tailed Unpaired t test. Sorting strategy in Supplementary Figure. 2c. Note, different scales for Y-axes in Fig. 2a–c highlight that all LECs are capable of antigen presentation albeit in different amplitudes based on their anatomical location. d Schematic of experimental timeline. e opt-SNE plot depicting 6 LEC metaclusters identified from ~2 million LECs isolated from n = 84 mice across nine timepoints each with two independent experiments. f Heat map depiction for expression of APC-related molecules mapped onto opt-SNE projection. Temporal quantification for g MHC-II, h CD40, i. CD86, j ICAM1, k PD-L1, and l PD-L2 across distinct LECs. Two- Way ANOVA with Fisher’s LSD test statistics for Fig. 2g–l are provided in Supplementary Table 1-6. All data have n ≥ 4 mice, two independent experiments. All data are presented as mean ± SEM.
Antigen presentation is modulated by costimulatory signals, so we examined if such signals were expressed by which LECs and when. Repeated exposures to Spn induces formation of protective lung CD4+ TRM cells19. We collected LECs from multiple time-points after Spn infections (Fig. 2d) and subjected them to a 17-parameter high-dimensional multispectral flow cytometry (MSFC) panel that included antibodies to MHC-II, costimulatory molecules (CD80, CD86, CD40, ICOS-L), cell adhesion molecules with costimulatory functions (ICAM1 and VCAM1), and coinhibitory molecules (PD-L1 and PD-L2). Approximately 2 million CD45−EpCAM+ LECs from 84 C57BL/6J mice across timepoints were concatenated, projected into 2-dimensional optimized t-distributed stochastic neighbor embedding (opt-SNE) space26 and subjected to unsupervised clustering using Phenograph algorithm27. This empowered us to (i) identify distinct LEC clusters based on expression profiles integrating all measurements rather than individual surface markers, and (ii) quantify the relative abundances of these clusters over time. Phenograph clustering identified 34 distinct LEC clusters that aggregated into 6 metaclusters corresponding to each LEC type (Fig. 2e and Supplementary Fig. 3a, b). One metacluster did not cleanly correspond to recognized LEC types and shared club and multiciliated features likely representing a club-to-multiciliated transitional state (clusters 14, 18, 19), consistent with our timeline encompassing lung epithelial damage and regeneration28. Opt-SNE visualization confirmed the identity of these metaclusters (Supplementary Fig. 3c)26. As expected, some clusters were products of dynamic signaling events and not all clusters were present simultaneously; instead within each metacluster certain clusters dominated at distinct time points (Supplementary Fig. 3d). These observations demonstrate that LECs undergo dynamic modulation of surface APC phenotypes during microbial infections concurrent with the formation of lung CD4+ TRM niches.
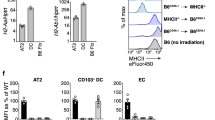
Next, we tracked the surface expression levels of T cell-directed molecules on the LEC metaclusters (Fig. 2e) and discovered that expression of costimulatory/ coinhibitory molecules on LECs was dynamic and highly cell type-specific (Fig. 2f–l and Supplementary Table 1–6). Club cells and multiciliated cells consistently expressed higher levels of T cell costimulatory CD40 (Fig. 2f, h) and CD86 (Fig. 2f, i), respectively, compared to any other LECs. SPClowMHChigh LECs expressed the highest levels of the T cell coinhibitory molecule PD-L1, followed by AT2 cells then all other LECs (Fig. 2f, k). Multiciliated cells were high expressers of PD-L2 (Fig. 2f, l). ICAM1 was elevated on all LECs during active inflammation, but especially on the AT2 cells (Fig. 2j). Throughout the time-course, little to no CD80, ICOS-L, or VCAM1 were observed on any LEC (Supplementary Fig. 3c). The modulation of APC molecules on LECs on day 8 was microbe-induced, since the absence of infection did not incite such changes (Supplementary Fig. 4). These data show that all LECs express costimulatory molecules in a cell-specific fashion. Columnar LECs of the conducting airways (including club and multiciliated cells) have inducible MHC-II and constitutive expression of costimulators (CD40 and CD86, respectively), whereas the lamellar body-containing alveolar LECs (AT2 cells and SPClowMHChigh LECs) express constitutive MHC-II with PD-L1 (both of which increase during infection).
LEC antigen presentation facilitates CD4+ TRM cell niches around conducting airways
We hypothesized that the anatomically segregated LEC antigen presentation pattern instructs the formation of CD4+ TRM niches within the bronchovascular bundles along the airways29. To test this, we first tracked the temporal dynamics of CD4+ T cells at timepoints coinciding with LEC antigen presentation using flow cytometry (as in Fig. 2d). To accurately discriminate between circulating versus lung CD4+ T cells, we used an in vivo antibody labeling approach30 and generated comprehensive snapshots of the dynamics of CD4+ T cell recruitment, expansion, and contraction leading to the formation of CD4+ TRM niches (Fig. 3a and Supplementary Fig. 5a). One Sp19F infection instigated a modest but transient increase in CD4+ T cells numbers by day 3 (Fig. 3a). We do not know the antigen specificity of these T cells, but at such an early time-point we suspect they were most likely not Spn-specific but were instead a mixture of naïve and unrelated cells that were recruited to the lung due to inflammation. Consistent with this, a single Sp19F infection did not induce lung CD4+ TRM cells (Supplementary Fig. 5b). A second Sp19F infection at day 7; however, elicited rapid recruitment of all conventional CD4+ T cell subsets including naïve T (TNaive), central memory T (TCM), effector memory T (TEM), and effector T (Teff) cells (Fig. 3a and Supplementary Fig. 5c). The CD4+ T cell numbers peaked by day 10, with dominating subsets comprised of CD4+ TNaive, TCM, and TEM cells along with activated and expanding Teff cells. At this point, ~17% of lung CD4+ Teff cells expressed IL-2Rα (CD25), suggesting dependence on IL-2 signaling. Between days 10 and 24, the lung CD4+ T cell compartment contracted until CD4+ TRM cells and a small pool of patrolling CD4+ TEM cells remained. CD4+ Teff cells were the highest PD-1 expressers, with most surface PD-1 on day 10 (Fig. 3b, c). Interestingly, LEC PD-L1 expression spiked on day 8 (Fig. 2k) preceding the CD4+ Teff PD-1 peak (Fig. 3c). Of note, only CD4+ TRM cells were PD-1high in the resting but experienced lungs at day 35 (Fig. 3d).

a Numbers of lung (i.v.CD45.2-) CD4+ T cell subsets at designated timepoints, n ≥ 5 mice/timepoint, two independent experiments, except day 10 (n = 3 mice). b Histogram for PD-1 on CD4+ T cells at day 10. c PD-1 levels on CD4+ T cells at designated timepoints, Two-way ANOVA with two-stage step-up method of Benjamini, Krieger, and Yekutieli to correct for multiple comparisons. FDR q value: *≤0.05 comparisons with TEM, #≤0.05 comparisons with CD25+ Teff cells, n = 6 mice/timepoint, two experiments, except day 10 (n = 3 mice). d Histogram for PD-1 on CD4+ T cells at day 35. e Representative immunofluorescent micrographs showing anatomical location of CD4+ cells (red) at designated timepoints. Autofluorescence(AF) identifies lung structures. Data represents n = 6 mice/timepoint, except day 14 (n = 5 mice), two experiments. f Quantification of Fig. 3e, Two-way ANOVA with two-stage step-up method of Benjamini, Krieger, and Yekutieli to correct for multiple comparisons. FDR q value: *≤0.05 comparisons with parenchyma, #≤0.05 comparisons with airway and Φ≤0.05 comparisons with day 8 within the niche, n = 6 mice/timepoint, except day 14 (n = 5 mice), two independent experiments. g Experimental timeline. h Representative immunofluorescent micrographs (top) and quantification (bottom) showing anatomical CD4+ cell distribution within experienced MHC-IIfl/fl and MHC-IIΔEpi lungs at day 56. Data represents n = 8 MHC-IIfl/fl and n = 6 MHC-IIΔEpi mice, two experiments. One-Way ANOVA with two-stage step-up method of Benjamini, Krieger, and Yekutieli to correct for multiple comparisons. FDR q value: *≤0.05. i Timeline of CD40L blockade. Number of lung (i.v.CD45.2−) CD4+- T cells, TRM (CD11ahighCD69+) cells, and CD103+ TRM cells at day 35 in experienced mice with MR-1 (CD40L blockade) or IgG control, n ≥ 5 mice, two independent experiments, One-Way ANOVA with Dunnett’s multiple comparison test. All data are presented as mean ± SEM.
We used immunofluorescence to localize CD4+ cells during the establishment of CD4+ TRM niches (Fig. 3e, f). The second Sp19F infection at day 7 elicited rapid recruitment of CD4+ cells on days 8–10 in the loose interstitium of the bronchovascular bundles, surrounding the coordinated arterial and airway branching trees of the lower respiratory tract (Fig. 3e, f and Supplementary Fig. 5c). Resolution of infection between days 14 and 24 was associated with contraction of CD4+ cells in parenchyma and bronchovascular bundles, except along the airway LECs where there appeared to be a stable CD4+ cell niche (Fig. 3e, f). By day 35, 4 weeks after infection, all remaining CD4+ cells were CD69+ TRM cells in these resting lungs (Fig. 3a), and they localized exclusively around the conducting airways (Fig. 3e, f). These observations suggest that the airway niche promotes survival and/or maintenance of recruited CD4+ T cells (perhaps via MHC-II plus the costimulatory CD40 and/or CD86 of the club and multiciliated cells) to form CD4+ TRM cells, while the parenchymal niches instigate contraction of CD4+ T cells (perhaps via LEC MHC-II plus coinhibitory molecules like PD-L1) during resolution of infection. If true, deletion of LEC MHC-II should disrupt the anatomical distribution of CD4+ TRM niches within the lungs.
To test this, we generated CD4+ TRM niches within MHC-IIfl/fl and MHC-IIΔEpi lungs and systematically quantified their anatomical distribution (Fig. 3g, h). The sustained absence of MHC-II was confirmed (Supplementary Fig. 5d). Consistent with the notion that LEC MHC-II facilitates the formation of CD4+ TRM cell niches within bronchovascular bundles, MHC-IIΔEpi lungs exhibited sparser CD4+ TRM niches along the airway epithelium and within bronchovascular interstitium (Fig. 3h). However, loss of LEC MHC-II did not change the parenchyma CD4+ T cell numbers, suggesting the involvement of other mechanisms in their contraction. For conducting airway LECs, MHC-II overlapped with CD40 better than CD86 (Fig. 2f–i), leading us to test whether the frequency of CD4+ TRM cells in the lungs was dependent on CD40 signals offered by club cells in addition to other canonical CD40+ cells. We examined this using the MR-1 antibody to block CD40-CD40L interaction during Spn-specific CD4+ Teff cell recruitment (Fig. 3i). Consistent with the requirement for CD40 signals for the formation/maintenance of CD4+ TRM cells, blockade of CD40-CD40L interactions significantly reduced lung CD4+ T cell, CD4+ TRM cell, and CD103+ CD4+ TRM cell numbers (Fig. 3i). Taken together, our results indicate that antigen presentation by LECs facilitates the formation of CD4+ TRM niches around the conducting airways, but is dispensable for their exclusion from the lung parenchyma.
CD4+ TRM cells display multipotent phenotypes that are constrained by LEC MHC-II
Residence of CD4+ TRM cells within barrier tissues as frontline adaptive immune sentinels involves inevitable and recurrent stimulations by cognate antigens over their lifespan. We next asked whether antigen presentation by LECs instructs CD4+ TRM cell activities during repeated memory recall encounters. To test this, we generated CD4+ TRM cells in MHC-IIΔEpi and MHC-IIfl/fl lungs as in Fig. 3g, followed by recurring exposures to a serotype-mismatched nonlethal strain of Spn serotype 23A (Sp23A) (Fig. 4a). Deletion of LEC MHC-II did not impede reactivation of CD4+ TRM cells nor affected rapid clearance of Spn (Supplementary Fig. 6a–d) and as such, these infections mimic homeostatic nonlethal inhalation of microbes to which adult lungs would already possess CD4+ TRM cells. Measurements of airway cellularity revealed no ongoing inflammation as evidenced by <1% neutrophil content of the bronchoalveolar lavage (BAL) fluid on day 105 (Supplementary Fig. 6e). Sustained deletion of LEC MHC-II was confirmed (Supplementary Fig. 6f).

a Schematic of experimental timeline and antibody panel used. b Left: Heat map depicting normalized expression levels of distinct molecules on lung (i.v.CD45.2−) memory (CD44+CD11a+) CD4+ T cells clusters on day 105. The lineage determining transcription factor (LDTF) status of each cluster is depicted. Right: Relative abundance of each cluster at day 105 in MHC-IIfl/fl and MHC-IIΔEpi lungs. Two-way ANOVA with two-stage step-up method of Benjamini, Krieger, and Yekutieli to correct for multiple comparisons. FDR p value: *≤ 0.05. c Left: Heat map depicting normalized expression levels of distinct molecules on blood intravascular (i.v.CD45.2+) memory (CD44+CD11a+) CD4+ T cell clusters on day 105. LDTF status of each cluster is depicted. Right: Relative abundance of each cluster at day 105 in MHC-IIfl/fl and MHC-IIΔEpi lungs. Two-way ANOVA with two-stage step-up method of Benjamini, Krieger and Yekutieli to correct for multiple comparisons. FDR p value: *≤ 0.05. d Relative multipotency indices for memory (CD44+CD11a+) CD4+ T cells in lung (circle) and blood (triangles) of MHC-IIfl/fl (white) and MHC-IIΔEpi mice (red) at day 105, One-way ANOVA with Holm-Sidak’s multiple comparison test. e Frequency of activated (CD69+) Tbet+, Gata3+ or RORγT single-positive (SP) TH17 cells in MHC-IIfl/fl and MHC-IIΔEpi lungs 8hpi Sp35B, Two-Way ANOVA with Fisher’s LSD test. For all experiments, positivity for a LDTF was determined using cutoffs identified from clusters negative for that LDTF. f Intracellular cytokine staining (ICS) profile of lung (i.v.CD45.2−) and blood (i.v.CD45.2+) CD4+ T cells isolated from lungs on day 105 and stimulated with PMA/Ionomycin ex vivo, two-way ANOVA with Geisser-Greenhouse correction and two-stage step-up method of Benjamini, Krieger, and Yekutieli to correct for multiple comparisons. p value: #≤ 0.05 comparison between lung and blood; *≤ 0.05 genotype-dependent comparison within lungs; Φ ≤ 0.05 genotype-dependent comparison within blood. All data have n ≥ 6 mice, two independent experiments. All data are presented as mean ± SEM.
To examine CD4+ TRM phenotypes in the absence of LEC MHC-II, we used a 21-parameter high-dimensional MSFC panel including antibodies to CD4+ TRM markers, T cell lineage-defining transcription factors (LDTFs), and markers of activation and proliferation (Fig. 4a). Analysis of total lung CD4+ T cell and CD4+ TRM cell numbers on day 105 revealed comparable frequencies between genotypes (Supplementary Fig. 6g). Next, we used Phenograph to identify distinct clusters of CD4+ T cells based on unique expression profiles, and quantified their relative abundance in genotype-dependent fashion. Phenograph clustering of concatenated lung (i.v.CD45.2−) CD44+CD11a+ CD4+ T cells identified 21 distinct memory CD4+ T cell clusters (Supplementary Fig. 7a), none being unique to either genotype and all being negative for CD62L. For further analyses, we focused on the 20 clusters at >0.5% frequency (Fig. 4b). Remarkably, 17 of the 20 memory CD4+ T cell clusters co-expressed ≥2 LDTFs (cluster 17 was positive for all LDTFs tested) (Fig. 4b). Among these, clusters 10 and 19 were CD69− and represented TEM cells while the remaining 15 clusters encompassed TRM cells comprised of 2 CD103+ Treg-31, 1 TH1/17-, 1 TH2/17-, 8 TH1/2-, and 3 TH1/2/17- lung TRM clusters at homeostasis. Thus, our results highlight the presence of complex lineage plasticity and multipotency within lung CD4+ TRM cells. Of note, clusters 9 and 12 were negative for all tested LDTFs; however, positivity for other LDTFs not included in our panel, like BCL6, cannot be excluded.
Quantification of relative frequencies revealed significant perturbations in 8 of the 20 memory CD4+ T cell clusters due to deletion of LEC MHC-II (Fig. 4b). While MHC-IIΔEpi mice exhibited significant expansion of TH1/2/17 TRM clusters 1 and 4, and the TH1/17 TRM cluster 8, they revealed a significant contraction in 3 TH1/2 TRM clusters (3, 5, and 7) and the TH1 TRM cluster 6 (Fig. 4b). Of note, all CD4+ TRM clusters enlarged in MHC-IIΔEpi mice were Ki67−, suggesting they were not the result of uninhibited proliferation at tissue homeostasis. Furthermore, neither of the Treg TRM cell clusters were affected by LEC MHC-II deletion, implying that the observed effects were probably independent of Treg-suppressive functions. Our findings, therefore suggest that LEC MHC-II imposes inhibitory constraints on CD4+ TRM cell plasticity and multipotency (defined as coexpression of ≥2 LDTFs).
Next, we investigated whether deletion of LEC MHC-II induces perturbations in the circulating CD4+ T cell compartment by performing Phenograph clustering on the intravascular (i.v.CD45.2+) CD44+CD11a+ CD4+ T cells collected from the lungs of the same mice. We identified 21 distinct clusters (Supplementary Fig. 7b), none being unique to either genotype. In stark contrast to the lungs, only 11 of the 21 blood memory CD4+ T cell clusters exhibited multipotency (Fig. 4c) with 10 of the 21 clusters being positive for only one of these LDTFs (Fig. 4c). Comparisons of relative frequencies for multipotency revealed stark differences in the permitted degree of plasticity conferred to memory CD4+ T cells based on: (i) the status of LEC MHC-II and (ii) the anatomical site of T cell residence. For example, while 81% of MHC-IIfl/fl lung CD4+ TRM cells exhibited multipotency (of which 20% were TH1/2/17), a significantly higher proportion (88%) of MHC-IIΔEpi lung CD4+ TRM cells coexpressed ≥2 LDTFs (of which 38% were TH1/2/17) (Supplementary Fig. 7c). Irrespective of their LEC MHC-II status, only ~61% of all blood memory CD4+ T cells displayed such plasticity (none of which were TH1/2/17) (Supplementary Fig. 7c). Comparing phenotypes of extravascular (tissue) and intravascular (circulating) CD4+ T cells in the lungs of MHC-IIfl/fl and MHC-IIΔEpi mice revealed that the tissue cells had greater complexity than the circulating pools (evidenced by Tbet+Gata3+RORγT+ triple-positive CD4+ T cells corresponding to TH1/2/17 skewing)(Supplementary Fig. 7c) with consistently high multipotency (Fig. 4d) that was exacerbated by the loss of LEC MHC-II (Fig. 4d and Supplementary Fig. 7c). Our results, thus (i) ascertain antigen presentation by LECs as a critical regulator of CD4+ TRM cell plasticity and (ii) emphasize the existence of distinct selective pressures against memory CD4+ T cell plasticity in circulation versus barrier tissues of the same immunocompetent host.
LEC MHC-II may govern CD4+ TRM cell plasticity by two ways: (i) by constraining plasticity of pre-existing TH17-specified CD4+ TRM cells during memory recalls or instead (ii) by regulating the development of multipotent CD4+ TRM cells from Teff cells29. To test these possibilities, we first generated CD4+ TRM cells in C57BL/6J, MHC-IIfl/fl, and MHC-IIΔEpi mice and sought to examine if newly established CD4+ TRM cells were exclusively TH17-specified (Supplementary Fig. 8a). Consistent with the notion that CD4+ TRM cells are not solely TH17-specified, we observed heterogeneity and plasticity in CD4+ TRM cells even at day 56 (Supplementary Fig. 8b, c). Furthermore, we detected modest but significant expansion of TH1/2/17 CD4+ TRM cells in MHC-IIΔEpi mice even without subsequent Sp23A memory recalls (Supplementary Fig. 8c–e), suggesting that CD4+ TRM cells are innately plastic and LEC MHC-II might regulate multipotency development from tissue-infiltrating Teff cells. To confirm this, we generated CD4+ TRM cells in MHC-IIfl/fl and MHC-IIΔEpi mice before deletion of LEC MHC-II, and then provided Sp23A memory recall exposures in presence of the lymph node egress inhibitor FTY720 (Supplementary Fig. 9a). This denied CD4+ T cells access to the systemic circulation (Supplementary Fig. 9b–d) so that only pre-established lung CD4+ TRM cells could contribute to the memory recall responses and plasticity depending on LEC MHC-II status. No differences in CD4+ TRM cell phenotypes between MHC-IIfl/fl and MHC-IIΔEpi lungs were observed due to LEC MHC-II deletion (Supplementary Fig. 9e–h). Together, these observations suggest that LEC MHC-II does not govern phenotypes of established TRM cells but rather constrains the multipotency of CD4+ TRM cells developing from Teff cells during primary and memory recall encounters.
LEC MHC-II directs skewing of lung CD4+ TRM cell responses
We next investigated the activity of the multipotent TRM cells on restimulation with a nonlethal mismatched Spn serotype (Sp35B) in order to elicit a heterotypic response. We profiled lung CD4+ T cells 8 hours after infection (Fig. 4a), when Spn-specific TRM cells already bolster neutrophil recruitment11,19. Concatenated lung CD62L−CD44+CD11a+ CD4+ T cells included 26 distinct clusters (Supplementary Fig. 10a), none being unique to either genotype. Unlike resting TRM cells, no CD69+ clusters in the infected lung were triple positive for Tbet, Gata3, and RORγT (Supplementary Fig. 10b, c), suggesting a rapid transition away from extensive multipotency towards more limited phenotypes with one or two dominant LDTF.
Quantification of relative frequencies revealed significant perturbations in 4 of 15 CD69+ T cell clusters (Supplementary Fig. 10b) within MHC-IIΔEpi lungs. Of these, TH17 clusters 3 and 8 and TH1 cluster 7 were enriched in MHC-IIΔEpi lungs, while the TH1/17 cluster 6 was reduced, compared to MHC-IIfl/fl littermates. Quantification of cumulative frequencies identified a significant expansion of TH17 cells with a concomitant reduction of the less-specified TH1/17 cells within the MHC-IIΔEpi lungs (Supplementary Fig. 10c). Cumulative frequencies of each individual transcription factor within RORγT+ T cells further suggested a bias towards TH17 and away from TH1 in the absence of LEC MHC-II (Fig. 4e), based on LDTF content. This was further supported by the strong negative correlation of Tbet+ TH17 cells with RORγT+ single-positive (SP) TH17 cells on memory recall (Supplementary Fig. 10d). Of note, these differences were independent of blood CD4+ T cell involvement since no genotype-dependent differences were observed in the pulmonary intravascular memory cells (i.v.CD45.2+CD62L−CD44+CD11a+CD4+) of the same mice (Supplementary Fig. 11). Thus, based on LDTFs, the absence of LEC MHC-II exaggerates lung TH17 cell responses, at the cost of TH1 activity.
To define T cell lineages by cytokine expression, we used intracellular cytokine staining with PMA/Ionomycin stimulated lung and blood CD4+ T cells from experienced MHC-IIΔEpi and MHC-IIfl/fl mice. Across conditions, the majority of these CD4+ T cells were either IFN-γ+ or IL-17A+, followed by IFN-γ+IL-17A+ dual expressor cells (Fig. 4f). Only very few cells expressed IL-5 or IL-13. Consistent with the absence of MHC-II on LECs leading to reprogrammed TRM cells, lung CD4+ T cells from MHC-IIΔEpi mice expressed increased IL-17A and decreased IFN-γ when stimulated (Fig. 4f). The latter finding was recapitulated by antigen-specific ex vivo stimulation of CD4+ T cells (Supplementary Fig. 12). Very consistently, TH1 activity was blunted in CD4+ T cells from lungs devoid of LEC MHC-II.
The absence of LEC MHC-II dysregulates barrier immune landscape
CD4+ TRM cells confer microbial clearance by accelerating innate immunity1,2. We examined if LEC MHC-II and its regulation of CD4+ TRM cell activities instructs innate immune cellular responses to memory recall infections. While both the MHC-IIfl/fl and MHC-IIΔEpi lungs exhibited comparable Sp35B clearance (Fig. 5a) and BAL cells (Fig. 5b), quantification of extravascular myeloid cells in the infected lung using flow cytometry revealed significant perturbations in the lung immune landscape of MHC-IIΔEpi mice within 8 hours of heterotypic infection (Fig. 5c, and Supplementary Fig. 13, 14). MHC-IIΔEpi lungs possessed higher frequencies of eosinophils and tissue-resident alveolar macrophages, but they exhibited reduced accumulation of multiple monocyte-related cells including Ly6C+ inflammatory monocytes, monocyte-derived interstitial macrophages, and monocyte-derived DCs, in addition to reduced CD11b+ conventional DCs and plasmacytoid DCs (Fig. 5c). These results suggest that deletion of LEC MHC-II has far-reaching consequences on barrier immunity. Of note, these changes were downstream of CD4+ TRM cell reactivation, since uninfected lungs exhibited no inflammation (Supplementary Fig. 6e) or decrements in monocyte-related cell compartments (Supplementary Fig. 15a, b), although eosinophil enrichment was apparent (Supplementary Fig. 15a, b). To connect CD4+ T cell clusters to the observed dysregulation in barrier immune landscape on memory recall, we performed correlation analyses for each CD69+CD4+ T cell lineage and all the myeloid cell changes from the same mice (Fig. 5d). Consistent with the notion that RORγT+ Treg cells suppress TH17 responses32, RORγT+ Treg cells inversely correlated with neutrophil responses. Furthermore, consistent with Tbet promoting monocyte influx33, TH1/17 exhibited direct correlation with Ly6C+ monocytes and monocyte-derived CD11b+ DCs. Interestingly, we observed strong and direct correlations between eosinophils and TH17 cells and lung neutrophils with LDTF-negative (TN) CD4+ TH cells, suggesting possible T cell influences on myeloid cells.

a Airway Sp35B burden 8hpi. b Total bronchoalveolar lavage (BAL) cellularity, macrophages, neutrophils (PMNs) and lymphocytes 8hpi Sp35B. c. Frequencies of major lung (i.v. CD45.2−) myeloid cell populations identified 8hpi Sp35B, two-tailed Mann–Whitney test. Note, the mice used in these experiments belonged to both sexes and were of various sizes at the experiment endpoint (~6-8 months old at euthanasia). This was reflected in differences in lung sizes and hence, the frequency data herein and absolute numbers in Supplementary Fig. 14 are both used to draw conclusions. d Heat map representation of two-tailed Pearson correlation analyses of frequencies of the lung (i.v. CD45.2−) activate (CD69+) CD4+ T cell lineages with frequencies of major myeloid cells identified in Fig. 5c in lungs 8hpi Sp35B. p-value: *≤ 0.05, **≤ 0.01, ***≤ 0.001. e Frequencies of intravascular (i.v.CD45.2+) immune cells depicted as fraction of i.v.CD45.2+ cells 8hpi Sp35B. two-tailed Mann–Whitney test. f Blood neutrophil-to-lymphocyte ratio (NLR), lymphocyte-to-monocyte ratio (LMR), eosinophil-to-monocyte ratio (EMR) and eosinophil-to-lymphocyte ratio (ELR) in i.v.CD45.2+ fraction of lungs 8hpi Sp35B. two-tailed Mann–Whitney test. All experiments have n ≥ 8 mice, 2 independent experiments. All data are presented as mean ± SEM.
Reduced lung monocytes and persistently high eosinophils in the MHC-IIΔEpi lungs evoke features reported in patients with checkpoint blockade immunotherapy (CBI)34,35,36,37 and checkpoint inhibitor pneumonitis (CIP)38. We probed the leukocyte frequencies within the blood fraction (i.v.CD45.2+) of MHC-IIfl/fl and MHC-IIΔEpi lungs for features proposed as robust clinical biomarkers for CBI34,35,36,37,39,40. Consistently, blood fraction of MHC-IIΔEpi lungs phenocopied features of CBI as manifested by presence of elevated eosinophil frequencies, reduced monocyte frequencies, elevated lymphocyte to monocyte ratios, elevated eosinophil-to-monocyte ratios, and elevated eosinophil-to-lymphocyte ratios during infection (Fig. 5e, f and Supplementary Fig. 15c, d). Our results thus demonstrate that memory recall of CD4+ TRM cells expanded in absence of LEC MHC-II triggers dysregulated barrier immunity phenocopying features of CBI, emphasizing the complex (and incompletely understood) intra- and interleukocyte interactions mediating barrier immunity.
LEC MHC-II functions in a post-translational coupling with PD-L1
The similarities of immune dysregulations in infected MHC-IIΔEpi mice and patients with CBI and CIP led us to further investigate checkpoint molecules on LECs. LEC MHC-II correlated strongly with LEC PD-L1 throughout all timepoints and across all LEC-types (Fig. 6a). Higher PD-L1 levels on MHC-IIhigh LECs were also confirmed in human lungs, which had similar positive correlations between the 2 signals (Fig. 6b). When we examined surface PD-L1 expression on LECs of MHC-IIΔEpi mice, we unexpectedly observed loss of this immune checkpoint on alveolar LECs during homeostasis and pneumonia (Fig. 6c and Supplementary Fig. 16a). This coupling also extended to other lung challenges (Supplementary Fig. 17), suggesting an evolutionarily conserved coupling between MHC-II and PD-L1 on LECs leading to an unanticipated LEC-specific interruption of checkpoint signaling. We aimed to elucidate the mechanism underlying the loss of PD-L1 on MHC-IIΔEpi LECs. There was a comparable expression of Pd-l1 mRNA in MHC-IIΔEpi and MHC-IIfl/fl LECs (Supplementary Fig. 16b), suggesting post-transcriptional effects. We posited that PD-L1 depended on LEC MHC-II for surface localization, and tested this using a monoclonal antibody that binds to PD-L1 extracellular domains41. Consistent with our hypothesis, while most PD-L1 of MHC-IIfl/fl alveolar LECs was surface exposed, almost all PD-L1 within MHC-IIΔEpi alveolar LECs was trapped intracellularly (Fig. 6d). In addition to failed PD-L1 delivery to the cell surface, distal LECs exhibited decreased content of intracellular PD-L1 (Supplementary Fig. 16c), consistent with PD-L1 instability and degradation in MHC-IIΔEpi LECs. These data suggest that surface presentation of the checkpoint molecule PD-L1 is entirely dependent upon the expression levels of MHC-II in LECs. This absence of LEC PD-L1 may contribute to phenotypes resulting from LEC MHC-II loss, as evidenced in PD-1 deficient mice42 with relevant pneumonia history (Fig. 6e) showing greater memory CD4+ TRM cell plasticity (Fig. 6f and Supplementary Fig. 18) and multipotency (Fig. 6g) in their lungs but not their blood, akin to MHC-IIΔEpi mice. Taken together, MHC-II on LECs emerges as having two-pronged effects on immunity, directing adaptive immunity via antigen presentation and regulating adaptive immunity via PD-L1.

a Scatterplot correlating MHC-II and PD-L1 on LECs across nine timepoints designated in Fig. 2d (n = 54 mice, two independent experiments). Two-tailed Pearson’s correlation coefficient (r) and p values denoted. b PD-L1 levels on human LECs (n = 7). Two-tailed Wilcoxon matched-pairs signed-rank test. Scatterplot correlating MHC-II and PD-L1 on human LECs (n = 7). One-tailed Pearson’s correlation coefficient (r) and p values denoted. c Histogram for surface PD-L1 levels on LECs from naïve and Sp19F-infected MHC-IIfl/fl and MHC-IIΔEpi mice 48hpi. n ≥ 4 mice, two independent experiments. d Contour plots for subcellular localization of native PD-L1 in alveolar LECs isolated from Sp19F-infected MHC-IIfl/fl and MHC-IIΔEpi mice 48hpi. n ≥ 8 mice, two independent experiments; mean ± SD. e Schematic of experimental timeline used. f Pie charts for mean frequencies of distinct lung (i.v.CD45.2−) CD4+ TRM cell- and blood (i.v.CD45.2+) CD4+ TEM cell- lineages in PD-1+/+ and PD-1−/− mice at day 85 as identified using manual gating, Two-way ANOVA with Fisher’s LSD test. p-value: #≤ 0.05 for comparison between lung and blood; *≤ 0.05 for genotype-dependent comparison within lungs; n ≥ 5 mice, two independent experiments. g Relative multipotency indices for lung (i.v.CD45.2−) CD4+ TRM cells (circles) and blood (i.v.CD45.2+) CD4+ TEM cells (triangles) in PD-1+/+ (white) and PD-1−/− (red) mice at day 85, n ≥ 5 mice, 2 independent experiments, One-way ANOVA with Holm-Sidak’s multiple comparison test. All data are presented as mean ± SEM.
Discussion
Herein, we show that diverse LECs, including a distinct SPClowMHChigh LEC, function as APCs to CD4+ T cells in anatomically segregated fashions (Supplementary Figure 19a). We discover that LEC MHC-II is dynamically regulated on all LECs in conjunction with CD4+ T cell recruitment and activation (Supplementary Figure 19b) and that LEC MHC-II imposes constraints on aberrant expansion and plasticity of CD4+ TRM cells (Supplementary Figure 19c) via the PD-L1:PD-1 signaling axis wherein epithelial PD-L1 depends on MHC-II. Further, we show that disruption of LEC MHC-II has significant consequences on the barrier immune landscape, thus, establishing LEC-CD4+ TRM cell immune synapses as core elements of barrier immunity. Epithelial antigen presentation, thus, plays a pivotal role in localizing and directing CD4+ TRM cell immunity.
MHC-II expression is high on AT2 cells and SPClowMHChigh LECs. SPClowMHChigh LECs possess features of traditional club cells and MHC-Ihigh club-like stem cells23, but they resemble AT2 cells in their high MHC-II, lamellar bodies, and abundance within the lung. They may represent an AT2 cell subset. Given their overlaps with stem cells of the lung23,43, and reported connections of MHC-II to epithelial stem cell biology in other barrier tissues15,17, the immunological synapse may be a component of tissue repair and SPClowMHChigh LECs may contribute to resolution, repair, or regeneration. If and how SPClowMHChigh LECs compare to Wnt-active AT2-stem cells44,45 and AT2-signaling cells46 remains to be determined. CD4+ TRM cells can regulate stem cell renewal, proliferation, differentiation fate, and tissue repair in the skin and the gut6,15. Similar roles may apply for MHC-II in regulating lung stem cell functions47.
Epithelial MHC-II has been connected to regeneration, but its immune significance is less established. Our results identify antigen presentation as a pan-epithelial feature with major roles in lung and CD4+ TRM biology. MHC-II on LECs was envisioned as restricted to AT2 cells14,48, but now is expanded to SPClowMHChigh LECs and to club cells and multiciliated cells during inflammation. Possible immune roles for epithelial MHC-II were controversial and speculative20,49,50,51. Here, we provide in vivo evidence of functional significance whereby deletion of LEC MHC-II led to decreased seeding and aberrant phenotypes of CD4+ TRM cells around conducting airways, with diminished recruitment of monocyte-derived cells during subsequent infections. Thus, LEC MHC-II is a critical tissue-localized cue for resident memory cells.
Our results identified a surprising degree of transcriptional plasticity and multipotency within CD4+ TRM cells, more than previously appreciated6,19,29. Adult humans daily inhale ~11,000 liters of air with its contaminants, regularly microaspirate, and experience frequent respiratory infections, a tremendous environmental exposure for lung memory cells. Factors constraining CD4+ TRM cell plasticity driven by this environmental pressure were unknown. Our studies reveal 3 major findings. (i) CD4+ TRM cells possess extensive flexibility, wherein many TRM cells co-express ≥2 classically antagonistic LDTFs. Such plasticity within TH17 effector cells has been reported52 but our work extends these paradigms to TRM cells and suggests a stem-cell-like uncommitted property for otherwise “primed” TRM cells that might allow flexibility to choose effector pathways suited to the nature of perceived threats3. Epigenetic accessibility of LDTFs to target genes defines T cell responses, and future studies using single-cell epigenetic and transcriptomic profiling of multipotent TRM cells are warranted5,53. Consistent with TH17 effector cells, we observed a tendency for TH17 TRM cell clusters to acquire a TH2 Gata3 signal at homeostasis52, but this was not sustained at the cytokine level post-restimulation. Whether Gata3 expression reflects bivalent epigenetic marks adjacent to Gata3 gene within TRM cells53 or indicates a poised TH2 transcriptome that would translate into TH2 cytokines upon licensing by alarmins such as IL-18 and IL-33 (which are precluded in our 8-h challenge model with Spn)6 needs further investigation. (ii) LECs impose constraints on the aberrant expansion of TRM cells co-expressing Tbet, Gata3, and RORγT via PD-L1:PD-1 signaling axis. Our study establishes PD-1 as a checkpoint molecule that restrains CD4+ T cell phenotypic boundaries in peripheral non-lymphoid tissues. In doing so, we distinguish these restraints as distinct from those imposed by CTLA-4 during initial priming within the lymph nodes in that they curtail expansion, but not de novo generation, of multipotent TRM cell clusters54. Given that MHC-II and PD-L1 expression in LECs is anatomically segregated and enriched in alveolar LECs, the physical locations of the most multipotent TRM cells within the lung becomes an important next question. (iii) Our findings show that the lungs provide a permissive environment for memory CD4+ T cell plasticity compared to the blood. We propose that this may reflect distinct selective pressures in barrier tissues where antigenic encounters are more diverse in terms of quantity, quality, and frequency.
Deletion of LEC MHC-II altered the barrier immune landscape. Reactivated CD4+ TRM cells expanded in the absence of LEC MHC-II show an intrinsic bias to deviate away from TH1 responses, precipitating consequences on host immunity that include increased alveolar macrophages and eosinophils accompanied by reduced monocytes, monocyte-derived interstitial macrophages, monocyte-derived DCs, CD11b+ cDCs, and pDCs within MHC-IIΔEpi lungs. While molecular mechanisms are likely multifactorial, it is reasonable to posit that reduced TH1 responses tilted the barrier immune landscape towards weaker monocyte recruitment and more eosinophilia33,55. This remodeled immunity phenocopies biomarkers of successful CBI34,35,36,37,39,40 and is in line with higher incidences of CIP in PD-1/PD-L1 CBI patients56. Our observations thus suggest that PD-1/PD-L1 CBI may interfere with epithelial constraints on CD4+ TRM cells and unleash unrestrained TRM cell activation leading to immunopathologies like CIP, colitis, IBD, and skin inflammation, all of which may involve CD4+ T cell plasticity7,8,9,56,57. PD-L1 blockade results in unrestrained immunopathology of the lungs in response to viral antigens58, and a similar axis for restraining CD8+ TRM cell-driven pathology in the human pancreas is exploited by PD-L1-expressing pancreatic macrophages59. The exact biological role for the exhaustion marker PD-1 on CD4+ TRM cells that are otherwise highly responsive to antigenic restimulation is incompletely understood1. Our results suggest that TRM cell PD-1 may act akin to a molecular switch that an organ like the lung can exploit to limit the plasticity of TRM cells arising there.
Our findings have implications that pervade fields beyond barrier epithelial and CD4+ TRM cell biology. High levels of MHC-II on AT2 cells14, keratinocytes17, and Lgr5+ intestinal stem cells15 connect antigen presentation to diverse tumor-initiating stem cells (TISCs)60,61,62. While PD-L1 expression on Lgr5+ ISCs is uncertain, keratinocytes, like AT2 cells and SPClowMHChigh LECs, do express PD-L163. Given our observations, it is tempting to posit that MHC-I, MHC-II, and PD-L1 co-expression on TISCs may jointly be responsible for evasion of early antitumor T cell immunosurveillance during tumorigenesis. Furthermore, high MHC-II and PD-L1 levels might also provide a tumor-conducive immunosuppressive microenvironment wherein expansion of antitumor CD4+ T cells would be restricted64. Indeed, murine lungs deficient in MHC-II within non-hematopoietic cells are resistant to metastatic tumorigenesis65. Studies investigating the tumorigenic potential of SPClowMHChigh LECs and elucidating the role of MHC-II in epithelial stem cells are merited. Detailed understanding of stem cell antigen presentation will help guide the development of innovative immunomodulatory agents and inform development of molecular targets that could be valuable biomarkers for early cancer detection and immunotherapy outcomes, such as MHC-II for melanoma66.
Taken together, our study identifies antigen presentation as a pan-epithelial feature that exhibits division of labor in anatomically segregated and temporally dynamic fashions within mammalian lungs. We identify epithelial antigen presentation as a tissue-resident regulator of CD4+ TRM cell locations, plasticity, and activities, and implicate disruption of epithelial restraints on CD4+ TRM cells as underlying checkpoint blockade induced immune dysregulation. Distinct epithelial cells in other barrier tissues may have similar rheostat functions in TRM cell immunity. Epithelial MHC is emerging as relevant to stem cell biology, cancer, and immunomodulatory therapy. Thus, considering epithelial antigen presentation may inform future strategies to curb immunopathology during chronic diseases and checkpoint blockade.
Methods
Mice
C57BL/6J (Stock# 000664), OT-II TCR transgenic (B6.Cg-Tg(TcraTcrb)425Cbn/J) (Stock # 004194), and B6.Cg-Pdcd1tm1.1Shr/J (PD-1−/−, Stock #028276)42 mice were obtained from The Jackson laboratories (USA) at 6 weeks old age, all on the C57BL/6 background. SPC-GFP mice20 were back-crossed more than 15 generations onto C57BL/6 and bred in Boston University animal facilities. Mice for targeted deletion of MHC-II on LECs were generated in-house at BU-ABSL2 facility. Briefly, male B6.129X1-H2-Ab1tm1Koni/J (Stock# 013181; C57BL/6 background) and female Nkx2-1tm1.1(cre/ERT2)Zjh/J mice (Stock# 014552; B6.129SF2 background) were obtained from The Jackson Laboratories (USA) and crossed to create Nkx2.1creERT2H2-Ab1fl/fl mice. Both founder mice lack I-E due to mutation in H2-Ea locus allowing deletion of H2-Ab1 to abrogate MHC-II entirely. Nkx2-1 is expressed in all LECs and some cells of telencephalon, thyroid, and pituitary67,68,69 and Nkx2-1-driven Cre efficiently targets all LECs70. All breeders were homozygous floxed for H2-Ab1. For experiments, all mice were homozygous for loxP sites flanking H2-Ab1 exon1 and were identified as Cre-positive or negative based on the presence or absence of Nkx2.1cre/ERT2. Cage and littermate controls of both sexes were used for the studies. BASC Reporter mice including the BASC viewer and the BASC v-race mice were on a mixed hybrid C57BL/6 x 129/SV background21. All mice were housed in specific pathogen-free environment on a 12-h light-dark cycle at 30–70%, humidity and temperature of 20–26 °C, with ad libitum access to standard chow and water. for all experiments, 7–14-week-old mice were used. Mice were euthanized using isoflurane overdose and death confirmed using pneumothorax before organ collections. Animal procedures were performed with compliance to all relevant ethical regulations for animal testing and research, in the United States in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (after review and approval by the Institutional Animal Care and Use Committee of Boston University) and in Germany according to the German animal protection law (after review and approval by the Regierungspräsidium Darmstadt, Veterinärdezernat of Hesse, Germany).
Human lung biopsy samples
De-identified pathologically “disease-free” samples from lung tissue wedge resections or lobectomies were used for experiments. None of the patients included received any checkpoint blockade therapy. The Boston University Medical Center Institutional Review Board reviewed and approved the study, deeming that it did not meet the definition of “human subjects research” (Protocol # H-32271).
Experimental pneumonia
Mice were anesthetized via intraperitoneal (i.p.) injection of ketamine and xylazine before infection. To induce experience, Streptococcus pneumoniae (Spn) suspended in sterile saline was instilled via a 24-gauge angiocatheter directed to the left lobe after surgical exposure of the trachea. Mice were infected intratracheally (i.t.) with 1–5 × 106 CFU of serotype 19F Spn (Strain EF3030) or saline and euthanized 48 h postinfection. To generate Spn-specific lung-resident CD4+ TRM cells, mice were infected intratracheally (i.t.) with 1–5 × 106 CFU of serotype 19F Spn (Strain EF3030) or saline one week apart, and then allowed to recover for 28–35 days19. Mice with such infection histories exhibited a lung-resident CD4+ TRM cell-dependent protection on memory recall challenge with a lethal serotype-mismatched Spn19. To test CD4+ TRM cell phenotypes on repeated memory recall, 2X Sp19F experienced mice were infected with 1–3 × 106 CFU of serotype 23A Spn (Sp23A) on Days 35 and 49 before allowing recovery for another 5 weeks. For final challenge, 3–5 × 106 CFU of serotype Sp35B Spn (Sp35B) was intratracheally instilled. Of note, Sp35B (like Sp19F and Sp23A) causes nonlethal pneumonia and was chosen to mimic nonlethal homeostatic bacterial exposures experienced by humans. For analyses of MHC-II and PD-L1 coupling, mice were infected with 50 PFU of Influenza A virus PR8 or 1 × 105 CFU of Klebsiella pneumoniae (ATCC43816).
Mouse experimental procedures
For in vivo DQ-Ovalbumin uptake and processing assay, 20 μg of DQ-Ovalbumin, DQ-OVA (Molecular Probes, Cat# D-12053) in 50 μL of sterile saline was instilled i.t. via angiocath directed to the left lung 3 h before euthanasia. For Tamoxifen administration, tamoxifen (Sigma) was dissolved in corn oil (Sigma) to 20 mg/mL stock concentration and stored at 4 °C. Mice were i.p. injected at 100 mg/kg of body weight for five consecutive days. Treated mice were allowed to rest for at least 2 weeks for tamoxifen to wash out before infecting them. For in vivo CD40L blockade, Mice received i.p. injections of 400 μg antimouse CD40L (Clone MR-1, BioXCell, Cat# BE0017-1) or IgG control (BioXCell, Cat# BE0091) antibody on day 6. This was followed by 100 μg i.n. instillations and 250 μg i.p. injections of the respective antibodies on days 7, 9, 11, and 13. For FTY720 administration, Mice were given 1 mg/kg of FTY720 (Sigma) every alternate day by intraperitoneal injections. For recombinant IFN-γ administration, anesthesized mice received a bolus of 100 ng of recombinant IFN-γ (R&D systems) in 100 μL intranasally.
Murine lung digestion for epithelial cell flow cytometry and FACS sorting
For high throughput flow cytometry of lung epithelial cells, elastase digestion of murine lung was performed. Briefly, euthanized mice were exsanguinated, trachea cannulated and lungs lavaged thrice with 1 mL Ca-Mg free DPBS containing 0.5 mM EDTA solution. The lungs were then inflated with 1 mL digestion media (RPMI 1640 containing 10% Dextran, 4.5 units/mL of Elastase (Worthington Biochemicals, Lakewood, NJ), and 150 μg/mL of DNase I) and plugged with 0.5 mL of 1% low melting temperature agarose in PBS. The heart-lung blocks were then placed on petri dishes and covered with ice to allow agarose to solidify for at least 5 min. Left lobes of the enzyme-instilled and plugged lungs were dissected and kept for enzymatic digestion in additional 2 mL of digestion media for 1 h at 37 °C at 200 rpm. The digested lungs were then minced finely using disposable razor blades, washed down with 1 mL digestion media, and transferred to 50 mL conical tubes. The slurry was then kept on a shaker for 20 mins at 37 °C at 200 rpm before adding 10mL of RPMI 1640 with 50% heat-inactivated FBS and 100 μL of 10 mg/mL DNase I. This suspension was then gently vortexed to allow cell aggregate dissociation and incubated on ice for 5 mins. Ten-milliliter RPMI 1640 was then added and the suspension was shaken vigorously at 300 rpm at 37 °C before sequential gravity-mediated filtering through 100 and 70 μm cell strainers. The single-cell suspensions were then centrifuged at 300 g for 10 mins to pellet cells before RBC lysis and resuspension in FACS buffer for flow cytometry as per usual procedures. Cells were blocked with TruStain αCD16/CD32 Fc-Block (BioLegend). Flow cytometry was performed on LSR II Flow Cytometer (BD Biosciences). High-dimensional multiparameter spectral flow cytometry was performed on Aurora (Cytek), SpectraFlo (Cytek) software was utilized for spectral unmixing of the data using an ordinary least square algorithm, and data were analyzed with FlowJo software (BD Biosciences). Gating strategies are provided in the Supplemental figures and were based on use of Fluorescence minus one (FMO) controls.
For data generated using the BASC reporter mice, dispase digestion was used21. Briefly, the lungs of euthanized mice were perfused with PBS, cannulated via the trachea, and instilled with 1 ml prewarmed dispase solution (Corning). The trachea was ligated, the lungs extracted from the thorax, and then finely minced using scissors. Two milliliters dispase and DNase I (20 U/ml, Roche) were added to the resulting slurry, and samples digested at 37 °C shaker for 20–30 min. Resulting cell suspensions were gently mixed and sequentially filtered through 100- and 40-μm strainers followed by wash with excess DMEM through the filter. The single-cell suspensions were centrifuged (5 min, 300 g), pellets resuspended in 1 mL pre-cooled FACS buffer. Cell suspensions were resuspended in MACS buffer, mixed with anti-CD45, anti-CD31, and anti-Ter119 microbeads (Miltenyi Biotec), and incubated at 4 °C for 15 min. After washing, cells were loaded onto pre-conditioned MS columns that had been placed in the magnetic field of a MACS separator and the flow-through containing unlabeled cells was collected. These enriched LECs were then blocked using CD16/CD32 Fc-Block (BD Biosciences), stained, and washed before flow cytometry was performed on LSRFortessa Flow Cytometer (BD Biosciences). Gating was based on the use of Fluorescence minus one (FMO) controls. The list of antibodies used in this study are provided in Supplementary Table 7.
For RNA-profiling, epithelial subsets from stained single-cell suspensions isolated from elastase digested lungs were sorted into RPMI 1640 with 20% FBS on ice using FACS-Aria II SORP (BD Biosciences) before proceeding to RNA extraction.
For TEM analyses, stained LEC cell suspensions were fixed using 2% paraformaldehyde in PBS before sorting using the MoFlo Astrios cell sorter (Beckman Coulter).
Murine lung digestion for leukocyte flow cytometry
To allow enumeration of extravascular versus the intravascular fraction of lung CD4+ T cells, anesthetized mice were retro-orbitally administered 2 μg anti-CD45.2 antibody 3 min prior to euthanasia. Lungs were collected in RPMI 1640 with 10% FBS before processing for flow cytometry. Single-cell suspensions were prepared by digestion of lungs in type 2 collagenase (Worthington Biochemicals, Lakewood, NJ) and DNase I19. Cells were blocked with TruStain αCD16/CD32 Fc-Block (BioLegend). Flow cytometry was performed on LSR II Flow Cytometer (BD Biosciences). High-dimensional multiparameter spectral flow cytometry was performed on Aurora (Cytek), SpectraFlo (Cytek) software was utilized for spectral unmixing of the data using ordinary least square algorithm and data were analyzed with FlowJo software (BD Biosciences). Gating strategies are provided in the Supplemental figures and were based on the use of Fluorescence minus one (FMO) controls. The list of antibodies used in this study is provided in Supplementary Table 7.
Lung processing for human lung flow cytometry
Pathologically deemed normal segments of lung biopsies obtained from tumors resected from patients were collected in RPMI 1640. The lung pieces were minced in 2 mL of Digestion media (RPMI containing 10% Dextran, 10 units/mL of elastase, and 150 μg/mL of DNase I) with a disposable razor blade to a fine slurry. The slurry was then washed down with 3 mL more digestion media and transferred to 50 ml conical tubes. The dish was washed with additional 5 mL digestion media and transferred to the 50 mL tube to have a total of 10 mL cell slurry. The suspension was shaken at 250 rpm for 1 h at 37 °C with intermittent vortexing every 20 mins after which 10 mL of RPMI 1640 with 50% heat-inactivated FBS and 200 μL of 10 mg/mL DNAse I was added. The suspension was vortexed to dissociate aggregates and kept on ice for 5 mins. Additional 10 mL RPMI 1640 media was added to bring the final liquid volume to 30 mL and the tube shaken at 300 rpm for 10 min at 37 °C. The lung minces were sequentially filtered through 100 and 70 μm cell strainers using gravity (no scraping). The single-cell suspensions then centrifuged at 300 g for 10 mins to pellet cells before RBC lysis and resuspension in FACS buffer for flow cytometry as per usual procedures. Cells were blocked with Human TruStain FcX- Block (BioLegend). Flow cytometry was performed on LSR II Flow Cytometer (BD Biosciences) and data were analyzed with FlowJo software (BD Biosciences). Gating strategies are provided in the Supplemental figures and are based on the use of Fluorescence minus one (FMO) controls. The list of antibodies used in this study is provided in Supplementary Table 7.
Intracellular cytokine, protein, and transcription factor staining for flow cytometry
For intracellular staining of transcription factors and Ki67, eBioscience Foxp3/ Transcription Factor Staining Buffer Set (Cat# 00-5523-00) was used as per manufacturer’s protocols. For intracellular cytokine staining and subcellular PD-L1 localization, eBioscience Intracellular Fixation & Permeabilization Buffer Set (Cat# 88-8824-00) was used as per manufacturer’s protocols. The list of antibodies used in this study is provided in Supplementary Table 7.
Transmission electron microscopy
The sorted cells or mouse lungs were fixed for 2 h in 2% glutaraldehyde plus 1% paraformaldehyde in 0.1 M Na Cacodylate buffer, pH 7.4. After washing in buffer, samples were postfixed in 1.5% osmium tetroxide, block stained in 1.5% uranyl acetate, and dehydrated in acetone. They were then embedded in Epon 812, sectioned and stained with uranyl acetate and lead citrate. Images were captured using Tecnai F20 microscope.
RNA extraction and Real-time PCR
RNA was extracted from sorted LECs using RNAeasy Micro Kit (Cat# 74004) as per manufacturer’s protocols and stored at −80 °C. qRT-PCR was performed using the RNA-to-Ct kit (Life Technologies, Cat# 4392938). Commercially available predesigned TaqMan gene expression assays for Spc (Mm00488144_m1), Scgb1a1 (Mm00442046_m1), Foxj1 (Mm01267279_m1), Aqp5 (Mm00437578_m1), Aw112010 (Mm01197675_m1), Sox2 (Mm03053810_s1), H2-K1 (Mm01612247_mH), Cdkn1a (Mm00432448_m1), Cd74 (Mm00658576_m1), H2-DMa (Mm00439226_m1), H2-DMb1 (Mm04213366_s1), H2-DMb2 (Mm00783707_s1), H2-Oa (Mm00468476_m1), H2-Ob (Mm00468801_m1), Cd274 (Mm03048248_m1), and 18S rRNA (Cat# 4319413E) from Applied Biosystems were used. The probe context sequence for these assays are detailed in Supplementary Table 8. The quantity of the detectable mRNA was calculated by normalizing to 18S rRNA from the respective sample and then expressed as fold change over mRNA levels of the whole lung of naive mice of pertinent genotype.
Ex vivo antigen presentation assay
FACS-sorted LECs from Sp19F-infected mice were co-cultured with OT-II CD4+ T cells (1:10 ratio) with 25μg/mL of OVA323-339 peptide (Anaspec) in media containing 25% DMEM (Gibco), 25% F-12 (Gibco), 50% RPMI 1640 (Gibco) supplemented with 10% Heat inactivated FBS, Penicillin/Streptomycin (100 U/mL, Gibco), 1X Non-essential amino acids (Gibco), 2 mM L-Glutamine (Glutamax, Gibco), 1 mM Na-pyruvate (Gibco), 10 mM HEPES (Gibco), 0.2 μM beta-mercaptoethanol, 10 μg/mL Insulin, 50 mM Hydrocortisone, 100 μg/mL Transferrin, 1 mM Na-selenite, and 1 mM beta-estradiol for 8 h at 37 °C 5% CO2 incubator. For assays with C57BL/6J LECs, EasySep™ Mouse CD4+ T Cell negative selection and Isolation Kit (StemCell Technologies, Cat# 19852) was used to enrich OT-II CD4+ T cells from spleens of OT-II TCR transgenic mice and 10 μg/mL of antimouse MHC-II antibody (Biolegend) was used for MHC-II blockade. For assays with MHC-IIΔEpi LECs, EasySep™ Mouse CD4+ T Cell negative selection and Isolation Kit (StemCell Technologies, Cat# 19852) was used to enrich OT-II CD4+ T cells from spleens of OT-II TCR transgenic mice. This was followed by MHC-II blockade of the enriched CD4+ T cell suspension to block any excess MHC-II expressing splenocytes before rigorous washing and coculture with LECs for 8 h.
Ex vivo stimulation and Intracellular Cytokine staining of CD4+ T cells
To allow identification of extravascular versus intravascular fraction of lung CD4+ T cells, anesthetized mice were retro-orbitally administered 2 μg anti-CD45 antibody 3 min prior to euthanasia. Single-cell suspension of lung leukocytes was prepared as described before.
For PMA/Ionomycin stimulation, 2 × 106 cells were stimulated ex vivo in 12 well plate with 250 ng/mL Phorbol Myristate Acetate (PMA)(LC Laboratories, Woburn, MA) and 1.5 μg/mL Ionomycin (Sigma, St. Louis, MO) in T cell stimulation media (RPMI 1640 (Gibco) supplemented with 10% Heat inactivated FBS, Penicillin/Streptomycin (100U/mL, Gibco), 1X Non-essential amino acids (Gibco), 2 mM L-Glutamine (Glutamax, Gibco), 1 mM Na-pyruvate (Gibco), 10 mM HEPES (Gibco), and 0.2 μM beta-mercaptoethanol) for 1 h at 37 °C and 5% CO2. Monensin (Biolegend, Cat# 420701) and Brefeldin A (Biolegend, Cat# 420601) both at 1X final concentration were added to the cell suspension for last 5 h at 37 °C and 5% CO2. Cells were then processed for intracellular cytokine staining, as per manufacturer’s protocols.
For Spn-specific stimulation, 2 × 106 lung cells were stimulated with beta-propiolactone killed Sp35B (ratio of 1:10 of cells:bacteria) or 1 mg/mL chicken egg ovalbumin (Sigma) ex vivo in 12 well plate for 3 h followed by addition of Monensin and Brefeldin A at 1X final concentration for 14 h at 37 °C and 5% CO2. Cells were then processed for intracellular cytokine staining, as per manufacturer’s protocols. The list of antibodies used in this study is provided in Supplementary Table 7.
Immunofluorescent staining
The trachea of euthanized mice were cannulated with an 18-gauge angiocath and 1 mL of Tissue-Tek Optimal Cutting Temperature (O.C.T.) compound (Sakura Finetek). Once the lungs were inflated, the left bronchus was tied using a suture, the lung washed in sterile HBSS before embedding in cryomolds with O.C.T. and flash freezing at −80 °C until sectioning. Frozen 8 μm thin coronal sections of the left lungs that contained the whole face of the lungs including the entire airway tree structure were collected for further analyses. Sections with folds and/or tears were rejected. The sections that met our inclusion criteria were first fixed, washed, and permeabilized (with 0.2% Triton X) before blocking with Blocking buffer (PBS with 10% normal donkey serum and 3% BSA) followed by overnight incubation with rabbit antimouse CD4 (abcam, Cat# ab183685) at 4 °C in a humidified chamber. Next day, sections were vigorously washed before incubation with Alexa 594 conjugated Affinipure donkey antirabbit IgG (Jackson Immunoresearch) at room temperature for 1 hr in a dark humidified chamber. All slides were counterstained with DAPI (Molecular Probes by Life Technologies, R37606) before mounting the sections with FluorSave (Millipore Calbiochem: 345789) and covering with coverslip for visualization. Of note, a “no anti-mouse CD4” treated primary control was used to identify true CD4+ events; a section of day 10 mouse lung (i.e., 3 days post 2x Sp19F infection when the lung possesses most CD4 T cells, Fig. 3A) was chosen for the “no anti-CD4 primary control” to ensure that the inclusion criteria for CD4+ events was stringent. Images were captured using a Leica DM4 microscope equipped with Leica DFC 7000T camera and processed using ImageJ 2.0.0-rc-69.
Quantification of anatomical distribution of CD4+ cells
Fifteen images of randomly selected fields per coronal section from each mouse lung were identified in a raster fashion, from the top left to bottom right, and were captured at 10x objective with Numerical Aperture of 0.30. Images included autofluorescence of lung tissue in the GFP channel to allow identification of lung anatomical structures. ImageJ was used for automated quantification of lung tissue area identified as airway (along the conducting airways), bronchovascular interstitium (interstitial area around major arteries within the bronchovasculature) and the parenchyma (alveoli) within the GFP channel which was permissible for tissue autofluorescence (AF). Once the areas of distinct lung structures were identified, ImageJ was used to computationally enumerate CD4+ events within the same manually selected, anatomically distinct areas of the imaged fields within the Alexa 594 channel to minimize the inclusion of artefacts as false-positive signals. This was done by setting a standardized minimum threshold value which excluded 99.9% of the background signal on the “no anti-CD4 primary control” slide. Images within Alexa 594 channel underwent watershed segmentation to differentiate overlapping CD4+ events. Once this was done for every one of the 15 images per lung, the number of CD4+ events per unit area of the identified lung structure (NX) was identified as (1):
where X = the anatomical structure of the lung wherein the CD4+ events were identified.
If at least 1 of the 15 images per mouse lung contained a structure of interest (i.e., an instance of X), the dynamics of NX, i.e D(NX) for that mouse lung was calculated as (2):
where n = number of images per lung containing instances of X
Since each group contained a total of at least four mice per time points collected across two experiments, the group dynamics of NX for the whole group of mice at their specific time points, i.e., G(D(NX)) was then calculated as (3):
where m = total number of mice per group for that timepoint.
Algorithmic analysis of single-cell fluorescence datasets
Data processing pipeline was established using the Omiq.ai cloud computation platform (Omiq). For temporal LEC MSFC data analysis, live CD45−EpCAM+ single-cell datasets containing equal quantities (25,000 events per animal) of cells from all experimental timepoints were concatenated and asinh transformed (cofactor = 6000). Datapoints were projected into two-dimensional space with opt-SNE algorithm26 incorporating fluorescence, forward scatter and side scatter parameters (perplexity = 50, theta = 0.5, opt-SNE endpoint = 7500; PCA preinitialization embedding). Intensities of each fluorescence parameter were overlaid to represent expression levels. Same dataset was clustered with Phenograph algorithm (k = 20, distance metric = euclidean)27. Groupings of clusters based on hierarchical clustering of median fluorescence intensities (MFI) across multiple protein markers were annotated and color-overlaid on the opt-SNE projection of multidimensional data. For T cell flow cytometry profiling, live CD45+CD4+CD19−CD45.2+ for lung intravascular (‘blood’) cells or live CD45+CD4+CD19−CD45.2− for lung extravascular (‘lung’) T cells were concatenated, asinh transformed (cofactor = 6000) and clustered with Phenograph (k = 20, distance metric = euclidean). CD44+CD11a+ clusters (as determined based on the MFI cutoff) were subsampled as memory T cell data. These data were re-clustered with Phenograph (k = 20, distance metric = euclidean) and projected into opt-SNE space (perplexity = 30, theta = 0.5, opt-SNE endpoint = 5000; PCA preinitialization embedding). Clusters were color-coded and overlaid on the opt-SNE projections. Each marker MFIs of Phenograph clustered datasets were organized into hierarchically clustered heatmaps. Clusters were classified as positive for specific lineage-defining transcription factors (LDTF) based on the MFI value cutoffs set by measuring the MFIs of LDTF-negative non-T cell populations sampled from the same dataset. This approach was preferred over the FMO-based cutoff calculation to alleviate the MFI difference caused by nonspecific binding of anti-LDTF antibodies to cells that do not express them. Frequencies of each cell type and MFI values for individual markers were calculated from corresponding clusters per each animal and data were plotted and compared using Prism 8.0 (Graphpad).
Quantification of Relative Multipotency Index
Relative multipotency indices for CD4+ CD44+CD11a+ memory T cells in different anatomical sites were calculated as (4):
Where memory CD4+ T cells = CD4+CD62L−CD44+CD11a+ CD4+ T cells, and LDTF = lineage-defining transcription factors.
i.v.CD45.2− CD4+ memory T cells were identified as the lung memory T cells while the i.v.CD45.2+ CD4+ memory T cells were identified as the blood memory T cells.
Statistical Analyses
Statistical analyses were performed using Prism 8.0 (GraphPad). Differences were deemed statistically significant if the p value or FDR q value was ≤0.05. Each figure legend communicates the number of mice used per experiment, the number of experiment replicates performed and the statistical tests used to make comparisons. For all figures, data are represented as mean ± SEM.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Original data generated in this study are available within the paper and associated supplementary files, with additional information available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Szabo, P. A., Miron, M. & Farber, D. L. Location, location, location: tissue resident memory T cells in mice and humans. Sci. Immunol. 4, eaas9673 (2019).
Masopust, D. & Soerens, A. G. Tissue-resident T cells and other resident leukocytes. Annu. Rev. Immunol. 37, 521–546 (2019).
Bluestone, J. A., Mackay, C. R., O’Shea, J. J. & Stockinger, B. The functional plasticity of T cell subsets. Nat. Rev. Immunol. 9, 811–816 (2009).
Krausgruber, T. et al. Structural cells are key regulators of organ-specific immune responses. Nature 583, 296–302 (2020).
O’Shea, J. J. & Paul, W. E. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science 327, 1098–1102 (2010).
Harrison, O. J. et al. Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science 363, eaat6280 (2019).
Hondowicz, B. D. et al. Interleukin-2-dependent allergen-specific tissue-resident memory cells drive asthma. Immunity. 44, 155–166 (2016).
Boyman, O. et al. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J. Exp. Med. 199, 731–736 (2004).
Kleinschek, M. A. et al. Circulating and gut-resident human Th17 cells express CD161 and promote intestinal inflammation. J. Exp. Med. 206, 525–534 (2009).
DuPage, M. & Bluestone, J. A. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat. Rev. Immunol. 16, 149–163 (2016).
Shenoy, A. T. et al. Lung CD4(+) resident memory T cells remodel epithelial responses to accelerate neutrophil recruitment during pneumonia. Mucosal. Immunol. 13, 334–343 (2020).
Wein, A. N. et al. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J. Exp. Med. 216, 2748–2762 (2019).
Low, J. S. et al. Tissue-resident memory T cell reactivation by diverse antigen-presenting cells imparts distinct functional responses. J. Exp. Med. 217, e20192291 (2020).
Cunningham, A. C. et al. Constitutive expression of MHC and adhesion molecules by alveolar epithelial cells (type II pneumocytes) isolated from human lung and comparison with immunocytochemical findings. J. Cell. Sci. 107, 443–449 (1994).
Biton, M. et al. T helper cell cytokines modulate intestinal stem cell renewal and differentiation. Cell 175, 1307–1320.e1322 (2018).
Koyama, M. et al. MHC class II antigen presentation by the intestinal epithelium initiates graft-versus-host disease and is influenced by the microbiota. Immunity 51, 885–898.e887 (2019).
Tamoutounour, S. et al. Keratinocyte-intrinsic MHCII expression controls microbiota-induced Th1 cell responses. Proc. Natl. Acad. Sci. USA. 116, 23643–23652 (2019).
Weiser, J. N., Ferreira, D. M. & Paton, J. C. Streptococcus pneumoniae: transmission, colonization and invasion. Nat. Rev. Microbiol. 16, 355–367 (2018).
Smith, N. M. et al. Regionally compartmentalized resident memory T cells mediate naturally acquired protection against pneumococcal pneumonia. Mucosal. Immunol. 11, 220–235 (2018).
Lo, B., Hansen, S., Evans, K., Heath, J. K. & Wright, J. R. Alveolar epithelial type II cells induce T cell tolerance to specific antigen. J. Immunol. 180, 881–888 (2008).
Salwig, I. et al. Bronchioalveolar stem cells are a main source for regeneration of distal lung epithelia in vivo. EMBO J. 38, e102099 (2019).
Kim, C. F. et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 121, 823–835 (2005).
Kathiriya, J. J., Brumwell, A. N., Jackson, J. R., Tang, X. & Chapman, H. A. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem Cell 26, 346–358.e344 (2020).
Hashimoto, K., Joshi, S. K. & Koni, P. A. A conditional null allele of the major histocompatibility IA-beta chain gene. Genesis. 32, 152–153 (2002).
Taniguchi, H. et al. A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron. 71, 995–1013 (2011).
Belkina, A. C. et al. Automated optimized parameters for T-distributed stochastic neighbor embedding improve visualization and analysis of large datasets. Nat. Commun. 10, 5415 (2019).
Levine, J. H. et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis Cell 162, 184–197 (2015).
Rawlins, E. L. et al. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell 4, 525–534 (2009).
Amezcua Vesely, M. C. et al. Effector TH17 cells give rise to long-lived TRM cells that are essential for an immediate response against bacterial infection. Cell 178, 1176–1188.e1115 (2019).
Anderson, K. G. et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 9, 209–222 (2014).
Ichikawa, T. et al. CD103(hi) Treg cells constrain lung fibrosis induced by CD103(lo) tissue-resident pathogenic CD4 T cells. Nat. Immunol. 20, 1469–1480 (2019).
Levine, A. G. et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 546, 421–425 (2017).
Zimmermann, J. et al. T-bet expression by Th cells promotes type 1 inflammation but is dispensable for colitis. Mucosal. Immunol. 9, 1487–1499 (2016).
Gibney, G. T., Weiner, L. M. & Atkins, M. B. Predictive biomarkers for checkpoint inhibitor-based immunotherapy. Lancet. Oncol. 17, e542–e551 (2016).
Tanizaki, J. et al. Peripheral Blood Biomarkers Associated with Clinical Outcome in Non-Small Cell Lung Cancer Patients Treated with Nivolumab. J. Thorac. Oncol. 13, 97–105 (2018).
Prelaj, A. et al. Predictive biomarkers of response for immune checkpoint inhibitors in non-small-cell lung cancer. Eur. J. Cancer 106, 144–159 (2019).
Hopkins, A. M. et al. Predicting response and toxicity to immune checkpoint inhibitors using routinely available blood and clinical markers. Br. J. Cancer 117, 913–920 (2017).
Suresh, K. et al. The alveolar immune cell landscape is dysregulated in checkpoint inhibitor pneumonitis. J. Clin. Invest. 130, 4305–4315 (2019).
Failing, J. J., Yan, Y., Porrata, L. F. & Markovic, S. N. Lymphocyte-to-monocyte ratio is associated with survival in pembrolizumab-treated metastatic melanoma patients. Melanom. Res. 27, 596–600 (2017).
Sekine, K. et al. Change in the lymphocyte-to-monocyte ratio is an early surrogate marker of the efficacy of nivolumab monotherapy in advanced non-small-cell lung cancer. Lung Cancer 124, 179–188 (2018).
Paterson, A. M. et al. The programmed death-1 ligand 1:B7-1 pathway restrains diabetogenic effector T cells in vivo. J. Immunol. 187, 1097–1105 (2011).
Keir, M. E., Freeman, G. J. & Sharpe, A. H. PD-1 regulates self-reactive CD8+ T cell responses to antigen in lymph nodes and tissues. J. Immunol. 179, 5064–5070 (2007).
Barkauskas, C. E. et al. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest. 123, 3025–3036 (2013).
Nabhan, A. N., Brownfield, D. G., Harbury, P. B., Krasnow, M. A. & Desai, T. J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 359, 1118–1123 (2018).
Zacharias, W. J. et al. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 555, 251–255 (2018).
Travaglini, K. J. et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 587, 619–625 (2020).
Basil, M. C. et al. The Cellular and Physiological Basis for Lung Repair and Regeneration: Past, Present, and Future. Cell Stem Cell 26, 482–502 (2020).
Hasegawa, K. et al. Fraction of MHCII and EpCAM expression characterizes distal lung epithelial cells for alveolar type 2 cell isolation. Respir. Res. 18, 150 (2017).
Cunningham, A. C., Zhang, J. G., Moy, J. V., Ali, S. & Kirby, J. A. A comparison of the antigen-presenting capabilities of class II MHC-expressing human lung epithelial and endothelial cells. Immunology 91, 458–463 (1997).
Debbabi, H. et al. Primary type II alveolar epithelial cells present microbial antigens to antigen-specific CD4+ T cells. Am. J. Physiol. Lung Cell Mol. Physiol. 289, L274–L279 (2005).
Gereke, M., Jung, S., Buer, J. & Bruder, D. Alveolar type II epithelial cells present antigen to CD4(+) T cells and induce Foxp3(+) regulatory T cells. Am. J. Respir. Crit. Care Med. 179, 344–355 (2009).
Tortola, L. et al. High-Dimensional T Helper Cell Profiling Reveals a Broad Diversity of Stably Committed Effector States and Uncovers Interlineage Relationships. Immunity 53, 597–613 (2020).
Wei, G. et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167 (2009).
Wei, S. C. et al. Negative Co-stimulation Constrains T Cell Differentiation by Imposing Boundaries on Possible Cell States. Immunity 50, 1084–1098 (2019). e1010.
Cohn, L., Homer, R. J., Niu, N. & Bottomly, K. T helper 1 cells and interferon gamma regulate allergic airway inflammation and mucus production. J. Exp. Med. 190, 1309–1318 (1999).
Wang, D. Y. et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol. 4, 1721–1728 (2018).
Postow, M. A., Sidlow, R. & Hellmann, M. D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 378, 158–168 (2018).
Wang, Z. et al. PD-1(hi) CD8(+) resident memory T cells balance immunity and fibrotic sequelae. Sci. Immunol. 4, eaaw1217 (2019).
Weisberg, S. P. et al. Tissue-Resident Memory T Cells Mediate Immune Homeostasis in the Human Pancreas through the PD-1/PD-L1 Pathway. Cell Rep. 29, 3916–3932.e3915 (2019).
Rowbotham, S. P. & Kim, C. F. Diverse cells at the origin of lung adenocarcinoma. Proc. Natl. Acad. Sci. U S A 111, 4745–4746 (2014).
Ratushny, V., Gober, M. D., Hick, R., Ridky, T. W. & Seykora, J. T. From keratinocyte to cancer: the pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Invest. 122, 464–472 (2012).
van der Flier, L. G. & Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 71, 241–260 (2009).
Ritprajak, P., Hashiguchi, M., Tsushima, F., Chalermsarp, N. & Azuma, M. Keratinocyte-associated B7-H1 directly regulates cutaneous effector CD8+ T cell responses. J. Immunol. 184, 4918–4925 (2010).
Wei, Y. et al. The local immune landscape determines tumor PD-L1 heterogeneity and sensitivity to therapy. J. Clin. Invest. 129, 3347–3360 (2019).
Kreisel, D. et al. Cutting edge: MHC class II expression by pulmonary nonhematopoietic cells plays a critical role in controlling local inflammatory responses. J. Immunol. 185, 3809–3813 (2010).
Johnson, D. B. et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat. Commun. 7, 10582 (2016).
Xu, Q., Tam, M. & Anderson, S. A. Fate mapping Nkx2.1-lineage cells in the mouse telencephalon. J. Comp. Neurol. 506, 16–29 (2008).
Rankin, S. A. & Zorn, A. M. Gene regulatory networks governing lung specification. J. Cell Biochem. 115, 1343–1350 (2014).
Hawkins, F. & Kotton, D. N. Embryonic and induced pluripotent stem cells for lung regeneration. Ann. Am. Thorac. Soc. 12(Suppl 1.), S50–S53 (2015).
Rawlins, E. L. & Perl, A. K. The a“MAZE“ing world of lung-specific transgenic mice. Am. J. Respir. Cell Mol. Biol. 46, 269–282 (2012).
Acknowledgements
We thank Riley Pihl and the Boston University School of Medicine Flow Cytometry Core Facility (BU-FCCF) for assistance with flow cytometry and FACS sorting. We thank Dr. Esther Bullitt for assistance with electron microscopy and Dr. Mary Williams for her expertize with electron microscopy of LECs. We would also like to thank Dr. Jason Rock, Dr. Anna Engler and Hanne Richardson for advice with tamoxifen procedures and Cheryl Spencer for help with acquisition of consented human lung samples. Finally, we would like to thank all the consenting donors for their generosity. This work was supported by NIH grants including HL147397 to EIA, HL142199 to KAB, HL147461 to FTK, HL136725 to MRJ, GM120060 and HL111449 to LJQ, AI115053, HL135756, and HL137081 to JPM and T32 HL007035 for support of trainees in addition to support by the German Research Foundation (DFG) Clinical Research Group KFO309 TP08, the Excellence Cluster Cardio-Pulmonary Institute (CPI), and the German Center for Lung Research (DLZ) to TB.
Author information
Authors and Affiliations
Contributions
A.T.S. and J.P.M. conceived the project, designed the experiments and supervised the study. A.T.S. performed experiments and analyzed the data with assistance from C.L.D.A., E.I.A., K.A.B., F.T.K., A.R., N.S.E., A.M.S., I.M.C.M., B.R.T., A.H., W.N.G. Experiments with BASC-reporter mice were performed by I.S. with guidance from T.B. A.C.B. provided guidance with multispectral cytometry and performed computational analyses including the Phenograph clustering and optSNE visualization on the data. H.K., T.B., M.R.J., L.J.Q., A.C.B. and J.P.M. contributed resources used in this study. A.T.S. and J.P.M. wrote the manuscript, which was edited and approved by C.L.D.A., E.I.A., K.A.B., I.S., F.T.K., A.R., N.S.E, A.M.S., I.M.C.M., B.R.T., A.H., W.N.G., H.K., T.B., M.R.J., L.J.Q. and A.C.B.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Elizabeth Mellins, Kingston Mills and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shenoy, A.T., Lyon De Ana, C., Arafa, E.I. et al. Antigen presentation by lung epithelial cells directs CD4+ TRM cell function and regulates barrier immunity. Nat Commun 12, 5834 (2021). https://doi.org/10.1038/s41467-021-26045-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-26045-w
This article is cited by
-
Single-cell division tracing and transcriptomics reveal cell types and differentiation paths in the regenerating lung
Nature Communications (2024)
-
Convergence of YAP/TAZ, TEAD and TP63 activity is associated with bronchial premalignant severity and progression
Journal of Experimental & Clinical Cancer Research (2023)
-
CD4+ T cell memory
Nature Immunology (2023)
-
Cationic crosslinked carbon dots-adjuvanted intranasal vaccine induces protective immunity against Omicron-included SARS-CoV-2 variants
Nature Communications (2023)
-
Artesunate targets cellular metabolism to regulate the Th17/Treg cell balance
Inflammation Research (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
